






Abstract
The application of high-throughput sequencing approaches including paired tumor/normal sampling with therapeutic intent has demonstrated that 8%-19% of pediatric CNS tumor patients harbor a germline alteration in a classical tumor predisposition gene (NF1, P53). In addition, large-scale germline sequencing studies in unselected cohorts of pediatric neuro-oncology patients have demonstrated novel candidate tumor predisposition genes (ELP1 alterations in sonic hedgehog medulloblastoma). Therefore, the possibility of an underlying tumor predisposition syndrome (TPS) should be considered in all pediatric patients diagnosed with a CNS tumor which carries critical implications including accurate prognostication, selection of optimal therapy, screening, risk reduction, and family planning. The Pediatric Cancer Working Group of the American Association for Cancer Research (AACR) recently published consensus screening recommendations for children with the most common TPS. In this review, we provide an overview of the most relevant as well as recently identified TPS associated with the most frequently encountered pediatric CNS tumors with an emphasis on pathogenesis, genetic testing, clinical features, and treatment implications.
The notion that some human cancers may have a hereditary basis arose from descriptions of cancer-prone kindreds, wherein, the spectrum of cancers was limited, and stereotypical and similar patterns reappeared in different families. The majority of cancer susceptibility genes encode tumor suppressors, where one allele is inactivated in the germline, and loss of the second allele (loss of heterozygosity [LOH]) causes tumor initiation. Less commonly, cancer susceptibility is conferred by the presence of activating mutations in growth-promoting oncogenes such as RET, including those encoding receptor tyrosine kinases (anaplastic lymphoma kinase [ALK]) and other intracellular signaling proteins.1
Traditionally, genetic testing is recommended when patients present with a particular tumor type with a known association, a significant family history, and/or physical features consistent with a specific tumor predisposition syndrome (TPS). However, with the advent of genome-wide approaches to establish a genetic diagnosis, including single-nucleotide polymorphism (SNP) arrays, multigene panels, and whole-exome/genome sequencing, the “incidental” detection of a tumor predisposing germline alteration is becoming increasingly common.2 Furthermore, the application of high-throughput sequencing approaches (including matched normal) with diagnostic/therapeutic intent in pediatric oncology, and large-scale sequencing studies has demonstrated a broader TPS spectrum than previously realized. Overall, approximately 8%-19% of pediatric CNS tumor patients harbor currently recognized germline cancer-predisposing mutations,3,4 with wide variability based on tumor type.
Our approach to TPS will be grouped by the specific tumors they cause, enabling clinicians involved in the care of patients with brain tumors to consider the appropriate differential diagnoses based on the specific tumor type (see Table 1). A brief description of the most relevant TPS associated with pediatric CNS tumors with an emphasis on pathogenesis, genetic background, and clinical approach will be provided in this chapter.
List of Pediatric Central Nervous System Tumor Types and Associated Tumor Predisposition Syndromes
| Tumor Type | Associated Syndromes | Genotype-Phenotype Correlations |
|---|---|---|
| Glioma | 1. Neurofibromatosis type 1 (NF1) 2. Constitutional mismatch repair deficiency (CMMRD) 3. Li-Fraumeni syndrome (LFS) | Higher risk with variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543-909). Lower risk was associated with NF1 variants occurring in HEAT-like repeat regions (HLR, residues 1825-2428). Higher risk with MSH6/PMS2 mutations. Individuals with deletions or truncating mutations exhibit a more severe phenotype, with brain tumors developing in the first decade. None known |
| Medulloblastoma (MB) | 1. Gorlin syndrome (GS) 2. Familial adenomatous polyposis coli 3. Fanconi anemia (FA) 4. Rubinstein-Taybi syndrome (related to germline mutations in CREBBP) 5. LFS | Lower risk of MB in PTCH1 mutation-positive individuals. PTCH1 alterations more frequent in iSHH-II compared to iSHH-I. 20-fold higher risk in SUFU mutation-positive individuals. SUFU alterations significantly enriched in iSHH-I which are associated with inferior outcomes. Patients develop predominantly WNT subgroup MB and share the very favorable prognosis of CTNNB1-mutated MB. Patients with mutations in codons 686-1217 of APC gene have a 13-fold increased risk of developing MBs. Brain tumors including MB and glioma were primarily found in patients carrying mutations in both copies of the FANCD1/BRCA2 gene or FANCN/PALB2. None known None known |
| Choroid plexus carcinoma | LFS | None known |
| Vestibular schwannoma | Neurofibromatosis type 2 (NF2) | Truncation mutations: severe phenotype with earlier onset, increased frequency of meningiomas, and worse disease-related mortality Missense mutation, large deletions, NF2 mosaic: milder form of NF2 |
| Meningioma | NF2 LFS Cowden syndrome Gorlin syndrome (SUFU mutations) Multiple endocrine neoplasia (MEN), Schwannomatosis and meningiomatosis associated with alterations in SWI/SNF family members (SMARCB1 and SMARCE1) | Incidence of meningiomas lower in patients with mutations in the 3′ region relative to mutations in the 5′ region of the NF2 gene. None known None known None known None known None known |
| Atypical teratoid rhabdoid tumor | Rhabdoid tumor predisposition syndrome (RTPS) | SMARCA4 is associated with worse prognosis compared to SMARCB1. |
| Pineoblastoma | Hereditary retinoblastoma DICER syndrome | None known None known |
| Subependymal giant cell astrocytoma | Tuberous sclerosis (TS) | None known |
| Hemangioblastoma | von Hippel-Landau syndrome | None known |
| Tumor Type | Associated Syndromes | Genotype-Phenotype Correlations |
|---|---|---|
| Glioma | 1. Neurofibromatosis type 1 (NF1) 2. Constitutional mismatch repair deficiency (CMMRD) 3. Li-Fraumeni syndrome (LFS) | Higher risk with variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543-909). Lower risk was associated with NF1 variants occurring in HEAT-like repeat regions (HLR, residues 1825-2428). Higher risk with MSH6/PMS2 mutations. Individuals with deletions or truncating mutations exhibit a more severe phenotype, with brain tumors developing in the first decade. None known |
| Medulloblastoma (MB) | 1. Gorlin syndrome (GS) 2. Familial adenomatous polyposis coli 3. Fanconi anemia (FA) 4. Rubinstein-Taybi syndrome (related to germline mutations in CREBBP) 5. LFS | Lower risk of MB in PTCH1 mutation-positive individuals. PTCH1 alterations more frequent in iSHH-II compared to iSHH-I. 20-fold higher risk in SUFU mutation-positive individuals. SUFU alterations significantly enriched in iSHH-I which are associated with inferior outcomes. Patients develop predominantly WNT subgroup MB and share the very favorable prognosis of CTNNB1-mutated MB. Patients with mutations in codons 686-1217 of APC gene have a 13-fold increased risk of developing MBs. Brain tumors including MB and glioma were primarily found in patients carrying mutations in both copies of the FANCD1/BRCA2 gene or FANCN/PALB2. None known None known |
| Choroid plexus carcinoma | LFS | None known |
| Vestibular schwannoma | Neurofibromatosis type 2 (NF2) | Truncation mutations: severe phenotype with earlier onset, increased frequency of meningiomas, and worse disease-related mortality Missense mutation, large deletions, NF2 mosaic: milder form of NF2 |
| Meningioma | NF2 LFS Cowden syndrome Gorlin syndrome (SUFU mutations) Multiple endocrine neoplasia (MEN), Schwannomatosis and meningiomatosis associated with alterations in SWI/SNF family members (SMARCB1 and SMARCE1) | Incidence of meningiomas lower in patients with mutations in the 3′ region relative to mutations in the 5′ region of the NF2 gene. None known None known None known None known None known |
| Atypical teratoid rhabdoid tumor | Rhabdoid tumor predisposition syndrome (RTPS) | SMARCA4 is associated with worse prognosis compared to SMARCB1. |
| Pineoblastoma | Hereditary retinoblastoma DICER syndrome | None known None known |
| Subependymal giant cell astrocytoma | Tuberous sclerosis (TS) | None known |
| Hemangioblastoma | von Hippel-Landau syndrome | None known |
List of Pediatric Central Nervous System Tumor Types and Associated Tumor Predisposition Syndromes
| Tumor Type | Associated Syndromes | Genotype-Phenotype Correlations |
|---|---|---|
| Glioma | 1. Neurofibromatosis type 1 (NF1) 2. Constitutional mismatch repair deficiency (CMMRD) 3. Li-Fraumeni syndrome (LFS) | Higher risk with variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543-909). Lower risk was associated with NF1 variants occurring in HEAT-like repeat regions (HLR, residues 1825-2428). Higher risk with MSH6/PMS2 mutations. Individuals with deletions or truncating mutations exhibit a more severe phenotype, with brain tumors developing in the first decade. None known |
| Medulloblastoma (MB) | 1. Gorlin syndrome (GS) 2. Familial adenomatous polyposis coli 3. Fanconi anemia (FA) 4. Rubinstein-Taybi syndrome (related to germline mutations in CREBBP) 5. LFS | Lower risk of MB in PTCH1 mutation-positive individuals. PTCH1 alterations more frequent in iSHH-II compared to iSHH-I. 20-fold higher risk in SUFU mutation-positive individuals. SUFU alterations significantly enriched in iSHH-I which are associated with inferior outcomes. Patients develop predominantly WNT subgroup MB and share the very favorable prognosis of CTNNB1-mutated MB. Patients with mutations in codons 686-1217 of APC gene have a 13-fold increased risk of developing MBs. Brain tumors including MB and glioma were primarily found in patients carrying mutations in both copies of the FANCD1/BRCA2 gene or FANCN/PALB2. None known None known |
| Choroid plexus carcinoma | LFS | None known |
| Vestibular schwannoma | Neurofibromatosis type 2 (NF2) | Truncation mutations: severe phenotype with earlier onset, increased frequency of meningiomas, and worse disease-related mortality Missense mutation, large deletions, NF2 mosaic: milder form of NF2 |
| Meningioma | NF2 LFS Cowden syndrome Gorlin syndrome (SUFU mutations) Multiple endocrine neoplasia (MEN), Schwannomatosis and meningiomatosis associated with alterations in SWI/SNF family members (SMARCB1 and SMARCE1) | Incidence of meningiomas lower in patients with mutations in the 3′ region relative to mutations in the 5′ region of the NF2 gene. None known None known None known None known None known |
| Atypical teratoid rhabdoid tumor | Rhabdoid tumor predisposition syndrome (RTPS) | SMARCA4 is associated with worse prognosis compared to SMARCB1. |
| Pineoblastoma | Hereditary retinoblastoma DICER syndrome | None known None known |
| Subependymal giant cell astrocytoma | Tuberous sclerosis (TS) | None known |
| Hemangioblastoma | von Hippel-Landau syndrome | None known |
| Tumor Type | Associated Syndromes | Genotype-Phenotype Correlations |
|---|---|---|
| Glioma | 1. Neurofibromatosis type 1 (NF1) 2. Constitutional mismatch repair deficiency (CMMRD) 3. Li-Fraumeni syndrome (LFS) | Higher risk with variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543-909). Lower risk was associated with NF1 variants occurring in HEAT-like repeat regions (HLR, residues 1825-2428). Higher risk with MSH6/PMS2 mutations. Individuals with deletions or truncating mutations exhibit a more severe phenotype, with brain tumors developing in the first decade. None known |
| Medulloblastoma (MB) | 1. Gorlin syndrome (GS) 2. Familial adenomatous polyposis coli 3. Fanconi anemia (FA) 4. Rubinstein-Taybi syndrome (related to germline mutations in CREBBP) 5. LFS | Lower risk of MB in PTCH1 mutation-positive individuals. PTCH1 alterations more frequent in iSHH-II compared to iSHH-I. 20-fold higher risk in SUFU mutation-positive individuals. SUFU alterations significantly enriched in iSHH-I which are associated with inferior outcomes. Patients develop predominantly WNT subgroup MB and share the very favorable prognosis of CTNNB1-mutated MB. Patients with mutations in codons 686-1217 of APC gene have a 13-fold increased risk of developing MBs. Brain tumors including MB and glioma were primarily found in patients carrying mutations in both copies of the FANCD1/BRCA2 gene or FANCN/PALB2. None known None known |
| Choroid plexus carcinoma | LFS | None known |
| Vestibular schwannoma | Neurofibromatosis type 2 (NF2) | Truncation mutations: severe phenotype with earlier onset, increased frequency of meningiomas, and worse disease-related mortality Missense mutation, large deletions, NF2 mosaic: milder form of NF2 |
| Meningioma | NF2 LFS Cowden syndrome Gorlin syndrome (SUFU mutations) Multiple endocrine neoplasia (MEN), Schwannomatosis and meningiomatosis associated with alterations in SWI/SNF family members (SMARCB1 and SMARCE1) | Incidence of meningiomas lower in patients with mutations in the 3′ region relative to mutations in the 5′ region of the NF2 gene. None known None known None known None known None known |
| Atypical teratoid rhabdoid tumor | Rhabdoid tumor predisposition syndrome (RTPS) | SMARCA4 is associated with worse prognosis compared to SMARCB1. |
| Pineoblastoma | Hereditary retinoblastoma DICER syndrome | None known None known |
| Subependymal giant cell astrocytoma | Tuberous sclerosis (TS) | None known |
| Hemangioblastoma | von Hippel-Landau syndrome | None known |
Astrocytoma
Neurofibromatosis Type 1 (NF1)
Epidemiology and clinical features.—NF1 is characterized by an autosomal dominant (AD) inheritance pattern, with 100% penetrance and significant clinical variability both between families and within families.5 Incidence is 1:3000 individuals, half of which are considered familial and half de novo gene mutations. Affected individuals develop a combination of dermatologic, skeletal, ophthalmic, and neurologic findings at typical ages of onset and 90% of affected children are clinically diagnosed by the age of 7 (diagnostic criteria in Table 2).6 However, given the overlap of clinical features with constitutional mismatch repair deficiency (CMMRD) and Legius syndrome (germline SPRED1 mutation), genetic diagnostic confirmation is of value for therapeutic considerations and screening of family members, as well as preimplantation genetic diagnosis or prenatal testing.7 Mutational analysis of the NF1 gene can identify 95% of causative mutations in patients with classic NF1.8
Diagnostic Criteria for Tumor Predisposition Syndromes
| Tumor Predisposition Syndrome | Diagnostic Criteria |
|---|---|
| Neurofibromatosis type 1 (NF1) | NIH criteria6: the presence of at least two of the following clinical features is required to confirm a diagnosis of NF1: 6 or more café-au-lait macules >5 mm in prepubertal individuals or >15 mm in post-pubertal individuals 2 or more neurofibromas of any type, or 1 plexiform neurofibroma Axillary and/or inguinal freckling Optic nerve glioma 2 or more small elevated hamartomas of the iris (Lisch nodules) A distinct osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortex with or without pseudoarthrosis A first-degree relative with NF1 according to the above criteria |
| Constitutional mismatch repair deficiency (CMMRD) syndrome | Indication for CMMRD testing in a cancer patient ≥3 points Malignancies/premalignancies: one is mandatory; if more than one is present in the patient, add the points9 Carcinoma from the LS spectruma at age <25 years—3 points Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 years—3 points WHO grade III or IV glioma at age <25 years—2 points NHL of T-cell lineage or sPNET at age <18 years—2 points Any malignancy at age <18 years—1 point Additional features: optional; if more than one of the following is present, add the points Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations >1 cm in the patient 2 points Diagnosis of LS in a first- or second-degree relative 2 points Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative 1 point A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL 2 points A sibling with any type of childhood malignancy 1 point Multiple pilomatricomas in the patient 2 points One pilomatricoma in the patient 1 point Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient 1 point Consanguineous parents 1 point Deficiency/reduced levels of IgG2/4 and/or IgA 1 point |
| Gorlin syndrome (GS) | The diagnosis of GS is made based on clinical findings using established criteria and requires the presence of two or more major criteria or one major criterion plus two or more minor criteria.10 Major criteria More than 2 basal cell carcinomas or one under the age of 20 years Odontogenic keratocysts of the jaw proven by histology Three or more palmar or plantar pits Bilamellar calcification of the falx cerebri Bifid, fused, or markedly splayed ribs First-degree relative with NBCCS Minor criteria Medulloblastoma Congenital malformations: cleft lip or palate, frontal bossing, coarse face, moderate or severe hypertelorism Other skeletal abnormalities: Sprengel deformity, marked pectus deformity, marked syndactyly of the digits Radiological abnormalities: bridging of the sella turcica, vertebral anomalies such as hemivertebrae, fusion, or elongation of the vertebral bodies, modeling defects of the hands and feet, or flame-shaped lucencies of the hands or feet Ovarian fibroma |
| Li-Fraumeni syndrome (LFS) | The risk of a TP53 mutation exceeds 20% in any individual with11: A tumor belonging to the LFS tumor spectrum (eg, soft tissue sarcoma, osteosarcoma, brain tumor, premenopausal breast cancer, adrenocortical carcinoma, leukemia, lung bronchoalveolar cancer) before age 46 years and at least one first- or second-degree relative with a LFS tumor (except breast cancer if the proband has breast cancer) before age 56 years or with multiple tumors; or Multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; or Adrenocortical carcinoma or choroid plexus tumor, regardless of family history. |
| Neurofibromatosis type 2 (NF2) | Bilateral vestibular schwannomas OR12 A first-degree relative with NF2 and: 1. A unilateral vestibular schwannoma, or 2. Two of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lens opacity Unilateral vestibular schwannoma AND any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities Multiple meningiomas AND 1. Unilateral vestibular schwannoma OR 2. Any 2 of schwannoma, glioma, neurofibroma, cataract |
| Tumor Predisposition Syndrome | Diagnostic Criteria |
|---|---|
| Neurofibromatosis type 1 (NF1) | NIH criteria6: the presence of at least two of the following clinical features is required to confirm a diagnosis of NF1: 6 or more café-au-lait macules >5 mm in prepubertal individuals or >15 mm in post-pubertal individuals 2 or more neurofibromas of any type, or 1 plexiform neurofibroma Axillary and/or inguinal freckling Optic nerve glioma 2 or more small elevated hamartomas of the iris (Lisch nodules) A distinct osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortex with or without pseudoarthrosis A first-degree relative with NF1 according to the above criteria |
| Constitutional mismatch repair deficiency (CMMRD) syndrome | Indication for CMMRD testing in a cancer patient ≥3 points Malignancies/premalignancies: one is mandatory; if more than one is present in the patient, add the points9 Carcinoma from the LS spectruma at age <25 years—3 points Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 years—3 points WHO grade III or IV glioma at age <25 years—2 points NHL of T-cell lineage or sPNET at age <18 years—2 points Any malignancy at age <18 years—1 point Additional features: optional; if more than one of the following is present, add the points Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations >1 cm in the patient 2 points Diagnosis of LS in a first- or second-degree relative 2 points Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative 1 point A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL 2 points A sibling with any type of childhood malignancy 1 point Multiple pilomatricomas in the patient 2 points One pilomatricoma in the patient 1 point Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient 1 point Consanguineous parents 1 point Deficiency/reduced levels of IgG2/4 and/or IgA 1 point |
| Gorlin syndrome (GS) | The diagnosis of GS is made based on clinical findings using established criteria and requires the presence of two or more major criteria or one major criterion plus two or more minor criteria.10 Major criteria More than 2 basal cell carcinomas or one under the age of 20 years Odontogenic keratocysts of the jaw proven by histology Three or more palmar or plantar pits Bilamellar calcification of the falx cerebri Bifid, fused, or markedly splayed ribs First-degree relative with NBCCS Minor criteria Medulloblastoma Congenital malformations: cleft lip or palate, frontal bossing, coarse face, moderate or severe hypertelorism Other skeletal abnormalities: Sprengel deformity, marked pectus deformity, marked syndactyly of the digits Radiological abnormalities: bridging of the sella turcica, vertebral anomalies such as hemivertebrae, fusion, or elongation of the vertebral bodies, modeling defects of the hands and feet, or flame-shaped lucencies of the hands or feet Ovarian fibroma |
| Li-Fraumeni syndrome (LFS) | The risk of a TP53 mutation exceeds 20% in any individual with11: A tumor belonging to the LFS tumor spectrum (eg, soft tissue sarcoma, osteosarcoma, brain tumor, premenopausal breast cancer, adrenocortical carcinoma, leukemia, lung bronchoalveolar cancer) before age 46 years and at least one first- or second-degree relative with a LFS tumor (except breast cancer if the proband has breast cancer) before age 56 years or with multiple tumors; or Multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; or Adrenocortical carcinoma or choroid plexus tumor, regardless of family history. |
| Neurofibromatosis type 2 (NF2) | Bilateral vestibular schwannomas OR12 A first-degree relative with NF2 and: 1. A unilateral vestibular schwannoma, or 2. Two of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lens opacity Unilateral vestibular schwannoma AND any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities Multiple meningiomas AND 1. Unilateral vestibular schwannoma OR 2. Any 2 of schwannoma, glioma, neurofibroma, cataract |
Abbreviations: LS, Lynch syndrome; NBCCS, nevoid basal cell carcinoma syndrome; NHL, non-Hodgkin lymphomas; sPNET, supratentorial primitive neuroectodermal tumors.
aColorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, bladder carcinoma.
Diagnostic Criteria for Tumor Predisposition Syndromes
| Tumor Predisposition Syndrome | Diagnostic Criteria |
|---|---|
| Neurofibromatosis type 1 (NF1) | NIH criteria6: the presence of at least two of the following clinical features is required to confirm a diagnosis of NF1: 6 or more café-au-lait macules >5 mm in prepubertal individuals or >15 mm in post-pubertal individuals 2 or more neurofibromas of any type, or 1 plexiform neurofibroma Axillary and/or inguinal freckling Optic nerve glioma 2 or more small elevated hamartomas of the iris (Lisch nodules) A distinct osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortex with or without pseudoarthrosis A first-degree relative with NF1 according to the above criteria |
| Constitutional mismatch repair deficiency (CMMRD) syndrome | Indication for CMMRD testing in a cancer patient ≥3 points Malignancies/premalignancies: one is mandatory; if more than one is present in the patient, add the points9 Carcinoma from the LS spectruma at age <25 years—3 points Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 years—3 points WHO grade III or IV glioma at age <25 years—2 points NHL of T-cell lineage or sPNET at age <18 years—2 points Any malignancy at age <18 years—1 point Additional features: optional; if more than one of the following is present, add the points Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations >1 cm in the patient 2 points Diagnosis of LS in a first- or second-degree relative 2 points Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative 1 point A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL 2 points A sibling with any type of childhood malignancy 1 point Multiple pilomatricomas in the patient 2 points One pilomatricoma in the patient 1 point Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient 1 point Consanguineous parents 1 point Deficiency/reduced levels of IgG2/4 and/or IgA 1 point |
| Gorlin syndrome (GS) | The diagnosis of GS is made based on clinical findings using established criteria and requires the presence of two or more major criteria or one major criterion plus two or more minor criteria.10 Major criteria More than 2 basal cell carcinomas or one under the age of 20 years Odontogenic keratocysts of the jaw proven by histology Three or more palmar or plantar pits Bilamellar calcification of the falx cerebri Bifid, fused, or markedly splayed ribs First-degree relative with NBCCS Minor criteria Medulloblastoma Congenital malformations: cleft lip or palate, frontal bossing, coarse face, moderate or severe hypertelorism Other skeletal abnormalities: Sprengel deformity, marked pectus deformity, marked syndactyly of the digits Radiological abnormalities: bridging of the sella turcica, vertebral anomalies such as hemivertebrae, fusion, or elongation of the vertebral bodies, modeling defects of the hands and feet, or flame-shaped lucencies of the hands or feet Ovarian fibroma |
| Li-Fraumeni syndrome (LFS) | The risk of a TP53 mutation exceeds 20% in any individual with11: A tumor belonging to the LFS tumor spectrum (eg, soft tissue sarcoma, osteosarcoma, brain tumor, premenopausal breast cancer, adrenocortical carcinoma, leukemia, lung bronchoalveolar cancer) before age 46 years and at least one first- or second-degree relative with a LFS tumor (except breast cancer if the proband has breast cancer) before age 56 years or with multiple tumors; or Multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; or Adrenocortical carcinoma or choroid plexus tumor, regardless of family history. |
| Neurofibromatosis type 2 (NF2) | Bilateral vestibular schwannomas OR12 A first-degree relative with NF2 and: 1. A unilateral vestibular schwannoma, or 2. Two of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lens opacity Unilateral vestibular schwannoma AND any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities Multiple meningiomas AND 1. Unilateral vestibular schwannoma OR 2. Any 2 of schwannoma, glioma, neurofibroma, cataract |
| Tumor Predisposition Syndrome | Diagnostic Criteria |
|---|---|
| Neurofibromatosis type 1 (NF1) | NIH criteria6: the presence of at least two of the following clinical features is required to confirm a diagnosis of NF1: 6 or more café-au-lait macules >5 mm in prepubertal individuals or >15 mm in post-pubertal individuals 2 or more neurofibromas of any type, or 1 plexiform neurofibroma Axillary and/or inguinal freckling Optic nerve glioma 2 or more small elevated hamartomas of the iris (Lisch nodules) A distinct osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortex with or without pseudoarthrosis A first-degree relative with NF1 according to the above criteria |
| Constitutional mismatch repair deficiency (CMMRD) syndrome | Indication for CMMRD testing in a cancer patient ≥3 points Malignancies/premalignancies: one is mandatory; if more than one is present in the patient, add the points9 Carcinoma from the LS spectruma at age <25 years—3 points Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 years—3 points WHO grade III or IV glioma at age <25 years—2 points NHL of T-cell lineage or sPNET at age <18 years—2 points Any malignancy at age <18 years—1 point Additional features: optional; if more than one of the following is present, add the points Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations >1 cm in the patient 2 points Diagnosis of LS in a first- or second-degree relative 2 points Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative 1 point A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL 2 points A sibling with any type of childhood malignancy 1 point Multiple pilomatricomas in the patient 2 points One pilomatricoma in the patient 1 point Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient 1 point Consanguineous parents 1 point Deficiency/reduced levels of IgG2/4 and/or IgA 1 point |
| Gorlin syndrome (GS) | The diagnosis of GS is made based on clinical findings using established criteria and requires the presence of two or more major criteria or one major criterion plus two or more minor criteria.10 Major criteria More than 2 basal cell carcinomas or one under the age of 20 years Odontogenic keratocysts of the jaw proven by histology Three or more palmar or plantar pits Bilamellar calcification of the falx cerebri Bifid, fused, or markedly splayed ribs First-degree relative with NBCCS Minor criteria Medulloblastoma Congenital malformations: cleft lip or palate, frontal bossing, coarse face, moderate or severe hypertelorism Other skeletal abnormalities: Sprengel deformity, marked pectus deformity, marked syndactyly of the digits Radiological abnormalities: bridging of the sella turcica, vertebral anomalies such as hemivertebrae, fusion, or elongation of the vertebral bodies, modeling defects of the hands and feet, or flame-shaped lucencies of the hands or feet Ovarian fibroma |
| Li-Fraumeni syndrome (LFS) | The risk of a TP53 mutation exceeds 20% in any individual with11: A tumor belonging to the LFS tumor spectrum (eg, soft tissue sarcoma, osteosarcoma, brain tumor, premenopausal breast cancer, adrenocortical carcinoma, leukemia, lung bronchoalveolar cancer) before age 46 years and at least one first- or second-degree relative with a LFS tumor (except breast cancer if the proband has breast cancer) before age 56 years or with multiple tumors; or Multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; or Adrenocortical carcinoma or choroid plexus tumor, regardless of family history. |
| Neurofibromatosis type 2 (NF2) | Bilateral vestibular schwannomas OR12 A first-degree relative with NF2 and: 1. A unilateral vestibular schwannoma, or 2. Two of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lens opacity Unilateral vestibular schwannoma AND any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities Multiple meningiomas AND 1. Unilateral vestibular schwannoma OR 2. Any 2 of schwannoma, glioma, neurofibroma, cataract |
Abbreviations: LS, Lynch syndrome; NBCCS, nevoid basal cell carcinoma syndrome; NHL, non-Hodgkin lymphomas; sPNET, supratentorial primitive neuroectodermal tumors.
aColorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, bladder carcinoma.
Optic pathway gliomas (OPGs) and brainstem low-grade gliomas (LGGs) are the most common intracranial neoplasms found in NF1, and an increased incidence of high-grade gliomas (HGGs) has been noted as well (50-fold higher when compared to the general population). Nearly a third of children with OPG have germline mutations in NF1. Conversely, OPGs are detectable in approximately 15% of NF1 patients, usually before the age of 7 years and bilateral OPGs are detected exclusively in NF1. Most of these tumors are WHO grade I pilocytic astrocytomas (PA), although most patients are diagnosed based on imaging without a biopsy. Patients with NF1 also often exhibit multiple T2 hyperintense lesions, mainly in the basal ganglia and brainstem which are referred to as unidentified bright objects (UBOs) and undergo spontaneous regression. Brainstem gliomas present in late childhood (mean age 7 years), exhibit mass effect on T2 and increased signal on T1 weighted images (unlike UBOs), are more indolent than sporadic brainstem gliomas, and may regress spontaneously. NF1-associated high-grade gliomas (NF1-HGG) are primarily diagnosed in adolescents and adults, mostly in hemispheric locations. They may arise de novo or through transformation of LGGs.13
Molecular genetics and pathogenesis.—The NF1 locus maps to chromosome 17q11.2 and encodes neurofibromin, a protein that harbors a GTPase-activating protein domain that functions to silence RAS in its activated form. Loss of neurofibromin due to biallelic inactivation of the NF1 gene leads to deregulated RAS activity, which initiates downstream signaling by activating the RAF/MEK/ERK and the Akt/mTOR pathways.14 About 90% of NF1 mutations are point mutations, with no definitive genotype-phenotype associations identified. A higher risk for developing OPGs has been reported in NF1 patients harboring variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543-909) while a lower risk was associated with NF1 variants occurring in HEAT-like repeat regions (HLR, residues 1825-2428).15
A comprehensive molecular profiling study in NF1 patients using whole-exome sequencing (WES) of tumor and matched blood germline DNA demonstrated that LGGs have a low mutational burden and primarily exhibit LOH in the NF1 region along with alterations in genes (FGFR1, PIK3CA) encoding component of the mitogen-activated protein (MAP) kinase pathway.16
The mutational landscape of gliomas in adolescents and adults is more complex, with tumors histologically diagnosed as PA, but classified as anaplastic astrocytomas with piloid features by DNA methylation profiling.17 NF1-associated HGGs share many of the same molecular alterations as sporadic HGGs, including mutations in TP53, CDKN2A, ATRX, PI3K, and genes involved in transcription/chromatin regulation.16,18
Clinical implications and treatment.—Patients with asymptomatic OPG or other LGG are managed conservatively, with imaging surveillance and close clinical/ophthalmological follow-up (American Association for Cancer Research [AACR] surveillance guidelines in Table 3).19 Surveillance neuroimaging in asymptomatic children with NF1 has not been shown to reduce the incidence of visual loss, with MRI recommended only for patients with ophthalmological findings suggestive of an OPG, such as proptosis, optic disc pallor, or vision loss.19
Surveillance Guidelines for Patients Diagnosed With a CNS Tumor Predisposition Syndrome
| Tumor Predisposition Syndrome | Tumor Surveillance Guidelines |
|---|---|
| Neurofibromatosis type 1 | Semi-annual ophthalmologic examination between ages of 2 and 8 years and annually thereafter until 20 years of age (screen for OPG).19 Ophthalmologic assessment should include measures of visual acuity and color vision; visual evoked potentials are not recommended. Optical coherence tomography (OCT) may be considered as an objective measure of axonal integrity/axonal loss and thickness of retinal nerve fiber layer. MRI brain/orbits if vision loss on 2 consecutive eye exams. Review of growth and sexual development to identify precocious puberty (ie, hypothalamic involvement). |
| Constitutional mismatch repair deficiency syndrome | Brain MRIs for CNS malignancies, CBCs for leukemia, and abdominal ultrasounds for lymphoma screening starting in infancy and every 6 months thereafter.20 Endoscopic and colonoscopic surveillance starting at 6 years of age and annually thereafter to detect gastrointestinal tumors. |
| Gorlin syndrome | Regular dermatology exams are recommended to monitor for the development of basal cell carcinomas.21 Annual panoramic radiographs of the jaw are recommended starting at age 8. Screening X-rays have been recommended to look for rib anomalies, calcification of the falx, and calcified ovarian fibromas. SUFU mutation carriers: every 4-month brain MRI through age 3 years and every 6-month brain MRI until the age of 5 years for medulloblastoma screening. PTCH1 mutation carriers: no radiographic screening recommended. |
| Li-Fraumeni syndrome | Brain tumor: annual brain MRI (first MRI with contrast; thereafter without contrast if previous MRI normal and no new abnormality).22 Complete physical examination every 3-4 months, including blood pressure, anthropometric measurements plotted on a growth curve (with attention to rapid acceleration in weight or height), Cushingoid appearance, signs of virilization (pubic hair, axillary moisture, adult body odor, androgenic hair loss, clitoromegaly, or penile growth), and full neurologic assessment. ACC: US of abdomen and pelvis every 3-4 months. In case of unsatisfactory US, blood tests may be performed every 3-4. months: total testosterone, dehydroepiandrosterone sulfate, and androstenedione. Soft tissue and bone sarcoma: Annual WBMRI. |
| Neurofibromatosis type 2 | AACR surveillance guidelines23: 1. Annual history and physical exam (including audiology with measurement of pure-tone thresholds and Word Recognition Scores). 2. Annual (consider twice yearly in first year since diagnosis or signs of rapid growth) brain MRI starting at 10 years of age. Screening may begin earlier in patients with high-risk genotypes or symptomatic diagnoses. 3. If baseline imaging shows no characteristic sites of involvement, reduce the frequency of screening to every 2 years. 4. Protocols should include high-resolution (1-3 mm slice thickness) imaging through the internal auditory meatus. 5. Surveillance spinal MRI is recommended at 24- to 36-month intervals beginning at 10 years of age. 6. Whole-body MRI may be obtained. |
| Rhabdoid tumor predisposition syndrome | SMARCB1 truncating alteration—MRI brain every 3 months to age 5 years.21 Consider WBMRI to age 5 years. Ultrasound abdomen every 3 months. SMARCB1 missense alteration—no screening recommended. SMARCA4 alteration—no brain imaging, abdominal ultrasound every 6 months may be considered (ovary). |
| Tumor Predisposition Syndrome | Tumor Surveillance Guidelines |
|---|---|
| Neurofibromatosis type 1 | Semi-annual ophthalmologic examination between ages of 2 and 8 years and annually thereafter until 20 years of age (screen for OPG).19 Ophthalmologic assessment should include measures of visual acuity and color vision; visual evoked potentials are not recommended. Optical coherence tomography (OCT) may be considered as an objective measure of axonal integrity/axonal loss and thickness of retinal nerve fiber layer. MRI brain/orbits if vision loss on 2 consecutive eye exams. Review of growth and sexual development to identify precocious puberty (ie, hypothalamic involvement). |
| Constitutional mismatch repair deficiency syndrome | Brain MRIs for CNS malignancies, CBCs for leukemia, and abdominal ultrasounds for lymphoma screening starting in infancy and every 6 months thereafter.20 Endoscopic and colonoscopic surveillance starting at 6 years of age and annually thereafter to detect gastrointestinal tumors. |
| Gorlin syndrome | Regular dermatology exams are recommended to monitor for the development of basal cell carcinomas.21 Annual panoramic radiographs of the jaw are recommended starting at age 8. Screening X-rays have been recommended to look for rib anomalies, calcification of the falx, and calcified ovarian fibromas. SUFU mutation carriers: every 4-month brain MRI through age 3 years and every 6-month brain MRI until the age of 5 years for medulloblastoma screening. PTCH1 mutation carriers: no radiographic screening recommended. |
| Li-Fraumeni syndrome | Brain tumor: annual brain MRI (first MRI with contrast; thereafter without contrast if previous MRI normal and no new abnormality).22 Complete physical examination every 3-4 months, including blood pressure, anthropometric measurements plotted on a growth curve (with attention to rapid acceleration in weight or height), Cushingoid appearance, signs of virilization (pubic hair, axillary moisture, adult body odor, androgenic hair loss, clitoromegaly, or penile growth), and full neurologic assessment. ACC: US of abdomen and pelvis every 3-4 months. In case of unsatisfactory US, blood tests may be performed every 3-4. months: total testosterone, dehydroepiandrosterone sulfate, and androstenedione. Soft tissue and bone sarcoma: Annual WBMRI. |
| Neurofibromatosis type 2 | AACR surveillance guidelines23: 1. Annual history and physical exam (including audiology with measurement of pure-tone thresholds and Word Recognition Scores). 2. Annual (consider twice yearly in first year since diagnosis or signs of rapid growth) brain MRI starting at 10 years of age. Screening may begin earlier in patients with high-risk genotypes or symptomatic diagnoses. 3. If baseline imaging shows no characteristic sites of involvement, reduce the frequency of screening to every 2 years. 4. Protocols should include high-resolution (1-3 mm slice thickness) imaging through the internal auditory meatus. 5. Surveillance spinal MRI is recommended at 24- to 36-month intervals beginning at 10 years of age. 6. Whole-body MRI may be obtained. |
| Rhabdoid tumor predisposition syndrome | SMARCB1 truncating alteration—MRI brain every 3 months to age 5 years.21 Consider WBMRI to age 5 years. Ultrasound abdomen every 3 months. SMARCB1 missense alteration—no screening recommended. SMARCA4 alteration—no brain imaging, abdominal ultrasound every 6 months may be considered (ovary). |
Abbreviations: AACR, American Association for Cancer Research; ACC, adrenocortical carcinoma; CBC, complete blood count; OPG, optic pathway gliomas; US, ultrasound; WBMRI, whole body magnetic resonance imaging.
Surveillance Guidelines for Patients Diagnosed With a CNS Tumor Predisposition Syndrome
| Tumor Predisposition Syndrome | Tumor Surveillance Guidelines |
|---|---|
| Neurofibromatosis type 1 | Semi-annual ophthalmologic examination between ages of 2 and 8 years and annually thereafter until 20 years of age (screen for OPG).19 Ophthalmologic assessment should include measures of visual acuity and color vision; visual evoked potentials are not recommended. Optical coherence tomography (OCT) may be considered as an objective measure of axonal integrity/axonal loss and thickness of retinal nerve fiber layer. MRI brain/orbits if vision loss on 2 consecutive eye exams. Review of growth and sexual development to identify precocious puberty (ie, hypothalamic involvement). |
| Constitutional mismatch repair deficiency syndrome | Brain MRIs for CNS malignancies, CBCs for leukemia, and abdominal ultrasounds for lymphoma screening starting in infancy and every 6 months thereafter.20 Endoscopic and colonoscopic surveillance starting at 6 years of age and annually thereafter to detect gastrointestinal tumors. |
| Gorlin syndrome | Regular dermatology exams are recommended to monitor for the development of basal cell carcinomas.21 Annual panoramic radiographs of the jaw are recommended starting at age 8. Screening X-rays have been recommended to look for rib anomalies, calcification of the falx, and calcified ovarian fibromas. SUFU mutation carriers: every 4-month brain MRI through age 3 years and every 6-month brain MRI until the age of 5 years for medulloblastoma screening. PTCH1 mutation carriers: no radiographic screening recommended. |
| Li-Fraumeni syndrome | Brain tumor: annual brain MRI (first MRI with contrast; thereafter without contrast if previous MRI normal and no new abnormality).22 Complete physical examination every 3-4 months, including blood pressure, anthropometric measurements plotted on a growth curve (with attention to rapid acceleration in weight or height), Cushingoid appearance, signs of virilization (pubic hair, axillary moisture, adult body odor, androgenic hair loss, clitoromegaly, or penile growth), and full neurologic assessment. ACC: US of abdomen and pelvis every 3-4 months. In case of unsatisfactory US, blood tests may be performed every 3-4. months: total testosterone, dehydroepiandrosterone sulfate, and androstenedione. Soft tissue and bone sarcoma: Annual WBMRI. |
| Neurofibromatosis type 2 | AACR surveillance guidelines23: 1. Annual history and physical exam (including audiology with measurement of pure-tone thresholds and Word Recognition Scores). 2. Annual (consider twice yearly in first year since diagnosis or signs of rapid growth) brain MRI starting at 10 years of age. Screening may begin earlier in patients with high-risk genotypes or symptomatic diagnoses. 3. If baseline imaging shows no characteristic sites of involvement, reduce the frequency of screening to every 2 years. 4. Protocols should include high-resolution (1-3 mm slice thickness) imaging through the internal auditory meatus. 5. Surveillance spinal MRI is recommended at 24- to 36-month intervals beginning at 10 years of age. 6. Whole-body MRI may be obtained. |
| Rhabdoid tumor predisposition syndrome | SMARCB1 truncating alteration—MRI brain every 3 months to age 5 years.21 Consider WBMRI to age 5 years. Ultrasound abdomen every 3 months. SMARCB1 missense alteration—no screening recommended. SMARCA4 alteration—no brain imaging, abdominal ultrasound every 6 months may be considered (ovary). |
| Tumor Predisposition Syndrome | Tumor Surveillance Guidelines |
|---|---|
| Neurofibromatosis type 1 | Semi-annual ophthalmologic examination between ages of 2 and 8 years and annually thereafter until 20 years of age (screen for OPG).19 Ophthalmologic assessment should include measures of visual acuity and color vision; visual evoked potentials are not recommended. Optical coherence tomography (OCT) may be considered as an objective measure of axonal integrity/axonal loss and thickness of retinal nerve fiber layer. MRI brain/orbits if vision loss on 2 consecutive eye exams. Review of growth and sexual development to identify precocious puberty (ie, hypothalamic involvement). |
| Constitutional mismatch repair deficiency syndrome | Brain MRIs for CNS malignancies, CBCs for leukemia, and abdominal ultrasounds for lymphoma screening starting in infancy and every 6 months thereafter.20 Endoscopic and colonoscopic surveillance starting at 6 years of age and annually thereafter to detect gastrointestinal tumors. |
| Gorlin syndrome | Regular dermatology exams are recommended to monitor for the development of basal cell carcinomas.21 Annual panoramic radiographs of the jaw are recommended starting at age 8. Screening X-rays have been recommended to look for rib anomalies, calcification of the falx, and calcified ovarian fibromas. SUFU mutation carriers: every 4-month brain MRI through age 3 years and every 6-month brain MRI until the age of 5 years for medulloblastoma screening. PTCH1 mutation carriers: no radiographic screening recommended. |
| Li-Fraumeni syndrome | Brain tumor: annual brain MRI (first MRI with contrast; thereafter without contrast if previous MRI normal and no new abnormality).22 Complete physical examination every 3-4 months, including blood pressure, anthropometric measurements plotted on a growth curve (with attention to rapid acceleration in weight or height), Cushingoid appearance, signs of virilization (pubic hair, axillary moisture, adult body odor, androgenic hair loss, clitoromegaly, or penile growth), and full neurologic assessment. ACC: US of abdomen and pelvis every 3-4 months. In case of unsatisfactory US, blood tests may be performed every 3-4. months: total testosterone, dehydroepiandrosterone sulfate, and androstenedione. Soft tissue and bone sarcoma: Annual WBMRI. |
| Neurofibromatosis type 2 | AACR surveillance guidelines23: 1. Annual history and physical exam (including audiology with measurement of pure-tone thresholds and Word Recognition Scores). 2. Annual (consider twice yearly in first year since diagnosis or signs of rapid growth) brain MRI starting at 10 years of age. Screening may begin earlier in patients with high-risk genotypes or symptomatic diagnoses. 3. If baseline imaging shows no characteristic sites of involvement, reduce the frequency of screening to every 2 years. 4. Protocols should include high-resolution (1-3 mm slice thickness) imaging through the internal auditory meatus. 5. Surveillance spinal MRI is recommended at 24- to 36-month intervals beginning at 10 years of age. 6. Whole-body MRI may be obtained. |
| Rhabdoid tumor predisposition syndrome | SMARCB1 truncating alteration—MRI brain every 3 months to age 5 years.21 Consider WBMRI to age 5 years. Ultrasound abdomen every 3 months. SMARCB1 missense alteration—no screening recommended. SMARCA4 alteration—no brain imaging, abdominal ultrasound every 6 months may be considered (ovary). |
Abbreviations: AACR, American Association for Cancer Research; ACC, adrenocortical carcinoma; CBC, complete blood count; OPG, optic pathway gliomas; US, ultrasound; WBMRI, whole body magnetic resonance imaging.
Only a third of patients with NF1-OPG require treatment, with the primary goal to preserve vision. Radiotherapy is generally avoided given the increased risk of secondary neoplasms24 and Moyamoya syndrome,25 with carboplatin- or vinblastine-based regimens most commonly used. Recently, molecular targeted therapy with MEK inhibitors has demonstrated impressive anti-tumor effects and is increasingly being considered as second-line therapy for NF1-associated LGGs.26 The current COG (Children’s Oncology Group) study ACNS1831 is a randomized phase 3 trial testing anti-tumor efficacy and visual outcomes during treatment with selumetinib (MEK inhibitor) compared with standard chemotherapy and may alter the current treatment paradigm for NF1-associated LGG (NCT03871257). NF1-associated HGGs share many molecular features and the poor prognosis of sporadic HGGs and are treated similarly.
Turcot Syndrome Type 1 (TST1) or CMMRD
Epidemiology and clinical features.—Patients with monoallelic mutations in DNA mismatch repair (MMR) genes (MLH1, MSH2, PMS2, or MSH6) develop hereditary non-polyposis colorectal carcinoma (HNPCC), also known as Lynch syndrome (LS). Patients with HNPCC account for 4% of all patients with colorectal cancers and harbor a 3% lifetime risk of developing a brain tumor, notably glioblastoma.27 CMMRD is an autosomal recessive (AR) disorder that results from germline biallelic (homozygous or compound heterozygous) mutations in one of the MMR genes. CMMRD should be considered for young children who present with hematological malignancies (T-cell lymphomas), malignant brain tumors, and gastrointestinal (GI) cancers, in conjunction with: (1) café-au-lait macules and/or other signs of NF1 and/or hypopigmented skin lesions; (2) consanguineous parents; (3) a family history of LS-related cancers; (4) a second malignant tumor; and (5) siblings with childhood cancer. The lack of a family history of LS-related cancers should not rule out the possibility of CMMRD, as some families could carry less penetrant mutations and thus not meet clinical criteria for a diagnosis of LS.9 Because CMMRD shares clinical features with neurofibromatosis type 1 (NF1), familial adenomatous polyposis (FAP), and Li-Fraumeni syndrome (LFS), it is important to keep this diagnosis in mind when evaluating children who present with one or more of these features.28 New diagnostic criteria were recently published (Table 2).29
Children with CMMRD are usually affected within the first two decades of life. HGGs are the most common brain tumors, however, medulloblastoma (MB) and supratentorial primitive embryonal tumors have also been reported, and usually present in the second decade of life. HGGs may arise from transformation of LGGs and there exist reports of long-term survivors, suggesting a more favorable prognosis relative to patients with sporadic HGGs.30
Some genotype-phenotype correlations have been reported in CMMRD. PMS2 mutations are identified in 60% of cases while 40% harbor MLH1, MSH2, or MSH6 mutations. The incidence of brain tumors and second malignancy is higher in patients with MSH6/PMS2 mutations as they survive first malignancy. There is some evidence that individuals with deletions or truncating mutations resulting in no functional protein exhibit a more severe phenotype, with hematologic and brain cancers developing in the first decade of life.30
Molecular genetics and pathogenesis.—MMR genes play essential roles in maintaining genome integrity by correcting errors that arise during DNA replication and mutations in MMR genes lead to “hypermutant” glioblastoma (median of >6000 nonsynonymous coding mutations per tumor), characterized by point mutations (single-nucleotide variations) and microsatellite instability (MSI) in which mutations repetitive sequences (microsatellites) are not adequately repaired.20
Clinical implications and treatment.—Establishing a molecular diagnosis of CMMRD can be accomplished in a stepwise manner beginning with testing for MSI and/or loss of expression of an MMR protein, followed by mutational analysis of the appropriate MMR genes.31 A surveillance protocol successfully identified 39 asymptomatic tumors in a cohort of 23 children with CMMRD, including 2 malignant gliomas.31 AACR tumor surveillance guidelines are provided in Table 3.32 Clinicians should be aware of the possibility of increased toxicity and reduced efficacy of chemotherapy, as well as increased risk of second primary malignant tumors in CMMRD patients.9 Glioblastomas associated with CMMRD harbor a high mutational load, and responses to immune checkpoint inhibitor therapy have been reported.10,33 In light of this, we recommend enrolling patients on clinical trials testing immune checkpoint inhibitors (such as NCT02359565, NCT02992964, or NCT03838042), if eligible.
Medulloblastoma (MB)
The prevalence of genetic predisposition varies across MB molecular subtypes with nearly 40% of patients with sonic hedgehog medulloblastoma (SHH-MB) harboring germline alterations in different genes (see Figure 1). A relatively lower prevalence (6%-8%) of APC germline alterations is exclusively noted in the WNT-MB group while rare BRCA2 or PALB2 alterations (1% prevalence) are found among patients with group 3 or group 4 MB.34 Despite significant advances in the identification of MB-associated TPS, the impact of germline mutations on clinical outcomes within molecular subgroups of MB or in comparison to sporadic MB patients harboring somatic mutations in the same set of genes is unclear, and therapeutic approaches generally remain similar.
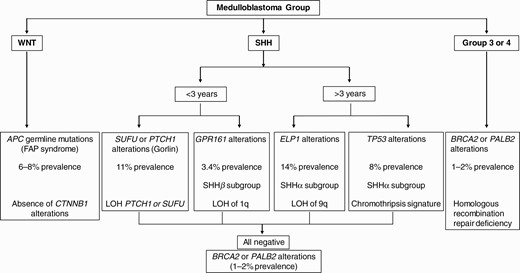
Approach to genetic testing in medulloblastoma.
Nevoid Basal Cell Carcinoma (BCC) Syndrome/Gorlin Syndrome (GS)
Epidemiology and clinical features.—GS is a multisystem, AD condition that affects approximately 1 in 57 000 individuals.35 GS is associated with developmental abnormalities as well as a predisposition to develop benign and malignant tumors, which include BCCs of the skin, odontogenic keratocysts (first sign seen in early childhood and cause dental complications), palmar and plantar dyskeratotic pits, intracranial calcification, melanomas, leukemias, lymphomas, breast cancer, and MB. The diagnosis of GS is made based on clinical findings using established criteria (see Table 2).21
The major symptoms differ depending on the underlying disease-causing gene. Patients with PTCH1 mutations have higher penetrance (90%) of BCC and jaw keratocysts relative to patients with SUFU mutations.36 MB is the most common CNS tumor in the setting of GS and develops earlier compared to sporadic tumors. Most patients are younger than 3 years of age, with tumor histology showing medulloblastoma with extensive nodularity (MBEN), which is almost pathognomonic for the syndrome in young children. Several reports have documented the development of intracranial meningiomas in patients with GS with or without prior craniospinal radiation therapy (RT).37,38
Molecular genetics and pathogenesis.—GS results from disruption of the SHH signaling pathway. To date, germline mutations in two different SHH-related genes are known to cause GS, albeit with different expression patterns. The most well-described gene is the SHH receptor PTCH1 located on chromosome 9q22.339 or the downstream regulator SUFU. Half of all mutations are considered sporadic, with the remaining transmitted from an affected parent. Loss of normal PTCH1 function relieves inhibition of smoothened (SMO), which results in signal transduction leading to translocation of Gli-1 transcription factors into the nucleus and transcription of target genes that control proliferation and differentiation by constitutive upregulation of the SHH pathway. PTCH mutations have been exclusively detected in the desmoplastic variant of MB, while LOH of PTCH is found in 50% of desmoplastic MBs. GS families with SUFU mutations may or may not fulfill the diagnostic criteria for GS, and de novo mutations are reported in 33% of patients.36
Clinical implications and treatment.—Ongoing management includes surveillance for disease-related complications and specific tumor-directed treatment. There is a lower (<2%) risk of developing MB in PTCH1 mutation-positive individuals, while the risk is up to 20-fold higher in SUFU mutation-positive individuals.36,40 RT is associated with an increased risk of developing BCCs within the irradiated fields and other tumors including meningiomas and HGGs.41 Infant SHH-MB and desmoplastic MBs in young children usually have a favorable outcome with radiation-sparing approaches which utilize intensive chemotherapy following surgery.42 DNA methylation profiling molecularly categorizes iSHH into two distinct groups—iSHH-I and iSHH-II. iSHH-I patients appear to have worse outcomes with regimens that do not include intraventricular chemotherapy43–45 in contrast to iSHH-II patients who have had excellent outcomes with chemotherapy-only regimens with or without intraventricular methotrexate (IVT-MTX).43–47PTCH1 alterations (mutations and focal deletions) are more frequent in iSHH-II (48%) compared to iSHH-I (35%).43 In contrast, deleterious SUFU alterations were significantly enriched in iSHH-I (32%) (including 5/6 pathogenic SUFU germline variants) and rare in iSHH-II (4%), supporting the notion that iSHH MBs (predominantly iSHH-I) harboring germline SUFU mutations are associated with inferior outcomes.43,48 These data can rationally inform the design of the next generation of molecularly risk-stratified clinical trial for infant MB wherein iSHH-II patients may receive a reduced-intensity regimen with systemic chemotherapy only, while iSHH-I patients should receive systemic chemotherapy combined with IVT-MTX. Small molecule inhibitors of the SHH pathway (vismodegib) have been tested for the treatment of recurrent SHH-MB.49 However, responses were short-lived and restricted to tumors harboring proximal (eg, PTCH1 or SMO mutations) abnormalities in the SHH pathway. A significant proportion of pediatric SHH-MB tumors are intrinsically resistant to SMO inhibitors because they harbor genetic alterations in the downstream effectors SUFU or GLI. Moreover, the disruption of SHH signaling associated with SMO inhibitors results in premature physeal fusion, limiting its use in skeletally immature patients. CX4945, a casein kinase 2 inhibitor that blocks signaling at the most terminal component GLI, is currently in a phase I/II trial for patients with recurrent/relapsed SHH-MB (PBTC-053).
Brain Tumor-Polyposis Syndrome 2 (BTPS 2 or Turcot Type 2; Familial Adenomatous Polyposis Coli)
Epidemiology and clinical features.—FAP is characterized by AD inheritance and the development of hundreds to thousands of polyps in the colon during adolescence with eventual progression to malignancy.50 Children with FAP are also at risk of developing MB, hepatoblastoma, and aggressive fibromatoses. MBs are the most common brain tumors in FAP patients (60%), followed by astrocytomas (14%) and ependymoma (10%). The lifetime risk of developing a primary CNS tumor is roughly 1% in the context of FAP, but the risk of developing MBs is 92 times higher than in the general population.51 Patients develop MBs during the second decade, predominantly WNT subtype, peaking at 15 years of age. The clinical characteristics of MBs occurring in the context of germline APC alterations include localized disease, frequent lateral location (ie, cerebellopontine angle or in the cerebellar peduncles, akin to sporadic WNT MBs), and classical histology.52 Genotype-phenotype correlations have been reported, as patients with mutations in codons 686-1217 have a 13-fold increase of developing MBs compared to FAP patients with other mutations.51
Molecular genetics and pathogenesis.—FAP is caused by germline mutations in the gene adenomatous polyposis coli (APC) located on chromosome 5q21-22.53APC serves as a ubiquitin ligase that regulates the protein stability of β-catenin which is integral to WNT signaling, controls transcription of cyclin D which associates with cyclin-dependent kinase (CDK)4/6 to negatively regulate RB1 activity.54APC mutations are rare in sporadic MB.
Clinical implications and treatment.—Since MB is rare in FAP, carriers are not routinely screened for these tumors.55 However, some groups recommend heightened clinical suspicion and a low threshold for neuroimaging in individuals with FAP with segment 2 APC gene mutation. Furthermore, the diagnosis of MB in FAP may precede the development of colonic adenocarcinoma which underscores the need for GI cancer surveillance in these patients. It is important to note that APC-mutated MBs are distinct from most WNT-activated MBs which harbor CTNNB1 mutations. Recent data indicate that MB associated with an APC germline pathogenic variant share the very favorable prognosis of CTNNB1-mutated MB.34,52 Combined with an increased risk of radiation-associated secondary neoplasms and the rarity of these tumors, it would be critical to consider enrollment on de-escalation therapeutic protocols with lower craniospinal radiation doses as currently being done for WNT MBs with a CTNNB1 pathogenic variant in the COG (NCT02724579) and PNET5 trials (NCT02066220).52
Fanconi Anemia (FA)
Epidemiology and clinical features.—FA has an incidence of approximately 1 in 250 000 births and is characterized by defective DNA repair resulting in progressive bone marrow failure, congenital abnormalities, and a predisposition for malignancy. Chromosomal breakage studies are sensitive, but not specific and should be followed by DNA sequencing deletion/duplication tests for all the known genes: FNCA, FANCB, FANCC, BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, BRIP1, FANCL, FANCM, PALB2, RAD51C, and SLX4.11 If an individual’s ethnic background is known to be associated with a particular FA mutation, targeted mutation analysis should be performed for that mutation. Most solid tumors in patients with FA have occurred when the patient was age 20 or older, however, brain tumors were reported in children below the age of 10 and sometimes preceded the diagnosis of FA.56 Furthermore, brain tumors including MB and glioma were found primarily in patients carrying mutations in both copies of the FANCD1/BRCA2 gene or FANCN/PALB2.57
Molecular genetics and pathogenesis.—All FA mutations have an AR inheritance pattern, except FANCB, which is X-linked. The FA complementary group of proteins plays a central role in DNA damage repair and homologous recombination. Biallelic FANCD1/BRCA2 mutations account for about 3%-4% of all FA patients and develop spontaneous chromosomal aberration at a high rate.58 FA-D1 cells are not biallelic for FANCD1/BRCA2 null alleles but have at least one hypomorphic FANCD1/BRCA2 allele expressing a FANCD1/BRCA2 protein with some residual activity.59 Such individuals present a more severe phenotype with early onset of cancer and the cumulative probability of developing a MB could be high as 85% in the first decade.60 The frameshift mutations c.886delGT and c.6174delT are associated with brain tumors.59
Clinical implications and treatment.—For any early-onset pediatric brain tumor with cutaneous, skeletal, or neurological abnormalities consistent with a diagnosis of FA or in case of severe unexpected toxicity from chemotherapy, genetic counseling is recommended.22,61,62 For individuals with mutations in the FANCD1/BRCA2 and FANCN/PALB2 genes, the identity of the patient’s mutations is essential for proper cancer surveillance and medical management.63 If a patient has biallelic FANCD1/BRCA2 mutations or a family history or clinical manifestations that are highly suggestive of FANCD1/BRCA2 mutations, additional tests such as a brain MRI and kidney ultrasound should be performed immediately to rule out any evidence of tumors. Individuals with FA undergoing treatment for MB are known to be highly sensitive to both RT and chemotherapy, with increased susceptibility for treatment-associated toxicities, especially from alkylator-based chemotherapy.12,22,62,64 Tumors harboring either BRCA1 or BRCA2 mutations have been demonstrated to have increased sensitivity to Poly ADP-ribose polymerase (PARP) inhibitors owing to defects in homologous recombination and reliance on alternative pathways to repair DNA damage that are targeted by these inhibitors. The concept of synthetic lethality is being exploited in the use of PARP inhibitors and may be useful in FA associated CNS tumors, analogous to their use in BRCA1/2 deficient breast, ovarian, and pancreatic cancers.65 In light of this, consideration may be given to metronomic chemotherapeutic strategies66–69 or clinical trials testing PARP inhibitors in combination with chemotherapy (NCT00994071).
G Protein-Coupled Receptor 161 (GPR161)
Recently, a novel TPS characterized by heterozygous protein-truncating germline mutation in GPR161 (genomic location: 1q24.2) was described.70 Afflicted patients develop infant SHH-MB (TP53 wild type, with methylation profile clustering with SHHβ subtype) as the primary clinical manifestation (median age of 1.5 years). Molecular tumor profiling demonstrated recurrent LOH of GPR161 as a second hit in the tumor resulting from segmental copy-neutral LOH of chromosome 1q which is conspicuously absent in GPR161 wild-type SHH-MB. Overall, the frequency of germline mutations among pediatric patients with SHH-MB is as follows: GPR161 3.4%, PTCH1 4.5%, and SUFU 5.6%.
ELP1
Germline heterozygous loss-of-function (LOF) variants across ELP1 represent the most common MB predisposition gene which was detected in 14% of pediatric patients with SHH-MB.71ELP1 is the largest component of the Elongator complex, and inactivation leads to translational deregulation due to Elongator deficiency and disruption of protein homeostasis. These patients are diagnosed at a median age of 6.3 years; earlier than germline TP53-associated SHH-MB but older than patients with germline SUFU- or PTCH1-associated MBs. ELP1-associated MBs are predominantly histologically diagnosed as desmoplastic nodular MB and are associated with a more favorable survival. These tumors clustered in the molecular SHHα subgroup based on DNA methylation profiling but were mutually exclusive with SHHα MB associated with germline and somatic TP53 mutation which are associated with inferior clinical outcomes. ELP1 associated MBs are characterized by somatic loss of chromosome arm 9q resulting in LOH of ELP1 and biallelic inactivation coupled with monoallelic loss of PTCH1 (9q22.32). Additional molecular aberrations include mutations or focal deletions in PTCH1 leading to biallelic inactivation along with amplifications of PPM1D and MDM4 (components that regulate p53 signaling).
Choroid Plexus Carcinoma (CPC)
CPC is considered an LFS-defining tumor72 with a high prevalence (~65%) of an underlying TP53 germline mutation among patients, even in the absence of a positive family history due to de novo mutations.
Li-Fraumeni Syndrome (LFS)
Epidemiology and clinical features.—LFS is an AD condition affecting 1 in 5000-10 000. Individuals with the disorder have a lifetime risk of 85%-100% of developing cancer, including sarcomas, breast cancer, adrenocortical carcinoma, leukemia, and brain tumors. There are established criteria for diagnosis (see Table 2).72 A geographical hotspot has been reported in the southern parts of Brazil and Paraguay.
Brain tumors are found in about 14% of the patients with TP53 germline mutations with an average of onset of 16 years.73 The frequency of brain tumors is high (21%) in patients with mutations in the L2 and L3 loop of TP53 that mediates the binding to the minor groove of DNA.73 Among associated CNS tumors, CPC affect LFS carriers in the first decade of life and MBs usually in the second, while malignant gliomas can occur throughout childhood, but more commonly in young adults. HGGs are the most common CNS tumor in the context of LFS. The prognostic significance of germline TP53 mutations in pediatric HGGs is unknown. 56% of all patients with SHH/TP53-mutated MBs harbor germline mutations in TP53.74TP53 mutant SHH-MBs harbor a unique molecular profile suggesting chromothripsis (“chromosome shattering”) as the initiating event and have unfavorable outcomes.75
Molecular genetics and pathogenesis.—LFS is linked to germline mutations in the tumor suppressor gene TP53 located at chromosome 17p13.1. Referred to as the “gatekeeper of the genome,” TP53 represents one of the key proteins that maintain genome integrity after DNA damage, hypoxia, and other stressors. Interestingly, whereas TP53 germline mutations are found in 70%-80% of families with classic LFS, they are only identified in 20%-40% of families with LFS-like syndrome.23 The CHK2 checkpoint homolog gene, CHEK2 (22q), also has been implicated in some families with classic LFS,76 while BRCA alterations are reported in the non-classic syndrome.77
Clinical implications and treatment.—Patients with HGG and a family history of LFS tumors, as well as patients diagnosed with a CPC or MB harboring somatic TP53 mutations should be screened for germline TP53 mutations.72 A strong association between germline TP53 and a rare IDH1 mutation (R132C) has been identified in LFS families78 and in two germline TP53 mutations identified in The Cancer Genome Atlas (TCGA) database,79 suggesting that patients with this mutation might be recommended for germline TP53 testing. Detection of a germline TP53 mutation has significant prognostic and therapeutic implications with individuals at increased risk for developing RT-induced secondary malignant tumors and myelodysplastic syndrome following specific chemotherapies.80 For LFS patients, the AACR recommends lifelong annual brain MRIs starting from birth.81 Aggressive surveillance protocols have identified several LGGs, perhaps, suggesting that some of LFS associated HGGs arise as secondary glioblastomas and may benefit from early intervention with impact on survival and quality of life of individuals with LFS.82 Immunohistochemistry (IHC) for TP53 has been proposed as a surrogate screening measure for the presence of TP53 alterations which confers resistance to chemotherapy and RT.83–85 TP53 immunopositivity has been reported as an adverse prognostic biomarker with 5-year survival rates for patients with TP53-immunopositive and negative CPCs being 0% and 82%, respectively.85 However, other studies have reported long-term radiation-free survivors treated with intensive myeloablative-chemotherapy containing regimens,86–88 including patients harboring germline TP53 alterations. Furthermore, a meta-analysis evaluated the role of RT in patients with CPC harboring germline TP53 alterations and reported an adverse impact on survival with the use of radiotherapy relative to irradiation sparing regimens.83
Vestibular Schwannomas (Bilateral) or Meningioma (Solitary)
Bilateral vestibular schwannomas (VS), arising at the vestibular branch of the eighth cranial nerve, are pathognomonic for the TPS neurofibromatosis type 2 (NF2). Though it shares a common name with NF1 for historical reasons, NF2 is an entirely different clinical entity in terms of its underlying cause, presentation, and clinical course.
Neurofibromatosis Type 2 (NF2)
Epidemiology and clinical features.—NF2 is transmitted in an AD manner with complete penetrance and an incidence of approximately 1:30 000 live births.89 Although the hallmark of NF2 is bilateral VSs, it is in fact a multiple neoplasia syndrome, resulting from a germline mutation in the NF2 tumor suppressor gene on chromosome 22q12. The diagnosis is made clinically based on the NIH-modified Baser criteria or Manchester criteria (see Table 2)90 or by identification of an NF2 mutation from the blood or identical mutation in 2 separate tumors from the same individual.91
The clinical course may vary greatly between families. Greater than half of all patients have no family history of NF2, and almost one-third of the de novo cases are due to mosaic mutations.92 Patients that are mosaic for NF2 present with a mild phenotype and harbor a reduced risk of transmitting NF2 to their offspring. Truncation mutations account for the majority, leading to a nonfunctional protein, and are associated with a severe phenotype with earlier onset, increased frequency of meningiomas, and worse disease-related mortality.93 Conversely, patients with missense mutation or large deletions typically have a milder form of NF2.94 Furthermore, the incidence of meningiomas appears to be lower in patients with mutations in the 3′ region compared to those harboring mutations in the 5′ region of the gene.95
The most common tumors seen in NF2 are vestibular and non-vestibular cranial and spinal nerve schwannomas, peripheral nerve schwannomas, meningiomatosis, intracranial or spinal meningiomas, and ependymomas. In the pediatric age group, the most frequent symptoms and signs of NF2 are related to underlying skin or spinal tumors.96
VSs occur in 95% of patients with NF2 and typically occur in late adolescence or adulthood. The risk of underlying NF2 is especially high in young patients presenting with a single VS or meningioma, and further workup and surveillance is necessary for these patients: approximately 20% of patients diagnosed with a unilateral VS or solitary meningioma and no other signs of NF2 under the age of 20 years have an underlying germline NF2 mutation.97 Non-vestibular cranial nerve schwannomas are found in up to 50% of NF2 patients.98 Malignant transformation of tumors in NF2 patients is very rare, and almost always associated with prior RT.
Molecular genetics and pathogenesis.—The NF2 tumor suppressor gene is located on chromosome 22. The NF2 gene product is a protein which is a member of the 4.1 family of cytoskeletal proteins termed “Merlin” (Moesin-ezrin-radixin-like protein).99 Inactivation of the NF2 gene can be detected in the majority of sporadic schwannomas100 and in 50%-60% of sporadic meningiomas. Merlin’s predominant tumor suppressive functions are attributable to its control of oncogenic gene expression through regulation of Hippo signaling.99
Clinical implications and treatment.—The diagnosis of NF2 may be delayed in patients presenting with a unilateral VS or solitary meningioma. Nearly 10% of individuals with unilateral VS will ultimately be diagnosed as having NF2, and the risk increases with younger age.97 Similarly, posterior lenticular opacities may predate the confirmation of any other lesions.101
AACR surveillance guidelines are outlined in Table 3, including imaging and audiologic surveillance.102 The primary goals of treatment in NF2 are to preserve hearing and other neurological functions. Traditional treatment options for growing or symptomatic tumors include radical or partial surgical resection and radiosurgery. More recently, treatment with bevacizumab has been found to result in imaging responses and hearing improvement in adults, and to a lesser extent in children, with NF2 and VS.103,104
Non-vestibular cranial nerve schwannomas are frequently multifocal. Therefore, surgery is often deferred unless they are unacceptably symptomatic, endangering function, or growing rapidly.105
The majority of spinal ependymomas in NF2 can be observed, but surgery may be indicated in symptomatic patients.
Atypical Teratoid Rhabdoid Tumor (ATRT)
Overall, ATRT accounts for 1%-2% of all pediatric brain cancer; and 20% of those occurring prior to 3 years of age.
Rhabdoid Tumor Predisposition Syndrome (RTPS)
Epidemiology and clinical features.—ATRTs are highly aggressive tumors that were historically associated with a dismal prognosis (median survival 6-12 months).106 However, the recent use of multimodal therapies has led to improved outcomes.107,108 Patients with RTPS develop ATRTs at a younger age (median of 7 months compared to 18 months for sporadic),108 can present with tumors at different anatomical locations synchronously or metachronously109–111 and are more likely to die from progressive disease compared to patients with sporadic ATRT.108,112 Individuals with RTPS have also been reported to develop schwannomas and meningiomas later in life.110
There is a phenotypic association between the mutation type and the development of RTPS or schwannomatosis. Patients with RTPS often have truncating mutations of the SMARCB1 gene, while mostly non-truncating mutations are seen in patients with schwannomatosis.113 In some cases, family members with the same germline mutation can develop either ATRT during early childhood or schwannomatosis if they survive until adulthood.114
Molecular genetics and pathogenesis.—The characteristic somatic genetic abnormality and the primary initiating event includes mutation of the INI1/SMARCB1 tumor suppressor gene, also known as INI1/hSNF5, located on chromosome 22q11.2.115SMARCB1 is part of a chromatin-remodeling complex involved in regulating gene expression, including genes involved in cell cycle control, DNA repair, and differentiation.116 Approximately 35% of patients with rhabdoid tumors have germline INI1/SMARCB1 alterations.110 While the majority of germline mutations are de novo, families with RTPS have been reported in which multiple siblings have been affected. Recent studies have reported a second disease locus, SMARCA4, which may be associated with worse prognosis compared to SMARCB1.117
Clinical implications and treatment.—The major tumor risk in RTPS is for the development of renal or extrarenal RTs with an aggressive phenotype. For CNS ATRTs, event free survival is better in patients with gross total resection.118 and the historically dismal survival rate appears to be improved with aggressive multi-modality therapy, including high-dose chemotherapy including methotrexate and radiotherapy.107,108 However, only 2 of 10 patients with germline SMRACB1 alterations survived beyond 2 years in the recently reported COG ACNS0333 study.108 This underscores the importance of evaluating novel drugs targeting altered signaling pathways in ATRT, some of which are currently in clinical trials and have demonstrated preliminary anti-tumor efficacy as monotherapy in the setting of relapsed/refractory disease, including the aurora kinase A (AURKA) inhibitor (Alisterib)119 and the EZH2 (enhancer of zeste homolog 2) inhibitor Tazemetostat.120 AACR surveillance recommendations are provided in Table 3.40 Although there are no studies to confirm the benefit of screening patients with germline INI1/SMARCB1 mutations, the aggressive natural history of the disease, apparently high penetrance, and well-defined age of onset for CNS RTs suggest that screening could prove beneficial.
Pineoblastoma
Pineoblastomas are highly malignant embryonal tumors that may be associated with germline mutations in the RB1 gene. Germline DICER1 mutations have also been reported, which are distinct from the DICER1 mutations predisposing to pleuropulmonary blastoma.121
RB1-Associated Pineoblastoma
Epidemiology and clinical features.—Individuals harboring germline RB1 mutations are at greatly increased risk to develop bilateral retinoblastoma (RB) tumors, generally during the first 5 years of life. Patients are also at increased risk (5%-15%)122 to develop tumors involving the pineal gland (so-called trilateral RB), synchronously or metachronously, with a median age at diagnosis of 23-48 months. The median interval between the diagnosis of bilateral RB and the diagnosis of the brain tumor is usually >20 months. Later in life, individuals with hereditary RB have a 10%-15% risk of developing second malignancies including sarcomas, melanomas, lung and bladder cancer.123
Molecular genetics and pathogenesis.—RB is caused by mutations in RB1, which resides at chromosomal locus 13q14.124RB1 encodes the RB protein (pRB), a potent tumor suppressor that regulates cell growth by inhibiting the function of the E2F family of transcription factors and preventing inappropriate S-phase entry.125 The germline RB1 mutation is inherited from a similarly affected parent in <20% of patients,126 with the majority developing de novo within one of the parent’s gametes or a cell of the developing embryo.127 Genetic testing for germline RB1 gene mutations can identify mutations in more than 90% of individuals with hereditary disease.128
Clinical implications and treatment.—The identification of germline RB1 mutations supports appropriate primary tumor treatment and preservation of vision, initiation of surveillance protocols, and genetic screening of other family members.129 Contemporary treatment approaches employing increased surveillance, intensive chemotherapy followed by consolidation with myeloablative chemotherapy and autologous hematopoietic progenitor cell rescue have resulted in improved 5-year overall survival (OS) (5% vs 48%),130 and improvement in median survival time (8 months vs 24 months).131 Most survivors reported in studies had small tumors (<15 mm), which were detected pre-clinically while screening.122 Therefore, screening with MRI has frequently been recommended at baseline and as often as every 6 months for 5 years for patients known or suspected to have a germline RB1 mutation with the hope of detecting trilateral RB at a subclinical stage.132 However, monitoring for second malignancies is of unclear benefit at the present time and needs to be studied further.129
DICER-Associated Pineoblastoma
The DICER1 gene is located at 14q32.13. It encodes a cytoplasmic endoribonuclease, which functions to split precursor molecules into microRNAs (miRNAs), thereby regulating the expression of cellular mRNA.133 Dysfunctional miRNA processing reduces the posttranslational regulation of mRNAs, contributing to tumorigenesis.134
Analysis of patients and families harboring germline DICER1 mutations has demonstrated the extended phenotype of malignancies associated with this TPS, including cystic nephroma, ovarian Sertoli-Leydig cell tumor, embryonal rhabdomyosarcoma, and pineoblastoma.133 DICER-associated CNS spindle cell sarcoma has been identified, with a cluster in South America.135 Close follow-up should be considered with particular attention to any changes in respiratory function, menstruation, and development of goiter which should raise suspicion for an underlying malignant etiology. AACR guidelines suggest that surveillance brain MRI may be considered, but the risk-benefit ratio is not yet known.136
Hemangioblastoma (HB)
HB are benign vascular tumors (WHO grade I) and represent the hallmark of the von Hippel-Lindau (VHL) syndrome. HBs are the presenting feature in about 60% of patients with VHL; conversely, 30% of all patients with cerebellar HBs have VHL.137
Subependymal Giant Cell Astrocytoma (SEGA)
SEGA is seen almost exclusively in the context of patients with tuberous sclerosis (TS) complex and conversely, SEGA is the only CNS tumor seen in TS.138
Conclusions
The explosion of new data from genomic studies has led to an exponential increase in our knowledge of hereditary predispositions to pediatric brain tumors. The possibility of an underlying TPS should be considered in all pediatric patients diagnosed with a CNS tumor. Recognizing a TPS is important for accurate diagnosis and prognosis, selection of optimal therapy, screening, risk reduction, and family planning. As more population data, functional modeling of specific variants, and deeply integrated clinical and molecular data are generated, many variants of unknown significance will be reclassified as benign polymorphisms or disease-causing mutations.
Funding
This work was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 to Memorial Sloan Kettering Cancer Center.
Conflict of interest statement. S.F.S. reports active consulting agreement with AstraZeneca (consulting fees received) and QED therapeutics. M.A.K. reports consultant agreements with AstraZeneca, Bayer, CereXis, QED Therapeutics and Recursion Pharma (consulting fees received). M.F.W. has nothing to declare.
References

{kind=link}