






Abstract
Paraneoplastic neurological syndrome (PNS) comprises a group of neurological disorders that result from a misguided immune response to the nervous system triggered by a distant tumor. These disorders frequently manifest before the diagnosis of the underlying neoplasm. Since the first reported case in 1888 by Oppenheim, the knowledge in this area has evolved rapidly. Several classic PNS have been described, such as limbic encephalitis, paraneoplastic cerebellar degeneration, encephalomyelitis, opsoclonus-myoclonus, sensory neuronopathy, Lambert-Eaton Myasthenic syndrome, and chronic gastrointestinal dysmotility. It is now recognized that PNS can have varied nonclassical manifestations that extend beyond the traditional syndromic descriptions. Multiple onconeural antibodies with high specificity for certain tumor types and neurological phenotypes have been discovered over the past 3 decades. Increasing use of immune checkpoint inhibitors (ICIs) has led to increased recognition of neurologic ICI-related adverse events. Some of these resemble PNS. In this article, we review the clinical, oncologic, and immunopathogenic associations of PNS.
Paraneoplastic neurological syndromes (PNS) comprise a group of neurological disorders that result from a misguided immune response to the nervous system triggered by a distant tumor.1,2 In 1888, Oppenheim reported empiric association of peripheral neuropathy and lung cancer highlighting the remote or indirect effect of cancer on the nervous system.3 This was followed by another description of association between cancer and neuropathy in 1890 by Auchè.4 Subsequently, Dr. Derek Denny-Brown, described 2 cases of sensory neuronopathy with myopathy (neuromyopathy) associated with bronchogenic pulmonary carcinoma.5 At that time, it was presumed this phenomenon may be related to a metabolic disorder associated with tumor. Later in the 1960s, Dr. Wilkinson and Dr. Zeromski investigated sera from patients with neuromyopathy associated bronchial carcinoma and found circulating antibodies against brain extracts6 as well as central nervous system neurons.7 In the 1980s, this perinuclear neuronal staining pattern was further characterized as anti-Hu (a.k.a. anti-neuronal nuclear antibody type 1 [ANNA1]) and details about neurological syndromes and oncologic associations were published by researchers from Sloan Kettering Cancer Center.8–10 In 1976, Dr. Trotter and colleagues identified an antibody against cerebellar Purkinje cells in serum of a patient with Hodgkin’s disease.11 Subsequently, anti-cerebellar Purkinje cells antibody was described in two patients with ovarian carcinoma and paraneoplastic cerebellar degeneration (PCD) by Dr. Greenlee and Dr. Brashear,12 and this was confirmed and further characterized by independent groups.13–16 Furthermore, in 1999, investigators from Sloan Kettering Cancer Center described the first serological biomarker of paraneoplastic encephalitis associated with testicular germ cell tumor, anti-Ma2-IgG.17 During this time, there were significant contributions from Mayo Clinic Neuroimmunology group, including discovery of P/Q-type voltage-gated calcium channel (VGCC) antibodies among patients with Lambert-Eaton Myasthenic syndrome. Another onconeural antibody anti-CV2 IgGwas initially described by Honnorat et al. in 1999,18 and was confirmed to be collapsin response-mediated protein 5 (CRMP5) in 2001.19 These early contributions and biomarker discoveries laid the foundation for the field of paraneoplastic neurological disorders. In the last two decades, many novel biomarkers have been discovered (Figure 1) and varied neurological syndromes have been characterized in association with paraneoplastic autoimmunity.
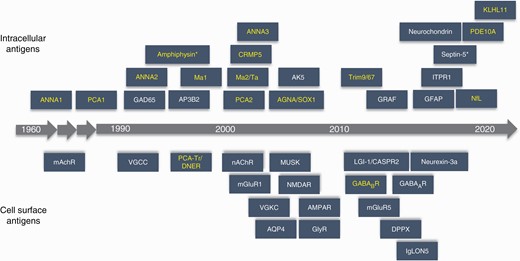
Autoantibody discovery timeline. Antibodies with high neoplastic association are in yellow. Abbreviations: AGNA/SOX1, anti-glial nuclear antibody against SRY (sex determining region Y)-box 1; AK5, adenylate kinase 5; AMPAR, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ANNA1 (anti-Hu), anti-neuronal nuclear antibody type 1; ANNA2 (anti-Ri), anti-neuronal nuclear antibody type 2; ANNA3, anti-neuronal nuclear antibody type 3; AP3B2, adapter protein 3, subunit B2; AQP4, aquaporin4; CASPR2, contactin-associated protein-like 2; CRMP5, collapsinresponse mediator protein 5; DPPX, dipeptidyl peptidase-like protein 6; GABAAR, gamma-aminobutyric acid type A receptor; GABABR, gamma-aminobutyric acid type B receptor; GAD65, glutamic acid decarboxylase 65; GFAP, glial fibrillary acidic protein; GlyR, glycine receptor; GRAF, GTPase regulator associated with focal adhesion kinase; IgLON5, immunoglobulin-like cell adhesion molecule 5; ITPR1, inositol 1,4,5-trisphophate receptor type 1; KLHL11, kelch-like protein 11; LGI-1, leucine-rich, glioma-inactivated 1; mAChR, muscarinic acetylcholine receptor; mGluR1, metabotropic glutamate receptor 1; mGluR5, metabotropic glutamate receptor 5; MuSK, muscle-specific tyrosine kinase; nAChR, nicotinic acetylcholine receptor; NfL, neurofilament; NMDA-R, N-methyl-d-aspartate receptor; PCA1 (anti-Yo) , Purkinje cell cytoplasmic antibody type 1; PCA2, Purkinje cell cytoplasmic antibody type 2; PCA-Tr/DNER, Purkinje cell cytoplasmic antibody type Tr or Delta/Notch-like epidermal growth factor-related receptor; PDE10A, phosphodiesterase 10A; Trim9/67, tripartite motif-containing protein 9 and 67; VGCC, voltage-gated calcium channel; VGKC, voltage-gated potassium channel. *Amphiphysin and septin-5 antibodies can have transient cell-surface expression.
A diagnostic criterion of PNS was recommended by a panel of neurologist with expertise in the diagnosis and management of these disorders in 2004 (Euronetwork PNS criteria).20 Based on these criteria, a definite PNS diagnosis required a classical syndrome, and identification of neoplasm within 5 years of PNS onset and/or the presence of well-characterized onconeural antibodies. For a patient presenting with a nonclassical syndrome the diagnosis of a definite PNS could be made if a well-characterized onconeural antibody was detected, or an onconeural antibody not considered well characterized was detected and cancer developed within 5 years of PNS onset, or PNS significantly improved after cancer treatment without concomitant immunotherapy. A recent population-based Italian study utilized these Euronetwork PNS diagnostic criteria and reported incidence of PNS was 0.89 per 100,000 person-years with a prevalence of 4.37 per 100,000.21
Herein, we review the pathogenesis, clinical syndromes, antibody and oncological associations, and management of PNS. Furthermore, we discuss the neurological complications of immune checkpoint inhibitors (ICIs) and their management.
Pathophysiology of Paraneoplastic Syndrome
The cellular or humoral immune response against antigens shared by the tumor and neural tissues is hypothesized to contribute to PNS pathogenesis. The majority of well-characterized onconeural antibodies target intracellular antigens. These neural antibodies targeting intracellular antigens are generally considered to be surrogate markers for a cytotoxic T-cell response rather than being directly pathogenic.22 On the other hand, the onconeural antibodies targeting cell-surface antigens are potentially directly pathogenic.
Upon cell death, cell-surface and intracellular proteins are phagocytosed and processed by the dendritic cells (DCs).23 Subsequently, the antigens are presented to the lymphocytes in the regional lymph nodes. Intracellular antigens lead to cell-mediated adaptive immune response. The antigens presented by the DCs activate autoantigen-specific CD8+ cytotoxic T-lymphocytes.24 The activated neural antigen-specific CD8+ T-lymphocytes are hypothesized to recognize and bind to the neural cells and, release proinflammatory cytokines and cytotoxic granules leading to cell death.25,26 Some studies have also demonstrated that antibodies targeting intracellular antigens such as ANNA1 (anti-Hu) or Purkinje cell cytoplasmic antibody type 1 (PCA1, a.k.a. anti-Yo) IgG can be taken by neurons and may be directly responsible for neuronal damage.27–30 However, direct neuronal cytotoxic effect of antibodies targeting intracellular antigens has not been consistently demonstrated in all in vitro studies.31,32 Intraventricular administration of anti-Hu antibodies in animal models did not lead to development of PNS.33 Immunohistopathological studies of patients with PNS have also demonstrated CD3+ and CD8+ T-lymphocyte predominant infiltrates in brain or dorsal root ganglia which supports cytotoxic T-cell-mediated neuronal loss, rather than antibody-mediated dysfunction.24,34–37
In contrast, the immune response against cell-surface protein is predominantly humoral- or B-cell-mediated. The complex interaction between B- and CD4+ T-lymphocytes leads to B-cell proliferation and differentiation to plasma cells and memory B cells. In some instances, B-lymphocytes can enter CNS where they undergo re-stimulation, maturation, and differentiation to plasma cells leading to intrathecal antibody synthesis.38,39 Unlike studies of intracellular antigen, various in vitro studies suggested that antibodies against cell-surface antigen can bind to cell-surface proteins on live neurons,38,40,41 and postmortem tissue examination demonstrated anti-IgG deposition around the neurons without substantial cell death.42 In animal studies, injection of N-methyl-d-aspartic acid receptor (NMDA-R) IgG, an antibody against cell-surface protein, led to alteration of glutamatergic transmission and enhance cortical excitability in Wistar rats.43,44 These findings demonstrated the pathogenic roles of antibodies against cell-surface protein.
Central Nervous System Syndromes
Paraneoplastic Limbic Encephalitis
Limbic system is involved in a variety of functions including memory, emotional behavior, mood processing, and olfaction. It consists of the hippocampus, amygdala, cingulate gyrus, hypothalamus, and the limbic cortex. Inflammation of these structures termed limbic encephalitis may result from autoimmune, inflammatory, or infectious etiologies. Paraneoplastic limbic encephalitis generally has a subacute onset. Common presenting features include anterograde amnesia, behavioral disturbances, and at times seizures.45 Cerebrospinal fluid (CSF) lymphocytic pleocytosis is common among patients with paraneoplastic limbic encephalitis.45 The MRI hallmark of this syndrome is increased T2 signal in medial temporal lobes involving the hippocampus and amygdala.46
Among patients presenting with limbic encephalitis, underlying onconeural antibody specificities may guide targeted neoplasm search. Neuropsychiatric dysfunction, sleep disorder and seizures, and metabotropic glutamate receptor 5 (mGluR5) antibody seropositivity are suggestive of underlying Hodgkin’s lymphoma.47 This syndrome was initially described by Ian Carr in his 15-year-old daughter in 1982.48 He named it “Ophelia syndrome” after a character from Shakespeare’s play “Hamlet.” Similarly, limbic encephalitis in association with Ma2-IgG or kelch-like protein 11 (KLHL11) IgG is suggestive of testicular or extra-testicular germ cell tumor.17,24,49,50 Various paraneoplastic antibodies associated with limbic encephalitis are listed in Table 1. Early diagnosis, treatment of the underlying tumor, and early initiation of immunosuppressive therapy have been associated with favorable outcomes.45,51
Paraneoplastic Neurological Syndromes, Clinical Features, and Associated Antibodies
| Paraneoplastic Syndromes | Clinical Feature | Associated Antibodies |
|---|---|---|
| Limbic encephalitis | Memory impairment, seizures, behavioral change, neuropsychiatric symptoms, mood and sleep disturbances | ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), mGluR5, ANNA2 (anti-Ri), PCA2, KLHL11, GABAB receptor, AMPAR, LGI1, CASPR2, ANNA3 |
| Encephalomyelitis | Encephalopathy, myelitis, and peripheral nerve or DRG involvement | ANNA1 (anti-Hu), CRMP5 (anti-CV2), amphiphysin, GFAP |
| Cerebellar degeneration | Ataxia (usually subacute onset), dysarthria, diplopia | PCA1 (anti-Yo), PCA2, PCA-Tr (DNER), mGluR1, ANNA1,2, Zic 4, VGCC (P/Q type), amphiphysin, Ma2, NfL, KLHL11 |
| Sensory neuronopathy | Asymmetric numbness, paresthesia, sensory ataxia, neuropathic pain | ANNA1 (anti-Hu), amphiphysin, PCA2, CRMP5 (CV2) |
| Opsoclonus myoclonus syndrome | Myoclonus and opsoclonus eye movements | ANNA2 (anti-Ri), ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), NMDA-R, GluD2 |
| Gastrointestinal dysmotility | Gastroparesis, constipation, pseudo-obstruction | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2 |
| Lambert-Eaton myasthenic syndrome | Muscle weakness, fatigue, and autonomic dysfunction and absent deep tendon reflexes | P/Q-type VGCC |
| Brainstem encephalitis | Ataxia, vertigo, diplopia, dysphagia, dysarthria | ANNA2 (anti-Ri), KLHL11, ANNA1 (anti-Hu), Ma2, PCA2, CRMP5 (CV2), amphiphysin, Zic 4 |
| Paraneoplastic myelopathy | Subacute myelopathy, usually tract-specific involvement | ANNA2 (anti-Ri), ANNA1 (anti-Hu), ANNA3, CRMP5 (anti-CV2), Amphiphysin, NfL |
| Paraneoplastic stiff person syndrome | Muscle spasms and exaggerated startle response | Amphiphysin, DPPX, GAD65, glycine receptor |
| Paraneoplastic chorea | Choreic movements with more extensive nervous system involvement | CRMP5 (anti-CV2), PDE10A, ANNA1 |
| Poyradiculoneuropathy | Painful asymmetric weakness and numbness, neuropathic pain | CRMP5 (anti-CV2), ANNA1 (anti-Hu), PCA2, and amphiphysin |
| Pan dysautonomia | Orthostatic hypotension, dry mouth, incontinence, gastroparesis, and cardiac arrhythmias | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2, ganglionic AChR antibodies |
| Peripheral nerve hyperexcitability | Cramps, myokymia, fasciculation | CASPR2, LGI1, CRMP5 (anti-CV2), Nectrin-1 |
| Myasthenia gravis | Oculobulbar weakness, dysarthria, dysphagia, fatigable proximal limb weakness | AChR binding antibody |
| Immune-mediated necrotizing myopathy | Proximal weakness with rare respiratory or cardiac involvement | HMGCR, SRP54 |
| Dermatomyositis | Proximal weakness, skin changes | SAE1, TIF1-γ, NXP2 |
| Paraneoplastic Syndromes | Clinical Feature | Associated Antibodies |
|---|---|---|
| Limbic encephalitis | Memory impairment, seizures, behavioral change, neuropsychiatric symptoms, mood and sleep disturbances | ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), mGluR5, ANNA2 (anti-Ri), PCA2, KLHL11, GABAB receptor, AMPAR, LGI1, CASPR2, ANNA3 |
| Encephalomyelitis | Encephalopathy, myelitis, and peripheral nerve or DRG involvement | ANNA1 (anti-Hu), CRMP5 (anti-CV2), amphiphysin, GFAP |
| Cerebellar degeneration | Ataxia (usually subacute onset), dysarthria, diplopia | PCA1 (anti-Yo), PCA2, PCA-Tr (DNER), mGluR1, ANNA1,2, Zic 4, VGCC (P/Q type), amphiphysin, Ma2, NfL, KLHL11 |
| Sensory neuronopathy | Asymmetric numbness, paresthesia, sensory ataxia, neuropathic pain | ANNA1 (anti-Hu), amphiphysin, PCA2, CRMP5 (CV2) |
| Opsoclonus myoclonus syndrome | Myoclonus and opsoclonus eye movements | ANNA2 (anti-Ri), ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), NMDA-R, GluD2 |
| Gastrointestinal dysmotility | Gastroparesis, constipation, pseudo-obstruction | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2 |
| Lambert-Eaton myasthenic syndrome | Muscle weakness, fatigue, and autonomic dysfunction and absent deep tendon reflexes | P/Q-type VGCC |
| Brainstem encephalitis | Ataxia, vertigo, diplopia, dysphagia, dysarthria | ANNA2 (anti-Ri), KLHL11, ANNA1 (anti-Hu), Ma2, PCA2, CRMP5 (CV2), amphiphysin, Zic 4 |
| Paraneoplastic myelopathy | Subacute myelopathy, usually tract-specific involvement | ANNA2 (anti-Ri), ANNA1 (anti-Hu), ANNA3, CRMP5 (anti-CV2), Amphiphysin, NfL |
| Paraneoplastic stiff person syndrome | Muscle spasms and exaggerated startle response | Amphiphysin, DPPX, GAD65, glycine receptor |
| Paraneoplastic chorea | Choreic movements with more extensive nervous system involvement | CRMP5 (anti-CV2), PDE10A, ANNA1 |
| Poyradiculoneuropathy | Painful asymmetric weakness and numbness, neuropathic pain | CRMP5 (anti-CV2), ANNA1 (anti-Hu), PCA2, and amphiphysin |
| Pan dysautonomia | Orthostatic hypotension, dry mouth, incontinence, gastroparesis, and cardiac arrhythmias | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2, ganglionic AChR antibodies |
| Peripheral nerve hyperexcitability | Cramps, myokymia, fasciculation | CASPR2, LGI1, CRMP5 (anti-CV2), Nectrin-1 |
| Myasthenia gravis | Oculobulbar weakness, dysarthria, dysphagia, fatigable proximal limb weakness | AChR binding antibody |
| Immune-mediated necrotizing myopathy | Proximal weakness with rare respiratory or cardiac involvement | HMGCR, SRP54 |
| Dermatomyositis | Proximal weakness, skin changes | SAE1, TIF1-γ, NXP2 |
Abbreviations: AMPAR, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; AChR, acetylcholine receptor; ANNA, anti-neuronal nuclear antibody; AP3B2, adaptor protein 3, subunit B2; CASPR2, contactin-associated protein 2; CLL, chronic lymphoid leukemia; CRMP5, collapsin response-mediated protein 5; DRG, dorsal root ganglia; DPPX, dipeptidyl peptidase-like protein-6; GABA, gamma-aminobutyric acid; GAD65, glutamic acid decarboxylase, 65 kDa isoform; GFAP, glial fibrillary acidic protein; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; IgG, immunoglobulin; GluD2, glutamate receptor δ2; KLHL11, kelch-like protein 11; LGI1, leucine-rich glioma-inactivated 1; mGluR, metabotropic glutamate receptor; NfL, neuronal intermediate filament light chain; NMDA-R, N-methyl-d-aspartic acid receptor; POEMS, polyneuropathy organomegaly endocrinopathy M-protein and skin changes; PCA, Purkinje cell cytoplasmic antibody; SCLC, small cell lung carcinoma; VGCC, voltage-gated calcium channel; SRP 54, signal recognition particle 54; SAE1, small ubiquitin-like modifier activating enzyme heterodimer 1; TIF1-γ, transcription intermediary factor 1-γ; NXP2, nuclear matrix protein 2.
Paraneoplastic Neurological Syndromes, Clinical Features, and Associated Antibodies
| Paraneoplastic Syndromes | Clinical Feature | Associated Antibodies |
|---|---|---|
| Limbic encephalitis | Memory impairment, seizures, behavioral change, neuropsychiatric symptoms, mood and sleep disturbances | ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), mGluR5, ANNA2 (anti-Ri), PCA2, KLHL11, GABAB receptor, AMPAR, LGI1, CASPR2, ANNA3 |
| Encephalomyelitis | Encephalopathy, myelitis, and peripheral nerve or DRG involvement | ANNA1 (anti-Hu), CRMP5 (anti-CV2), amphiphysin, GFAP |
| Cerebellar degeneration | Ataxia (usually subacute onset), dysarthria, diplopia | PCA1 (anti-Yo), PCA2, PCA-Tr (DNER), mGluR1, ANNA1,2, Zic 4, VGCC (P/Q type), amphiphysin, Ma2, NfL, KLHL11 |
| Sensory neuronopathy | Asymmetric numbness, paresthesia, sensory ataxia, neuropathic pain | ANNA1 (anti-Hu), amphiphysin, PCA2, CRMP5 (CV2) |
| Opsoclonus myoclonus syndrome | Myoclonus and opsoclonus eye movements | ANNA2 (anti-Ri), ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), NMDA-R, GluD2 |
| Gastrointestinal dysmotility | Gastroparesis, constipation, pseudo-obstruction | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2 |
| Lambert-Eaton myasthenic syndrome | Muscle weakness, fatigue, and autonomic dysfunction and absent deep tendon reflexes | P/Q-type VGCC |
| Brainstem encephalitis | Ataxia, vertigo, diplopia, dysphagia, dysarthria | ANNA2 (anti-Ri), KLHL11, ANNA1 (anti-Hu), Ma2, PCA2, CRMP5 (CV2), amphiphysin, Zic 4 |
| Paraneoplastic myelopathy | Subacute myelopathy, usually tract-specific involvement | ANNA2 (anti-Ri), ANNA1 (anti-Hu), ANNA3, CRMP5 (anti-CV2), Amphiphysin, NfL |
| Paraneoplastic stiff person syndrome | Muscle spasms and exaggerated startle response | Amphiphysin, DPPX, GAD65, glycine receptor |
| Paraneoplastic chorea | Choreic movements with more extensive nervous system involvement | CRMP5 (anti-CV2), PDE10A, ANNA1 |
| Poyradiculoneuropathy | Painful asymmetric weakness and numbness, neuropathic pain | CRMP5 (anti-CV2), ANNA1 (anti-Hu), PCA2, and amphiphysin |
| Pan dysautonomia | Orthostatic hypotension, dry mouth, incontinence, gastroparesis, and cardiac arrhythmias | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2, ganglionic AChR antibodies |
| Peripheral nerve hyperexcitability | Cramps, myokymia, fasciculation | CASPR2, LGI1, CRMP5 (anti-CV2), Nectrin-1 |
| Myasthenia gravis | Oculobulbar weakness, dysarthria, dysphagia, fatigable proximal limb weakness | AChR binding antibody |
| Immune-mediated necrotizing myopathy | Proximal weakness with rare respiratory or cardiac involvement | HMGCR, SRP54 |
| Dermatomyositis | Proximal weakness, skin changes | SAE1, TIF1-γ, NXP2 |
| Paraneoplastic Syndromes | Clinical Feature | Associated Antibodies |
|---|---|---|
| Limbic encephalitis | Memory impairment, seizures, behavioral change, neuropsychiatric symptoms, mood and sleep disturbances | ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), mGluR5, ANNA2 (anti-Ri), PCA2, KLHL11, GABAB receptor, AMPAR, LGI1, CASPR2, ANNA3 |
| Encephalomyelitis | Encephalopathy, myelitis, and peripheral nerve or DRG involvement | ANNA1 (anti-Hu), CRMP5 (anti-CV2), amphiphysin, GFAP |
| Cerebellar degeneration | Ataxia (usually subacute onset), dysarthria, diplopia | PCA1 (anti-Yo), PCA2, PCA-Tr (DNER), mGluR1, ANNA1,2, Zic 4, VGCC (P/Q type), amphiphysin, Ma2, NfL, KLHL11 |
| Sensory neuronopathy | Asymmetric numbness, paresthesia, sensory ataxia, neuropathic pain | ANNA1 (anti-Hu), amphiphysin, PCA2, CRMP5 (CV2) |
| Opsoclonus myoclonus syndrome | Myoclonus and opsoclonus eye movements | ANNA2 (anti-Ri), ANNA1 (anti-Hu), Ma2, CRMP5 (anti-CV2), NMDA-R, GluD2 |
| Gastrointestinal dysmotility | Gastroparesis, constipation, pseudo-obstruction | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2 |
| Lambert-Eaton myasthenic syndrome | Muscle weakness, fatigue, and autonomic dysfunction and absent deep tendon reflexes | P/Q-type VGCC |
| Brainstem encephalitis | Ataxia, vertigo, diplopia, dysphagia, dysarthria | ANNA2 (anti-Ri), KLHL11, ANNA1 (anti-Hu), Ma2, PCA2, CRMP5 (CV2), amphiphysin, Zic 4 |
| Paraneoplastic myelopathy | Subacute myelopathy, usually tract-specific involvement | ANNA2 (anti-Ri), ANNA1 (anti-Hu), ANNA3, CRMP5 (anti-CV2), Amphiphysin, NfL |
| Paraneoplastic stiff person syndrome | Muscle spasms and exaggerated startle response | Amphiphysin, DPPX, GAD65, glycine receptor |
| Paraneoplastic chorea | Choreic movements with more extensive nervous system involvement | CRMP5 (anti-CV2), PDE10A, ANNA1 |
| Poyradiculoneuropathy | Painful asymmetric weakness and numbness, neuropathic pain | CRMP5 (anti-CV2), ANNA1 (anti-Hu), PCA2, and amphiphysin |
| Pan dysautonomia | Orthostatic hypotension, dry mouth, incontinence, gastroparesis, and cardiac arrhythmias | ANNA1 (anti-Hu), CRMP5 (anti-CV2), PCA2, ganglionic AChR antibodies |
| Peripheral nerve hyperexcitability | Cramps, myokymia, fasciculation | CASPR2, LGI1, CRMP5 (anti-CV2), Nectrin-1 |
| Myasthenia gravis | Oculobulbar weakness, dysarthria, dysphagia, fatigable proximal limb weakness | AChR binding antibody |
| Immune-mediated necrotizing myopathy | Proximal weakness with rare respiratory or cardiac involvement | HMGCR, SRP54 |
| Dermatomyositis | Proximal weakness, skin changes | SAE1, TIF1-γ, NXP2 |
Abbreviations: AMPAR, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; AChR, acetylcholine receptor; ANNA, anti-neuronal nuclear antibody; AP3B2, adaptor protein 3, subunit B2; CASPR2, contactin-associated protein 2; CLL, chronic lymphoid leukemia; CRMP5, collapsin response-mediated protein 5; DRG, dorsal root ganglia; DPPX, dipeptidyl peptidase-like protein-6; GABA, gamma-aminobutyric acid; GAD65, glutamic acid decarboxylase, 65 kDa isoform; GFAP, glial fibrillary acidic protein; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; IgG, immunoglobulin; GluD2, glutamate receptor δ2; KLHL11, kelch-like protein 11; LGI1, leucine-rich glioma-inactivated 1; mGluR, metabotropic glutamate receptor; NfL, neuronal intermediate filament light chain; NMDA-R, N-methyl-d-aspartic acid receptor; POEMS, polyneuropathy organomegaly endocrinopathy M-protein and skin changes; PCA, Purkinje cell cytoplasmic antibody; SCLC, small cell lung carcinoma; VGCC, voltage-gated calcium channel; SRP 54, signal recognition particle 54; SAE1, small ubiquitin-like modifier activating enzyme heterodimer 1; TIF1-γ, transcription intermediary factor 1-γ; NXP2, nuclear matrix protein 2.
Paraneoplastic Encephalomyelitis
Paraneoplastic encephalomyelitis is a multifocal neurological syndrome, with multi-level involvement of neuroaxis.20 These patients usually have an involvement of peripheral nerves or myenteric plexus, suggesting encephalomyelitis is not exclusively limited to the central nervous system. Furthermore, multifocal neurological syndromes without encephalitis, such as paraneoplastic myeloneuropathy or paraneoplastic subacute combined degeneration have also been described.52 Patients presenting with a clinical syndrome predominantly involving one part of neuroaxis, should be described according to the focal syndrome rather than “encephalomyelitis.” Onconeural antibody biomarkers associated with encephalomyelitis include ANNA1 (anti-Hu), ANNA2 (ant-Ri), amphiphysin, and CRMP5 (CV2) (Table 2).2,10,53–57 This is a refractory syndrome with relatively poor long-term prognosis. In a series of 200 ANNA1 (anti-Hu)-IgG-associated encephalomyelitis cases, more than 50% of cases were severely disabled at the time of diagnosis.58 Higher mortality among these cases is associated with worse functional status at the time of diagnosis, involvement of more areas of the nervous system, age >60 years, and lack of treatment.58 Early treatment of underlying malignancy was found to be an independent predictor of at least stabilization of the neurological disability for 6 months or more. Because of the presumed role of cellular immunity and reported refractoriness to first-line immunotherapies, more aggressive immunosuppression in the form of early initiation of second-line agents such as cyclophosphamide should be considered.
Antibodies With High Cancer Association, Associated Antigens, and Oncological Associations
| Antibodies | Associated Antigens | Oncological Associations |
|---|---|---|
| ANNA-1 (anti-Hu) | nELAVL (Hu) | SCLC, neuroblastoma in children |
| ANNA-2 (anti-Ri) | NOVA 1, 2 (Ri) | Lung and breast adenocarcinoma |
| ANNA-3 | Unknown | SCLC |
| AGNA-1 | SOX1 | SCLC |
| PCA-1 (anti-Yo) | CDR2 or CDR2L | Ovarian, fallopian, endometrial, and breast adenocarcinoma |
| PCA-2 | MAP1B | SCLC |
| PCA-Tr | DNER | Hodgkin lymphoma |
| Amphiphysin | Amphiphysin | Breast adenocarcinoma, SCLC |
| CRMP5 (anti-CV2) | CRMP5 | SCLC, thymoma |
| Ma2 | PNMA2 | Testicular germ cell tumor, non-small cell lung cancer |
| KLHL11-IgG | KLHL11 | Testicular germ cell tumor |
| mGluR5 | mGluR5 | Hodgkin lymphoma |
| PDE10A | PDE10A | Adenocarcinomas (lung, renal and pancreatic) |
| NfL | Neuronal intermediate filament light chain | Neuroendocrine tumor |
| Antibodies | Associated Antigens | Oncological Associations |
|---|---|---|
| ANNA-1 (anti-Hu) | nELAVL (Hu) | SCLC, neuroblastoma in children |
| ANNA-2 (anti-Ri) | NOVA 1, 2 (Ri) | Lung and breast adenocarcinoma |
| ANNA-3 | Unknown | SCLC |
| AGNA-1 | SOX1 | SCLC |
| PCA-1 (anti-Yo) | CDR2 or CDR2L | Ovarian, fallopian, endometrial, and breast adenocarcinoma |
| PCA-2 | MAP1B | SCLC |
| PCA-Tr | DNER | Hodgkin lymphoma |
| Amphiphysin | Amphiphysin | Breast adenocarcinoma, SCLC |
| CRMP5 (anti-CV2) | CRMP5 | SCLC, thymoma |
| Ma2 | PNMA2 | Testicular germ cell tumor, non-small cell lung cancer |
| KLHL11-IgG | KLHL11 | Testicular germ cell tumor |
| mGluR5 | mGluR5 | Hodgkin lymphoma |
| PDE10A | PDE10A | Adenocarcinomas (lung, renal and pancreatic) |
| NfL | Neuronal intermediate filament light chain | Neuroendocrine tumor |
Abbreviations: AGNA, anti-glial nuclear antibody; ANNA, anti-neuronal nuclear antibody; CLL, chronic lymphoid leukemia; CRMP5, collapsin response-mediated protein 5; DNER, Delta/Notch-like growth factor-related receptor; nELAVL, neuronal embryonic lethal abnormal vision-like; GABA, gamma-aminobutyric acid; IgG, immunoglobulin; KLHL11, kelch-like protein 11; mGluR, metabotropic glutamate receptor; PCA, Purkinje cell cytoplasmic antibody; PDE10A, phosphodiesterase 10A; PERM, progressive encephalomyelitis with rigidity and myoclonus; SCLC, small cell lung carcinoma; SOX1; SRY-box transcription factor 1.
Antibodies With High Cancer Association, Associated Antigens, and Oncological Associations
| Antibodies | Associated Antigens | Oncological Associations |
|---|---|---|
| ANNA-1 (anti-Hu) | nELAVL (Hu) | SCLC, neuroblastoma in children |
| ANNA-2 (anti-Ri) | NOVA 1, 2 (Ri) | Lung and breast adenocarcinoma |
| ANNA-3 | Unknown | SCLC |
| AGNA-1 | SOX1 | SCLC |
| PCA-1 (anti-Yo) | CDR2 or CDR2L | Ovarian, fallopian, endometrial, and breast adenocarcinoma |
| PCA-2 | MAP1B | SCLC |
| PCA-Tr | DNER | Hodgkin lymphoma |
| Amphiphysin | Amphiphysin | Breast adenocarcinoma, SCLC |
| CRMP5 (anti-CV2) | CRMP5 | SCLC, thymoma |
| Ma2 | PNMA2 | Testicular germ cell tumor, non-small cell lung cancer |
| KLHL11-IgG | KLHL11 | Testicular germ cell tumor |
| mGluR5 | mGluR5 | Hodgkin lymphoma |
| PDE10A | PDE10A | Adenocarcinomas (lung, renal and pancreatic) |
| NfL | Neuronal intermediate filament light chain | Neuroendocrine tumor |
| Antibodies | Associated Antigens | Oncological Associations |
|---|---|---|
| ANNA-1 (anti-Hu) | nELAVL (Hu) | SCLC, neuroblastoma in children |
| ANNA-2 (anti-Ri) | NOVA 1, 2 (Ri) | Lung and breast adenocarcinoma |
| ANNA-3 | Unknown | SCLC |
| AGNA-1 | SOX1 | SCLC |
| PCA-1 (anti-Yo) | CDR2 or CDR2L | Ovarian, fallopian, endometrial, and breast adenocarcinoma |
| PCA-2 | MAP1B | SCLC |
| PCA-Tr | DNER | Hodgkin lymphoma |
| Amphiphysin | Amphiphysin | Breast adenocarcinoma, SCLC |
| CRMP5 (anti-CV2) | CRMP5 | SCLC, thymoma |
| Ma2 | PNMA2 | Testicular germ cell tumor, non-small cell lung cancer |
| KLHL11-IgG | KLHL11 | Testicular germ cell tumor |
| mGluR5 | mGluR5 | Hodgkin lymphoma |
| PDE10A | PDE10A | Adenocarcinomas (lung, renal and pancreatic) |
| NfL | Neuronal intermediate filament light chain | Neuroendocrine tumor |
Abbreviations: AGNA, anti-glial nuclear antibody; ANNA, anti-neuronal nuclear antibody; CLL, chronic lymphoid leukemia; CRMP5, collapsin response-mediated protein 5; DNER, Delta/Notch-like growth factor-related receptor; nELAVL, neuronal embryonic lethal abnormal vision-like; GABA, gamma-aminobutyric acid; IgG, immunoglobulin; KLHL11, kelch-like protein 11; mGluR, metabotropic glutamate receptor; PCA, Purkinje cell cytoplasmic antibody; PDE10A, phosphodiesterase 10A; PERM, progressive encephalomyelitis with rigidity and myoclonus; SCLC, small cell lung carcinoma; SOX1; SRY-box transcription factor 1.
Paraneoplastic Cerebellar Degeneration
PCD usually presents with subacute progressive cerebellar ataxia.59 MRI brain may be normal in the early course, but most cases have cerebellar atrophy later in the disease. Onconeural antibodies typically associated with PCD include PCA1 (anti-Yo), Purkinje cell cytoplasmic antibody type Tr (PCA-Tr), and mGlur-1. PCA-Tr targets Delta/Notch-like epidermal growth factor-related receptor (DNER), a transmembrane protein that is expressed in the dendrites of Purkinje cells. The mGlur-1 receptor is also located in dendrites and plays a role in rapid calcium signaling.60 Whereas, PCA1 (anti-Yo) antibody targets intracellular CDR2 and/or CDR2-L protein.1,61,62 The majority of PCD associated with PCA1 (anti-Yo) have relatively poor outcomes. Interestingly, some studies have reported better prognosis of PCA1 (anti-Yo) patients with breast cancer compared to those with gynecologic cancer.63
Paraneoplastic Brainstem Encephalitis
Paraneoplastic brainstem encephalitis manifests as diplopia, ataxia, vertigo, cranial neuropathy, dysarthria, dysphagia, and at times impaired level of consciousness. A majority of paraneoplastic brainstem encephalitis cases have inflammatory CSF. MRI brain in early disease course may be unremarkable or may show T2 hyper-intensity. However, as disease progresses patients’ brainstem atrophy becomes more apparent.
Onconeural antibodies associated with brainstem encephalitis are ANNA2 (anti-Ri), KLHL11, Ma2, CRMP5 (CV2), ANNA1 (anti-Hu), and amphiphysin. Laryngospasm and/or jaw dystonia are relatively distinguishable neurological presentations reported in 25% of ANNA2 (anti-Ri) paraneoplastic brainstem encephalitis cases.64 Whereas sensory neural hearing loss and tinnitus are early presenting features of KLHL11-IgG-associated neurological syndrome.50
A considerable proportion of patients with brainstem encephalitis continue to have progressive decline, despite treatment of underlying cancer and aggressive immunotherapy. However, among both Ma2 and KLHL11-IgG seropositive cases detection and treatment of testicular germ cell tumor has been associated with favorable clinical prognosis.49,65
Paraneoplastic Opsoclonus-Myoclonus Syndrome
Opsoclonus-myoclonus syndrome (OMS) also known as “dancing eye syndrome,” consists of spontaneous, arrhythmic, conjugate high-frequency oscillations of the eyes with horizontal, vertical, and torsional saccades that are associated with myoclonus of the head, trunk, or extremities.66 Opsoclonus is diagnosed clinically based on back-to-back multidirectional conjugate saccades without an inter-saccadic interval. In children, OMS age of onset ranges from 12 to 36 months.66,67 Approximately, 50% of pediatric OMS cases are associated with neuroblastoma.66 Glutamate receptor δ2 IgG has been identified as a serological biomarker in a subset of pediatric OMS cases.68 Among adult patients, paraneoplastic OMS has been associated with small cell lung carcinoma, breast adenocarcinoma, ovarian teratoma, and testicular germ cell tumor.69,70 Onconeural antibodies associated with adult paraneoplastic OMS include ANNA2 (anti-Ri), ANNA1 (anti-Hu), Ma2, CRMP5 (CV2), and NMDA-R antibodies.70 Treatment includes identification and management underlying of neoplasm, and early initiation of immunotherapy. Symptomatic treatments include clonazepam, levetiracetam, baclofen, and gabapentin.69 In comparison with idiopathic OMS, paraneoplastic OMS has a more refractory course. However, complete or partial neurological recovery after removal of underlying tumor has been reported.71
Paraneoplastic Myelopathies
Most of the cases have subacute or insidious onset myelopathic signs and symptoms, with a progressive course.72,73 MRI spine usually demonstrates longitudinally extensive (>3 vertebral segments) tract or gray matter-specific T2 hyperintense signal abnormalities.72,73 Lateral and dorsal columns and central gray matter are commonly involved. Gadolinium enhancement is seen in approximately 50% of the cases.72 The majority of cases have lymphocyte-predominant pleocytosis and elevated protein in the CSF. Supernumerary oligoclonal band have been reported in 30%-75% of patients.72,73 Onconeural antibodies associated with paraneoplastic myelopathies are amphiphysin, CRMP5 (CV2), ANNA1 (anti-Hu), ANNA2 (anti-Ri), ANNA3, and PCA2.72–74 Furthermore, paraneoplastic AQP4-IgG myelitis in association with non-small cell lung and breast cancers have also been reported.75
Paraneoplastic myelopathies are usually associated with poor outcome. The majority (52%) of cases reported in a large series from our institution required gait aid for ambulation.72 The median time from symptom onset to wheelchair dependence was 9 months (range 1-21 months).
Paraneoplastic Stiff Person Syndrome
“Stiff man” syndrome was first described in a series of male patients in 1956.76 With description of female cases the disease nomenclature was changed to gender-neutral “Stiff person syndrome” (SPS).77 SPS is characterized by fluctuating muscle stiffness, superimposed spasms, and exaggerated startle. The muscle spasms principally involve the trunk and lower limbs, typically symmetric, more proximal in distribution, and associated with an increased sensitivity to external stimuli.78 SPS spectrum disorders (SPSD) comprise of classic SPS, focal or segmental SPS such as stiff limb syndrome, progressive encephalomyelitis with rigidity and myoclonus (PERM), and overlapping syndromes.79 Glutamic acid decarboxylase (GAD) 65 IgG, glycine receptor (GlyR) IgG, amphiphysin IgG, and dipeptidyl peptidase-like protein 6 (DPPX) IgG are antibodies associated with SPSD.80,81 GAD65 and glycine receptor antibodies are usually associated with non-paraneoplastic SPSD. A minority of DPPX-IgG cases have underlying hematological malignancies. Amphiphysin IgG has a strong paraneoplastic association with breast cancer and small cell lung cancer.82,83 Treatment of paraneoplastic SPS is focused on symptomatic treatment, immunotherapy, and treatment of underlying tumor.84 Symptomatic treatment includes use of muscle relaxants such as benzodiazepines, baclofen, tizanidine, and botulinum toxin injections to help with the muscle spasms.78,84
Peripheral Nervous System Syndromes
Paraneoplastic Sensory Neuronopathy
Sensory neuronopathy is caused by damage to the neurons in dorsal root ganglia.85 Patients usually present with burning pain and numbness in upper and/or lower limbs, usually in an asymmetric pattern. Symptoms progress rapidly over weeks. Involvement of the face, abdomen, or trunk often provides a diagnostic clue. On examination, there is a loss of sensation to all modalities with marked impairment of proprioception, sensory ataxia, and deep tendon reflexes are diminished or absent. The electrophysiological evaluation shows absent or reduced sensory responses globally with normal to minimal abnormalities of the motor responses.86 CSF analysis commonly demonstrates elevated protein, pleocytosis, and the presence of oligoclonal bands. Small cell lung cancer is the most common cancer associated with this phenotype. Common onconeural antibody associations include ANNA1 (anti-Hu), amphiphysin, CRMP5 (CV2), and PCA2 antibodies.87 Other common cancer associations include thymoma and breast cancer.58 Even though majority of cases have a refractory course, combined therapeutic approach focused management of underlying cancer and early immunosuppressive therapy initiation have demonstrated most significant clinical benefit.88
Paraneoplastic Polyradiculoneuropathy
Polyradiculoneuropathy is characterized by clinical and electrophysiological involvement of nerve roots and proximal nerves.89 In some cases, MRI spine and/or lumbar plexus demonstrates T2 hyper-intensity and/or post-contrast gadolinium enhancement of nerve roots and plexus. CRMP5 (CV2), PCA2, ANNA1 (anti-Hu), and amphiphysin are the onconeural antibodies commonly associated with paraneoplastic polyradiculoneuropathy.89–91 CRMP5 CV2 IgG-associated neuropathy comprises a distinct clinical phenotype of subacute painful asymmetric polyradiculoneuropathy (both proximal and distal limb weakness and sensory involvement) with perivascular inflammation on nerve biopsy. It is commonly associated with small cell lung cancer and thymoma.89 Neuropathy has been described as the most frequent neurological presentation with the PCA2-IgG seropositivity. The majority of PCA2 neuropathy cases had polyradiculoneuropathy phenotype followed by sensory neuronopathy.91 A considerable proportion of amphiphysin IgG seropositive patients with breast cancer also present with symmetric axonal polyradiculoneuropathy with or without co-existing SPS.90
Autonomic Neuropathy
Autonomic neuropathy as a paraneoplastic syndrome can present in isolation or may co-exist with other central/peripheral nervous system manifestations. Chronic gastrointestinal dysmotility syndrome and acute pandysautonomia are well-characterized paraneoplastic phenotypes.92 Orthostatic hypotension, dry mouth, incontinence, and cardiac arrhythmias are other presenting symptoms/signs. Small cell lung cancer and ANNA1 (anti-Hu) and/or CRMP5 (CV2) antibodies are common oncological and serological associations, respectively. Autoimmune autonomic ganglionopathy associated with ganglionic AChR antibodies has also been rarely described in association with cancer.93,94
Peripheral Nerve Hyperexcitability Syndromes
Autoimmune peripheral nerve hyperexcitability syndromes can be of paraneoplastic origin, the two most commonly described phenotypes are Isaac syndrome and Morvan’s syndrome. Isaac syndrome is clinically characterized by generalized muscle stiffness (present even during sleep) and cramps, myokymia, fasciculation, and often associated with autonomic dysfunction-like sialorrhea and hyperhidrosis.95 Electromyography (EMG) characteristically shows myokymia and/or neuromyotonia. Patients with Morvan’s syndrome have CNS involvement (encephalopathy, sleep dysfunction) in addition to features of peripheral nerve hyperexcitability. Contactin-associated protein-like 2 (CASPR2) and/or Leucine-rich glioma-inactivated 1 (LGI1) IgG are neural-specific antibodies associated with this phenotype, while thymoma is the commonly associated tumor. Antibodies against Netrin-1 receptor have also been described in association with neuromyotonia and myasthenia gravis (MG) in patients with thymoma.96
Neuromuscular Junction Disorders
MG is an autoimmune disorder with antibodies against acetylcholine receptors (AChR) and other related proteins, attacking the post-synaptic membrane in the neuromuscular junction.97 Patients usually present with predominant oculobulbar weakness in the form of double vision, droopy eyelids, and often swallowing difficulty along with fatigable proximal limb weakness. In about 10%-15% of cases, MG occurs as a paraneoplastic phenomenon in association with thymoma and rarely lung cancer.97,98
Lambert-Eaton myasthenic syndrome is another rare neuromuscular junction disorder associated with presynaptic membrane P/Q-type VGCC with very distinctive clinical and electrophysiological features.99,100 Muscle weakness, fatigue, and autonomic dysfunction and absent deep tendon reflexes are the most common findings. More than 50% of cases are associated with cancer, most commonly small cell lung cancer. Diagnosis requires a high level of awareness and can initiate prompt screening for cancer.101
Paraneoplastic Myopathies
Immune-mediated necrotizing myopathy (IMNM) is a relatively rare autoimmune myopathy often associated with signal recognition particle 54 (SRP54) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) autoantibodies. Patients usually present with acute or subacute onset progressive proximal weakness with rare respiratory or cardiac involvement. Histopathologically, there tends to be minimal or no inflammation on muscle biopsy as compared to other immune myopathies.102 While statin use is the most-recognized risk factor for HMGCR-IgG-mediated syndrome, a minority of IMNM also have paraneoplastic associations.103 Malignancy tends to occur more frequently in seronegative cases.104
Dermatomyositis is another neurological presentation historically considered to have a paraneoplastic association.105 Certain myositis antibodies like anti-small ubiquitin-like modifier activating enzyme heterodimer 1 (anti-SAE1), anti-transcription intermediary factor 1-γ (anti-TIF1-γ), and anti-nuclear matrix protein 2 (anti-NXP2) antibodies are associated with an increased risk of cancer.106 Cancer is usually recognized within 3 years of myositis diagnosis, with most diagnosed within 12 months.
Neurological Immune-Related Adverse Events Associated With Immune Checkpoint Inhibitors
In current clinical practice, three classes of ICIs are being utilized including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death receptor 1 (PD-1), and programmed cell death ligand 1 (PDL-1) inhibitors.107 ICIs are being utilized for a variety of cancer indications including melanoma, non-small cell lung carcinoma, renal cell carcinoma, Hodgkin’s lymphoma, urothelial cancer, Merkel cell carcinoma, and extensive-stage small cell lung cancer.108
ICIs have been associated with a variety of immune-related adverse events (irAEs). All organ systems can be affected. The commonly involved organ systems are the lungs, gastrointestinal tract, liver, endocrine, and kidney. The involvement of the nervous system is less common.109 The neurologic iRAEs may resemble PNS phenotype, or preexisting PNS may be worsened by ICI administration. However, neurologic irAEs (even those resembling classic PNS phenotype) differ from PNS, as the former is caused by drug-induced disruption of immunologic self-tolerance and/or potentiation of autoimmunity. The median onset of neurologic irAEs is typically 2-16 weeks after the ICI initiation.110 The risk of irAEs appears to 3-fold higher during the first 4 weeks after ICI treatment compared to later timepoints.111
Pathophysiology
Physiologically, CTLA-4, PD-1, and PDL-1 are the immune checkpoint proteins that inhibit autoreactive T-lymphocytes preventing the occurrence of autoimmune disorder. In many cancers, the tumor cells enhance this inhibitory signal to escape the anti-tumor immune response. Therefore, blocking these molecules reactivates T-lymphocytes to destroy tumor cells. However, inhibition of the immune checkpoint proteins may lead to the development of immune-mediated complications as well as potentiation of the paraneoplastic disorders.112,113
The majority of the n-irAEs are presumed to be secondary to three potential mechanisms: (1) immune response against common autoantigens expressed by the cancer and neural tissue, (2) exacerbation of preexisting autoimmunity, and (3) direct toxic effect of therapeutic monoclonal antibody.114
Cancer antigen-driven paraneoplastic mechanism is supported by a PCD mouse model of subcutaneously implanted breast cancer treated CTLA-4 inhibitor.115 Additionally, autopsies of patients who developed fatal myocarditis after receiving CTLA-4 and PD-1 inhibitors have demonstrated robust T-cell and macrophage infiltration in myocardium, and selective clonal T-cell population infiltrating myocardium have been found to be identical to those present in tumor cells.116
Reports of MS exacerbation following administration of ICIs are good examples of n-irAEs secondary to preexisting autoimmunity.117
ICI-related hypophysitis in association with CTLA-4 inhibitors is a good example of potential role of these monoclonal antibodies in directly causing irAEs.114 However, expression of these checkpoint proteins does not seem to be restricted to pituitary gland and their expression in other neural tissues have also been described.
Central Nervous System irAEs
Various CNS disorders have been reported in association with ICIs, including aseptic meningitis, encephalitis, myelitis, cerebellar ataxia, sarcoidosis, and Vogt-Konyagi-Harada-like syndrome.2,113,118–120 Moreover, exacerbation as well as de novo multiple sclerosis have also been reported in association with ICI use, particularly ipilimumab (CTLA-4 inhibitor).121,122 CNS irAEs resembling classic PNS phenotypes include limbic encephalitis,123–125 cerebellar ataxia,126 OMS,127 and encephalomyelitis.128 Limbic encephalitis is one of the most common n-irAEs resembling PNS phenotype.2 The onconeural antibodies which were reported in association with ICI use include autoantibodies against ANNA1 (anti-Hu), Ma2, SOX1, and CRMP5 (CV2).2,112,118
Peripheral Nervous System irAEs
ICIs are also associated with peripheral nervous system involvements including myopathies, neuromuscular junction disorders as well as neuropathies.118,129 IMNM, and myositis have been reported in association with CTLA-4, PD-1, PDL-1 inhibitors or combination therapies.129 Of note, a considerable proportion of ICI-related myopathies (irMyopathies) may have concomitant myocarditis.129 Patients with ICI-related MG (irMG) may have co-existing features of myositis on histopathological assessments.118 Lambert-Eaton myasthenic syndrome in association with P/Q-type VGCC antibodies has also been reported among patients with squamous cell and small cell lung cancer.130,131
A wide range of neuropathies have been reported as a complication of ICI, including Guillain-Barre syndrome, sensory ganglionopathy, Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), sensorimotor polyneuropathy, axonal polyradiculoneuropathy, mononeuritis multiplex, cranial neuropathy, and phrenic nerve palsy.118,129 Elevated CSF protein has been reported in most of the cases, and more than 50% of cases have CSF pleocytosis.
Management of Paraneoplastic Syndromes
Tumor Detection
Detection and treatment of tumor are critical in the management of the PNS. Evaluation of a patient with suspected neurological paraneoplastic disorder requires a detailed history regarding risk factors of cancer (eg, smoking, family history of cancers), constitutional symptoms (eg, weight loss, prolonged fever), enlarged lymph nodes, hemoptysis. Computed tomography of the chest, abdomen, and pelvis, mammography, and ovarian and testicular ultrasonography should be considered if a PNS is suspected. Fluorodeoxyglucose positron emission tomography (FDG-PET) should be performed if the initial tests are unrevealing. FDG-PET considerably improves the sensitivity for tumor detection in the setting of paraneoplastic autoimmunity.132–134 If initial cancer screenings are negative, surveillance imaging at 6-month to yearly intervals should be considered to look for the emergence of tumor especially in patients with a positive onconeural antibody.2
Immunotherapy
There have been no data from placebo-controlled trials for immunotherapy in PNS. Discussion here will focus on our approach to patients with PNS. The major principles for PNS management are treatment of the underlying neoplasm and immunosuppression.135
In our practice, intravenous methylprednisolone (IVMP), intravenous immunoglobulin (IVIG), and plasma exchange (PLEX) are commonly utilized as acute immunotherapies either as single therapies or in combinations such as IVMP and PLEX or IVMP and IVIG.136,137 In most PNS cases, in the inpatient setting, we utilize IVMP 1 g daily over 5 days, unless there is a contraindication. We monitor clinical response for 3-5 days after the completion of IVMP before considering IVIG (0.4 g/kg/day for 5 days) or PLEX (5 sessions over 7-14 days). In the outpatient setting, we commonly utilize a 6-week or 12-week IVMP or IVIG regimens (Table 3). Ideally, we re-evaluate within a week of the last infusion. In addition to patient reported change in symptoms, objective measurements such as neurological examination, formal neurocognitive test, neuroimaging (MRI, PET brain), and electrodiagnostic studies, are used for evaluation of the treatment response. In most PNS cases, disease stabilization is considered as a favorable response.138,139
Immunotherapies for Management of Paraneoplastic Syndromes According to Mayo Clinic Experience (Non-Evidence-Based)
| Treatment | Route, Dose, and Duration | Mechanism of Action | Common Side Effects |
|---|---|---|---|
| Acute treatments | |||
| Methylprednisolone | IVMP 1000 mg/day for 3-5 days 6-week trial: IVMP 1000 mg/day for 3 days then weekly for 6 weeks 12-week trial: IVMP 1000 mg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Bind to glucocorticoid receptor in cytoplasm, then process through non-genomics and genomics results in reducing chemokine, cytokine, and leukocyte migration to the target tissue | Hyperglycemia, insomnia, psychosis, cushingoid appearance, osteoporosis, increased risk of infection |
| Plasma exchange | 1.5× plasma volume, 5-7 sessions alternate days (over 10-14 days) | Removal of pathological antibodies, immune complex, complements, and cytokines | Complication from catheter insertion, hemodynamic instability, infection |
| Intravenous immunoglobulin | IV 0.4 g/kg/day for 5 days 6-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 6 weeks 12-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Neutralize pathogenic antibodies and block Fc receptors on macrophages preventing phagocytosis | Headache, aseptic meningitis, thromboembolic events, acute renal failure, and anaphylaxis due to IgA deficiency |
| Maintenance therapy | |||
| Cyclophosphamide | IV (0.6-1.0 g/m2 monthly for 6 months) Oral (Typical dosing: 2 mg/kg, dosing based on GFR) | Alkylating agent that predominantly depletes T cells | Infection, malignancy (lymphoma, skin cancers, and others), hemorrhagic cystitis, infertility, alopecia, and nausea and vomiting |
| Rituximab | Initial loading dose: 1000 mg once, followed by another 1000 mg dose 2 weeks later. Maintenance dosing: 1000 mg every 6 months | Depletes CD20+ B cells | Allergic reaction, opportunistic infection, reactivation of tuberculosis infection or hepatitis B infection |
| Azathioprine | Start at 1.5 mg/kg/day. If tolerated, increase to 2 mg/kg/ day. Further increases in dose depend on the MCV or monitoring results. Typical goal dose: 2-3 mg/kg/d. | Inhibiting purine synthesis through metabolite 6-mercaptopurine leading to decreased B- and T-cell proliferation | Hypersensitivity reactions, Infection, nausea, anemia, rash, transaminitis, pancreatitis |
| Mycophenolate mofetil | Start at 500 mg twice per day. If tolerated, then increase to 1000 mg twice per day. Typical goal dose: 2000 mg per day | Inhibiting of inosine monophosphate dehydrogenase leading to decreased purine synthesis and DNA formation causing decreased B- and T-cell proliferation | Infection, increased risk of malignancy (lymphoma, skin cancers, etc.), diarrhea, hypertension, hepatitis, and renal failure |
| Treatment | Route, Dose, and Duration | Mechanism of Action | Common Side Effects |
|---|---|---|---|
| Acute treatments | |||
| Methylprednisolone | IVMP 1000 mg/day for 3-5 days 6-week trial: IVMP 1000 mg/day for 3 days then weekly for 6 weeks 12-week trial: IVMP 1000 mg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Bind to glucocorticoid receptor in cytoplasm, then process through non-genomics and genomics results in reducing chemokine, cytokine, and leukocyte migration to the target tissue | Hyperglycemia, insomnia, psychosis, cushingoid appearance, osteoporosis, increased risk of infection |
| Plasma exchange | 1.5× plasma volume, 5-7 sessions alternate days (over 10-14 days) | Removal of pathological antibodies, immune complex, complements, and cytokines | Complication from catheter insertion, hemodynamic instability, infection |
| Intravenous immunoglobulin | IV 0.4 g/kg/day for 5 days 6-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 6 weeks 12-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Neutralize pathogenic antibodies and block Fc receptors on macrophages preventing phagocytosis | Headache, aseptic meningitis, thromboembolic events, acute renal failure, and anaphylaxis due to IgA deficiency |
| Maintenance therapy | |||
| Cyclophosphamide | IV (0.6-1.0 g/m2 monthly for 6 months) Oral (Typical dosing: 2 mg/kg, dosing based on GFR) | Alkylating agent that predominantly depletes T cells | Infection, malignancy (lymphoma, skin cancers, and others), hemorrhagic cystitis, infertility, alopecia, and nausea and vomiting |
| Rituximab | Initial loading dose: 1000 mg once, followed by another 1000 mg dose 2 weeks later. Maintenance dosing: 1000 mg every 6 months | Depletes CD20+ B cells | Allergic reaction, opportunistic infection, reactivation of tuberculosis infection or hepatitis B infection |
| Azathioprine | Start at 1.5 mg/kg/day. If tolerated, increase to 2 mg/kg/ day. Further increases in dose depend on the MCV or monitoring results. Typical goal dose: 2-3 mg/kg/d. | Inhibiting purine synthesis through metabolite 6-mercaptopurine leading to decreased B- and T-cell proliferation | Hypersensitivity reactions, Infection, nausea, anemia, rash, transaminitis, pancreatitis |
| Mycophenolate mofetil | Start at 500 mg twice per day. If tolerated, then increase to 1000 mg twice per day. Typical goal dose: 2000 mg per day | Inhibiting of inosine monophosphate dehydrogenase leading to decreased purine synthesis and DNA formation causing decreased B- and T-cell proliferation | Infection, increased risk of malignancy (lymphoma, skin cancers, etc.), diarrhea, hypertension, hepatitis, and renal failure |
Abbreviations: IVMP, intravenous methylprednisone; IV, intravenous; IVIG, intravenous immunoglobulin; MCV, mean corpuscular volume.
Immunotherapies for Management of Paraneoplastic Syndromes According to Mayo Clinic Experience (Non-Evidence-Based)
| Treatment | Route, Dose, and Duration | Mechanism of Action | Common Side Effects |
|---|---|---|---|
| Acute treatments | |||
| Methylprednisolone | IVMP 1000 mg/day for 3-5 days 6-week trial: IVMP 1000 mg/day for 3 days then weekly for 6 weeks 12-week trial: IVMP 1000 mg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Bind to glucocorticoid receptor in cytoplasm, then process through non-genomics and genomics results in reducing chemokine, cytokine, and leukocyte migration to the target tissue | Hyperglycemia, insomnia, psychosis, cushingoid appearance, osteoporosis, increased risk of infection |
| Plasma exchange | 1.5× plasma volume, 5-7 sessions alternate days (over 10-14 days) | Removal of pathological antibodies, immune complex, complements, and cytokines | Complication from catheter insertion, hemodynamic instability, infection |
| Intravenous immunoglobulin | IV 0.4 g/kg/day for 5 days 6-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 6 weeks 12-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Neutralize pathogenic antibodies and block Fc receptors on macrophages preventing phagocytosis | Headache, aseptic meningitis, thromboembolic events, acute renal failure, and anaphylaxis due to IgA deficiency |
| Maintenance therapy | |||
| Cyclophosphamide | IV (0.6-1.0 g/m2 monthly for 6 months) Oral (Typical dosing: 2 mg/kg, dosing based on GFR) | Alkylating agent that predominantly depletes T cells | Infection, malignancy (lymphoma, skin cancers, and others), hemorrhagic cystitis, infertility, alopecia, and nausea and vomiting |
| Rituximab | Initial loading dose: 1000 mg once, followed by another 1000 mg dose 2 weeks later. Maintenance dosing: 1000 mg every 6 months | Depletes CD20+ B cells | Allergic reaction, opportunistic infection, reactivation of tuberculosis infection or hepatitis B infection |
| Azathioprine | Start at 1.5 mg/kg/day. If tolerated, increase to 2 mg/kg/ day. Further increases in dose depend on the MCV or monitoring results. Typical goal dose: 2-3 mg/kg/d. | Inhibiting purine synthesis through metabolite 6-mercaptopurine leading to decreased B- and T-cell proliferation | Hypersensitivity reactions, Infection, nausea, anemia, rash, transaminitis, pancreatitis |
| Mycophenolate mofetil | Start at 500 mg twice per day. If tolerated, then increase to 1000 mg twice per day. Typical goal dose: 2000 mg per day | Inhibiting of inosine monophosphate dehydrogenase leading to decreased purine synthesis and DNA formation causing decreased B- and T-cell proliferation | Infection, increased risk of malignancy (lymphoma, skin cancers, etc.), diarrhea, hypertension, hepatitis, and renal failure |
| Treatment | Route, Dose, and Duration | Mechanism of Action | Common Side Effects |
|---|---|---|---|
| Acute treatments | |||
| Methylprednisolone | IVMP 1000 mg/day for 3-5 days 6-week trial: IVMP 1000 mg/day for 3 days then weekly for 6 weeks 12-week trial: IVMP 1000 mg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Bind to glucocorticoid receptor in cytoplasm, then process through non-genomics and genomics results in reducing chemokine, cytokine, and leukocyte migration to the target tissue | Hyperglycemia, insomnia, psychosis, cushingoid appearance, osteoporosis, increased risk of infection |
| Plasma exchange | 1.5× plasma volume, 5-7 sessions alternate days (over 10-14 days) | Removal of pathological antibodies, immune complex, complements, and cytokines | Complication from catheter insertion, hemodynamic instability, infection |
| Intravenous immunoglobulin | IV 0.4 g/kg/day for 5 days 6-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 6 weeks 12-week trial: IVIG 0.4 g/kg/day for 3 days then weekly for 5 weeks, then every 2 weeks for 6 weeks | Neutralize pathogenic antibodies and block Fc receptors on macrophages preventing phagocytosis | Headache, aseptic meningitis, thromboembolic events, acute renal failure, and anaphylaxis due to IgA deficiency |
| Maintenance therapy | |||
| Cyclophosphamide | IV (0.6-1.0 g/m2 monthly for 6 months) Oral (Typical dosing: 2 mg/kg, dosing based on GFR) | Alkylating agent that predominantly depletes T cells | Infection, malignancy (lymphoma, skin cancers, and others), hemorrhagic cystitis, infertility, alopecia, and nausea and vomiting |
| Rituximab | Initial loading dose: 1000 mg once, followed by another 1000 mg dose 2 weeks later. Maintenance dosing: 1000 mg every 6 months | Depletes CD20+ B cells | Allergic reaction, opportunistic infection, reactivation of tuberculosis infection or hepatitis B infection |
| Azathioprine | Start at 1.5 mg/kg/day. If tolerated, increase to 2 mg/kg/ day. Further increases in dose depend on the MCV or monitoring results. Typical goal dose: 2-3 mg/kg/d. | Inhibiting purine synthesis through metabolite 6-mercaptopurine leading to decreased B- and T-cell proliferation | Hypersensitivity reactions, Infection, nausea, anemia, rash, transaminitis, pancreatitis |
| Mycophenolate mofetil | Start at 500 mg twice per day. If tolerated, then increase to 1000 mg twice per day. Typical goal dose: 2000 mg per day | Inhibiting of inosine monophosphate dehydrogenase leading to decreased purine synthesis and DNA formation causing decreased B- and T-cell proliferation | Infection, increased risk of malignancy (lymphoma, skin cancers, etc.), diarrhea, hypertension, hepatitis, and renal failure |
Abbreviations: IVMP, intravenous methylprednisone; IV, intravenous; IVIG, intravenous immunoglobulin; MCV, mean corpuscular volume.
The decision for long-term immunotherapy can be challenging. In patients with classic PNS and typical cancer-onconeural antibody associations, who present within 2 years from the onset of symptoms, we consider a long-term immunotherapy in the early disease course. For patients who presented late and are not having active disease progression, we usually re-evaluate the clinical response after treatment of underlying tumor and 12-week course of acute therapy, before considering the further escalation of immunological treatment.
Among patients with onconeural antibodies targeting an intracellular epitope, our preferred options for chronic immunosuppression are azathioprine, mycophenolate, and cyclophosphamide which target both T- and B-lymphocytes because the pathogenesis of these PNS is presumed to involve cellular immune response. Severity and progression are the factors that we use to determine the agent of choice. In aggressive cases, oral or IV cyclophosphamide is the preferred option. A prospective study demonstrated that twice as many patients who were treated with a combination of PLEX and cyclophosphamide demonstrated an improvement in disability compared to PLEX alone; however, severe leukopenia was seen in 4 of 10 patients who received cyclophosphamide.139 Among PNS with cell-surface antibodies, such as GABA-B-R IgG or mGlur5, we prefer rituximab as chronic immunotherapy because these antibodies are thought to be directly pathogenic. Therefore, suppression of antibody production by targeting B cells is potentially beneficial (Table 3).
Management of n-irAEs
The American Society of Clinical Oncology and the Society for Immunotherapy of Cancer recommend holding ICI in all CTCAE grade 3-4 toxicity and initiation of corticosteroids.140,141 Most n-irAE with PNS phenotype are classified as grade 3 or 4.2 In our practice, all severe n-irAE patients are treated with IVMP 1 g/day for 3-5 days or oral prednisone 60-80 mg/day depending on the n-irAE phenotype.107 In the patients with favorable response to corticosteroids, 4- to 6-week course of prednisone taper is considered. In the refractory cases, early initiation of IVIG, plasmapheresis, and other immunosuppression medications such as rituximab should be considered.107,141 The majority of the n-irAE patients with classic paraneoplastic phenotype and onconeural antibody seropositivity usually have a refractory course and require more aggressive immunosuppression.142 In few of these cases, we have utilized cyclophosphamide with limited success.112
Retreating with ICI potentially increases the risk of n-irAE relapse.107 We try to avoid re-treatment among patients with n-irAEs resembling classic paraneoplastic syndrome due to risk of irreversible neurological deterioration.142 If an ICI has to be resumed, we treat the patients with corticosteroids until the symptom resolution or stabilization and observe for 2-8 weeks before restarting an ICI.
Conclusion
In the past century, the number of serological biomarkers and associated phenotypes has considerably expanded. Furthermore, as cancer indications of ICIs grow, we are likely to encounter more patients with clinical phenotypes resembling PNS in our neurologic practice. Recognition of various clinical phenotypes, onconeural antibodies, and their neoplastic association may help clinicians diagnose these rare but refractory conditions early and treat them appropriately.
Funding
None declared.
Conflict of interest statement. Dr. J.J., Dr. P.P., Dr. S.O.M., and Dr. S.T. have no disclosures. Dr. S.J.P. is a named inventor on filed patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker. He has a patent pending for Septin 5, MAP1B, and KLHL11-IgGs as markers of neurological autoimmunity and paraneoplastic disorders. He has consulted for Alexion and Medimmune. He has received research support from Grifols, Medimmune, and Alexion. All compensation for consulting activities is paid directly to Mayo Clinic. D.D. has received research support from Center of Multiple Sclerosis and Autoimmune Neurology, Center for Clinical and Translational Science, and Grifols Pharmaceuticals. He has consulted for UCB and Astellas Pharmaceuticals. All compensation for consulting activities is paid directly to Mayo Clinic. Dr. D.D. has a patent pending for KLHL11-IgG as a marker of neurological autoimmunity.
Role of the funder/sponsor. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.

{kind=link}