






Abstract
Diffuse intrinsic pontine gliomas (DIPG), within diffuse midline gliomas are aggressive pediatric brain tumors characterized by histone H3-K27M mutation. Small-molecule inhibitors for the EZH2-H3K27 histone methyltransferase have shown promise in preclinical animal models of DIPG, despite having little effect on DIPG cells in vitro. Therefore, we hypothesized that the effect of EZH2 inhibition could be mediated through targeting of this histone modifying enzyme in tumor-associated microglia.
Primary DIPG tissues, and cocultures between microglia and patient-derived DIPG or -pediatric high-grade glioma (pHGG) cell lines, were used to establish the H3-K27M status of each cell type. Antisense RNA strategies were used to target EZH2 gene expression in both microglia and glioma cells. Microglia anti-tumoral properties were assessed by gene expression profile, tumor cell invasion capacity, microglial phagocytic activity, and associated tumor cell death.
In primary DIPG tissues, microglia do not carry the H3-K27M mutation, otherwise characteristic of the cancer cells. Activation of a microglial tumor-supportive phenotype by pHGG, independently of their H3-K27M status, is associated with a transient H3K27me3 downregulation. Repression of EZH2 in DIPG cells has no impact on tumor cell survival or their ability to activate microglia. However, repression of EZH2 in microglia induces an anti-tumor phenotype resulting in decreased cancer cell invasion capability, increased microglial phagocytosis, and tumor-related cell death.
These results indicate that microglia, beyond the tumor cells, contribute to the observed response of DIPG to EZH2 inhibition. Results highlight the potential importance of microglia as a new therapeutic avenue in DIPG.
Methylation of H3K27 is important for DIPG-induced activation of microglia.
EZH2 inhibition in microglia leads to robust anti-tumoral effects in pediatric HGG.
Microglia may represent a novel therapeutic avenue in the treatment of DIPG.
Here, we demonstrate the potential of microglia to exert anti-tumoral effects on pediatric high-grade glioma cells regardless of their H3K-27M mutational status. We define, for the first time, an epigenetic mechanism that regulates DIPG-induced microglia activation that can be specifically targeted by EZH2 inhibition to reprogram these cells. EZH2 inhibition has previously been shown to exert promising therapeutic benefits in animal models of DIPG, even though it has little effect on DIPG tumor cells in vitro. This work further highlights that EZH2 inhibition may exert its clinical benefit by targeting the tumor microenvironment rather than the tumor cells themselves. Given the poor prognosis in pHGGs, especially DIPG, and lack of effective treatment, our results provide further evidence for the use of EZH2 inhibitors in the clinic as well as uncovering microglia as potential cells of interest in the treatment of these devastating diseases.
Pediatric diffuse midline gliomas are a group of diseases that affect the brainstem, spinal cord, and thalamus. One subtype of this group, pediatric diffuse midline gliomas H3-K27M mutant, formerly known as diffuse intrinsic pontine glioma (DIPG), is an incredibly aggressive pediatric brain tumor which originates in the pons.1 DIPG strikes at the heart of childhood, with a median age at diagnosis of 7 years. DIPG prognosis is dismal with over 90% of cases dying within 2 years of diagnosis. The median overall survival of children diagnosed with DIPG is 9–12 months.2 These outcomes make DIPG one of the most devastating and challenging pediatric cancers. The pons, part of the brainstem, is involved in many of the body's most vital functions, and therefore the location of the DIPG tumor majorly limits possible interventions. In addition, DIPG tumors are diffusely infiltrative and by the time they are diagnosed, extensive tumor cell infiltration to surrounding normal tissue is often observed. These anatomic characteristics of DIPG exclude surgical resection as a therapeutic option. The standard of care for DIPG patients remains fractionated radiation therapy, which only mitigates neurological symptoms temporarily and extends progression-free and overall survival by a few months at best.3 This notable lack of significant progress for the treatment of DIPG has promoted an interest in the molecular characterization of this tumor type, in search of new potential molecular therapeutic targets.4
A molecular breakthrough for DIPG has been the identification of prevalent somatic mutations in histone H3 resulting in the substitution of lysine 27 with methionine (H3-K27M) in genes encoding histone H3 variants.5,6 H3-K27M mutations are found at large in gliomas occurring in midline locations and 2016 WHO classification even recognized this mutation as an intrinsic characteristic of diffuse midline gliomas such as DIPG.1 H3-K27M mutations are associated with a poorer prognosis in pediatric patients compared to H3 wild-type DIPG tumors.7,8 H3-K27M has been shown to inhibit the function of the histone-lysine N-methyltransferase Enhancer of zeste homolog 2 (EZH2) within the polycomb repressive complex 2 (PRC2), a multiprotein complex involved in the maintenance of transcriptional silencing, through the trimethylation of K27 of histone H3 (H3K27me3).9,10 H3-K27M mutations lead to hypomethylation of H3K27, which promotes a more accessible chromatin state characterized by H3K27 acetylation and aberrant gene expression likely to contribute to DIPG oncogenesis.9,11–13 Although DIPG has been shown to exhibit global loss of H3K27me3, PRC2 activity is still detected in H3-K27M patient cells and H3K27me3 peaks remain at hundreds of gene loci.11,12 The paradoxical global reduction of H3K27me3, but its increase at specific genomic loci, has been proposed to reprogram the epigenetic landscape and gene expression of H3-K27M expressing cells and drive DIPG tumorigenesis. In fact, evolutionary reconstruction of DIPG tumors suggests that the H3-K27M mutation is probably the first oncogenic event.14
The discovery that H3-K27M mutation, that alters the ability of EZH2 within the PRC2 complex to regulate methylation of H3K27 and thereby the transcriptome, has a profound impact on diffuse midline glioma biology, led to interest in targeting this histone modifying enzyme to combat these pediatric tumors.15 Small-molecule inhibitors for EZH2 have been tested as a potential treatment for DIPG.16,17 These studies led to two important findings. Firstly, when EZH2 inhibitors were used to treat DIPG cells in vitro, they either had no effect on these cells or required long exposure times to exhibit anti-tumoral activity. Secondly, inhibition of EZH2, either genetically or pharmacologically, led to robust reduction in tumor growth and significant extended survival in preclinical animal models of DIPG in vivo in contrast to what has been observed in vitro. Therefore, we decided to investigate whether microglia in the DIPG tumor microenvironment could contribute to the response of these tumors to EZH2 inhibition that ultimately explain the observed discrepancy between EZH2 inhibitor efficacy in vitro and in vivo in the context of DIPG.
In gliomas, animal models and analysis of human biopsies reveal that microglia, resident immune cells of the central nervous system, are found in large numbers in the region surrounding or within the core of the tumor mass. A close interplay is reported between microglia and high-grade glioma cancer cells, convincingly in adult malignancy, and targeting these cells is considered an important avenue for therapeutic intervention.18–20 Indeed, glioma cancer cells are capable of repressing the immune properties of microglia and can drive them into becoming tumor-supportive and immunosuppressive cells, and thereby promote brain tumor progression.21 Remarkably, we and others have reported that the activation of microglia toward selective phenotypes exerting unique functions, such as the tumor trophic states rely heavily on epigenetic mechanism.22,23 Indeed, for pediatric high-grade glioma, neoplastic tissue from human DIPG patients, as well as mouse xenografts generated by the injection in the pons of cells established from the biopsies of those same patients, are both reported to include in the tumour microenvironment cells positive for microglial markers.24–26 However, the contributions of microglia per se to the progression of DIPG, the molecular, potentially epigenetic, mechanisms leading to DIPG-induced microglial activation, and the benefit of targeting microglia to combat DIPG tumor, all remain to be determined.
Materials and Methods
Cell Culture and Treatments
Microglia BV2 (gift from Guy Brown, University of Cambridge), microglia HMC3 (ATCC, Cat. #CRL-3304), primary patient-derived pHGG SF188 cells (gift from Stefan M. Pfister, German Cancer Research Center), primary patient-derived DIPG SF8628 (MERCK, Cat. #SCC127), and primary-patient derived DIPG cells, SU-DIPG-XIII (gift from Michelle Monje, Stanford University) have been used in this study (details of the molecular classification of each tumor cells line can be found in Supplementary Table 1, and details of culture conditions in the Supplementary Methods). Cells were checked for mycoplasma with Venor™GeM mycoplasma detection kit (Minerva Biolabs). Cells passage number were taken under consideration. SF188, SF8628, and BV2 microglia were treated with 3µM of GSK343 (Sigma, Cat. #SML0766) or EPZ-6438 (Selectbiochem, Cat. #S7128) for indicated timepoints.
Immunofluorescence on Human Patient Tissue
Experiments with human tissue were carried out with ethical approval from the South Central-Hampshire B Research Ethics Committee (19/SC/0217) with tissues obtained from BRAIN UK. Three DIPG patient tissue samples with H3.3-K27M confirmed using either mutation specific antibody or sequencing were used. Three glioblastoma IDH wildtype patient tissues were used as controls. All samples were without prior treatment. Details on the staining protocol and antibodies used can be found in Supplementary Table 2 and Supplementary Method information.
For full list of methods please see Supplementary Information.
Results
DIPG-induced Microglia Activation Is Associated with the Transient Down-regulation of H3K27me3
To gain an understanding on possible epigenetic mechanisms that may underly the activation of microglia in the context of DIPG, we brought our attention to the trimetylation of histone H3 at lysine 27 (H3K27me3), considering as introduced (1) that this particular residue in histone H3 is found to be mutated in the vast majority of diffuse midline gliomas, including DIPG,1,5,6 (2) that this specific histone posttranslational modification placed by the EZH2 H3K27 histone methyltransferase containing PCR2 is associated with long term transcriptional repression,10 and (3) that inhibition of the EZH2 enzyme has been reported to impact on DIPG tumor growth and even survival in mouse models.15
For these investigations, two pediatric high-grade glioma cell lines were selected based on their histone H3 status: SF8628 human DIPG cell line that harbors the histone H3.3 Lys 27-to-methionine (K27M) mutation (RRID:CVCL_IT46) and SF188 human pediatric high-grade glioma that is wild-type for histone H3 (RRID:CVCL_6948, both originating from the Brain Tumor Research Center Tissue Bank at the University of California, San Francisco).12 The mutational status of both cell lines was confirmed by immunofluorescent and immunoblot analysis (Figure 1A and B). In agreement with previous reports, immunofluorescent and immunoblot analysis reveals a global loss of H3K27me3 expression levels in the SF8628 DIPG cells harboring the H3-K27M mutation, as compared to the SF188 DIPG cells that are wild-type for this residue on histone H3 (Supplementary Figure 1A and B). Concurrent higher levels of acetylation of H3K27 (H3K27ac), that can be compensatory histone mark for the lack of H3K27me3, was also observed by immunofluorescent and immunoblot analysis of H3-K27M SF8628 cells.27,28 (Supplementary Figure 1C and D). BV2 cells, murine microglia used to test the capability of these pediatric HGG cells to activate a microglial tumor supportive phenotype, were shown to be wild-type for histone H3 (Figure 1A) and to express a significant amount of the trimethylated form of H3K27, as well as its acetylated form (Supplementary Figure 1B and D). The use of microglial cells which do not carry the H3-K27M mutation for our experimentation is supported by the fact that H3-K27M is a somatic mutation, the observation that DIPG tumors are composed of a mix of cells positive and negative for the H3-K27M, with also variable expression levels of H3K27me3 and H3K27ac, and that single cell RNA-sequencing analyses performed on fresh H3-K27M glioma biopsies indicate that myeloid cells in the tumor mass were negative for H3-K27M.25,26 Immunohistochemistry analysis of tumor tissues from three different DIPG patients confirmed that DIPG cancer cells are H3-K27M positive, whereas IBA1-expressing microglia in their microenvironment do not carry the mutation (Figure 1C and D). Control tissue obtained from three different glioblastoma patients exhibited no observable staining for the H3-K27M, as well as the presence of microglia in the tumor microenvironment (Supplementary Figure 1E).
![Microglia do not carry the H3K-27M mutation in the DIPG microenvironment. (A) Confocal microscopy of SF188 and SF8628 glioma cells immunostained with H3K-27M mutation and DAPI, scale bar, 50 µm. (B) Immunoblot analysis of H3K-27M mutation and β-actin (ACTB) (right margin, molecular size, in kilodaltons [kDa]). Quantification of immunoblots are shown relative to β-actin expression. (C) and (D) Confocal microscopy of human DIPG tumors obtained from three individual patients with immunostaining for H3K-27M mutation, IBA1 and DAPI, scale bar, 30 µm. P value: ***<.001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/noa/3/1/10.1093_noajnl_vdab096/1/m_vdab096f0001.jpeg?Expires=1750211845&Signature=Axoj4WcVvC1jvtCcK8kGp9xmwFb5BwEr-rgvQ-nwmBQ6rzZb9pVC32VDZbizePULlCEGf8UIJdm6sIo93YTdD9x9GVd-D6CMNO3l4HICJBQTiT0NQHCOnvlDa4ye~pyho~Q2hVwkUyb9vxh~7Wn579RmjO8es1GweyVIEOBqmXeSYZANyO2WOh642FPLGo2BniNcCchmFM5gxqG1JnV0XIqM4Eb8Ays-Lg5pU5LIHZRJUn38Olgl1G~lg4391mJpFr4JVu27isDun3kGbOV~BRsZVsUuG~38Ig7YF5JMkFZv8xxG0dSqrIFlHbPx-3nK3727T6hJdlBdVN2J0Txypg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microglia do not carry the H3K-27M mutation in the DIPG microenvironment. (A) Confocal microscopy of SF188 and SF8628 glioma cells immunostained with H3K-27M mutation and DAPI, scale bar, 50 µm. (B) Immunoblot analysis of H3K-27M mutation and β-actin (ACTB) (right margin, molecular size, in kilodaltons [kDa]). Quantification of immunoblots are shown relative to β-actin expression. (C) and (D) Confocal microscopy of human DIPG tumors obtained from three individual patients with immunostaining for H3K-27M mutation, IBA1 and DAPI, scale bar, 30 µm. P value: ***<.001.
We show that independent of their histone H3 status, that is wild-type H3 versus H3-K27M mutant, pediatric HGG cells were able to polarize BV2 microglia toward a tumor-supportive phenotype, as demonstrated by their ability to in turn support the invasion capabilities into matrigel of the HGG cancer cells (Figure 2A). Next, we examined nitric oxide synthase-2 (Nos2) gene expression as induction of its expression is associated with the microglial pro-inflammatory phenotype, whereas its expression is found to be substantially suppressed upon exposure to glioma cells.29,30 Glioma cells can even efficiently reduce LPS-induced expression of NOS2 in microglia.31,32 Therefore, the acquisition of a microglial tumor-supportive phenotype was further illustrated by the suppression of expression of the Nos2 gene, that encodes for inducible nitric oxide synthase, a molecular characteristic of microglial cells polarized toward an immune repressive and tumor promoting activation state29,30 (Figure 2B). Of significant interest, several genes previously reported to be differentially expressed in TAMs isolated from primary human DIPG tumors compared to TAMs isolated from adult glioblastoma25 were also differentially expressed in our coculture system (Supplementary Figure 2). These genes included the potent pro-tumoral factors Tgfb1 and Ccl4 providing further evidence that microglia produce factors known to influence tumor cell states (Supplementary Figure 2).
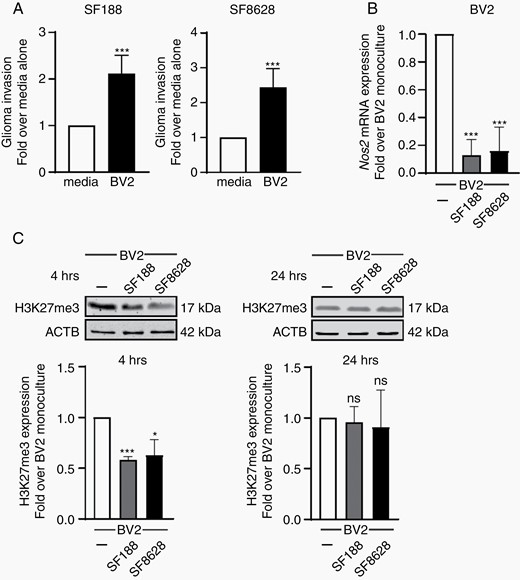
DIPG-induced microglia activation is associated with the transient downregulation of H3K27me3. (A) Quantification of the invasion capacity of SF188 and SF8628 glioma cells into matrigel coated transwells with BV2 microglia or 10% complete DMEM as control in the lower compartment; results are presented relative to 10% media only control, set as 1. (B) qPCR analysis of Nos2 expression in BV2 microglia grown as monocultures or with SF188 or SF8628 glioma cells as segregated co-cultures for 4 h, results are shown relative to BV2 monocultures, set as 1. (C) Immunoblot analysis of BV2 microglia cells for H3K27me3 and β-actin following segregated co-culture with SF188 and SF8628 glioma cells. Results shown relative to monoculture set as 1. P value: *<.05, and ***<.001.
Remarkably, immunoblot analysis revealed that the induction of this microglial phenotype with tumor trophic functions upon exposure to pediatric HGG cells, once more independently of the status of their lysine K27 residue on histone H3, is associated with a significant and rapid down-regulation in the expression level of H3K27me3 in microglia (Figure 2C). Of note, this downregulation appears to be transient, as microglial H3K27me3 returned to control level within 24 h (Figure 2C). Epigenetic changes, including transient posttranslational histone modifications, have been involved in cell phenotype regulation and reprogramming and are reported to be part of the mechanisms regulating microglial cellular plasticity.23,33
Repression of EZH2 Gene Expression in H3-K27M DIPG Cancer Cells Does Not Impact on Their Survival or Capability to Activate Microglia
The discovery that H3-K27M mutation in DIPG tumors that alters the ability of EZH2 within the PRC2 complex to regulate methylation of H3K27 and thereby the transcriptome has led to interest in targeting this histone modifying enzyme to combat these pediatric tumors. The potential beneficial effects of treatment with EZH2 chemical inhibitors, such as EPZ-6438/tazemetostat, and GSK343 or its derivatives, for pediatric glioma with mutated histone H3-K27M has been therefore explored. Disappointingly, only extended treatment (up to a week) with the EZH2 inhibitors were able to show in vitro beneficial effects on DIPG H3-K27M cells, with reduced proliferation and colony forming activity. However, targeting EZH2 functions in vivo, using the same drugs or a genetic approach appeared to lead to a robust reduction in tumor growth and significant extended survival in DIPG preclinical mouse models. Considering (1) the remarkable difference in response to EZH2 inhibition in vitro and in vivo, and (2) our discovery that pediatric HGG-induced microglial activation involves the regulation of H3K27me3 expression levels, we hypothesized that other cells in the DIPG tumor microenvironment, that is microglia, could contribute to the response of these tumors to EZH2 inhibition.
In order to address, which cells, microglia or DIPG cells could mediate the beneficial effect of EZH2 inhibition in vitro, we undertook to repress EZH2 gene expression, selectively in each of these cell types using a small-interfering RNA approach. RNA interference mediated gene silencing of EZH2 using a pool of siRNAs validated for limited off-target effects, was selected instead of pharmacological inhibition. Efficient knock-down of EZH2 gene expression was achieved in both SF188 pHGG and SF8628 DIPG cells, as illustrated by decreased EZH2 mRNA, as well as EZH2 protein expression in both cell lines after 24 h (Figure 3A and B). However, despite an efficient knockdown of EZH2 gene expression in these pediatric HGG cells, global H3K27me3, as well as H3K27ac expression levels remained chiefly unaffected, suggesting a low turnover for these histone posttranslational modifications in these cancer cells (Figure 3B). Of note, H3K27M expression levels were not affected by the reduction in EZH2 expression levels (Figure 3C). These observations could support previous reports that only extended treatment (up to a week) with EZH2 inhibitors were able to show in vitro beneficial effects on DIPG H3K27M cells, with reduced proliferation and colony forming activity,17 whereas shorter treatment periods (up to 72 h) with these EZH2 inhibitors were found to have no significant cytotoxic effect in vitro on pediatric high-grade glioma cells.34,35 We therefore assessed cell proliferation and cell death in our experimental model by immunoblotting. We measured proliferating cell nuclear antigen, PCNA, a nuclear nonhistone protein that is necessary for DNA synthesis, which is elevated during the G1/S phase of the cell cycle as well as and Poly (ADP-ribose) polymerase, PARP, that is inactivated by caspase-3 cleavage during apoptotic cell death. Strikingly, we found that neither of these biological processes were affected by EZH2 silencing (Figure 3D and E). Furthermore, repressing EZH2 gene expression in the H3-K27M SF8628 DIPG cells or in the wild-type H3 SF188 pHGG did not impact their ability to activate BV2 microglia toward a tumor-supportive phenotype. This is illustrated by the similar suppression of Nos2 gene expression in the microglial cells exposed to these pHGG cells transfected with a siRNA targeting EZH2 or a scramble siRNA control (Figure 3F). In addition, the effects of chemical EZH2 inhibition were also assessed in the H3-K27M SF8628 DIPG cells and the wild-type H3 SF188 pHGG treated with 3 µM GSK343 or EPZ-6438 (also known as Tazemetostat). Both cell lines were found to continue to proliferate at the same rate, as measured by the EdU assay, regardless of treatment with either of the EZH2 inhibitors (Supplementary Figure 3A and B). The absence of effect on proliferation was further confirmed by immunoblotting for the proliferation marker PCNA. Likewise, immunoblot analysis for cleaved PARP, that serves as a marker of cells undergoing apoptosis, revealed an absence of promotion of cell death upon GSK343 or EPZ-6438 treatment (Supplementary Figure 3C and D).
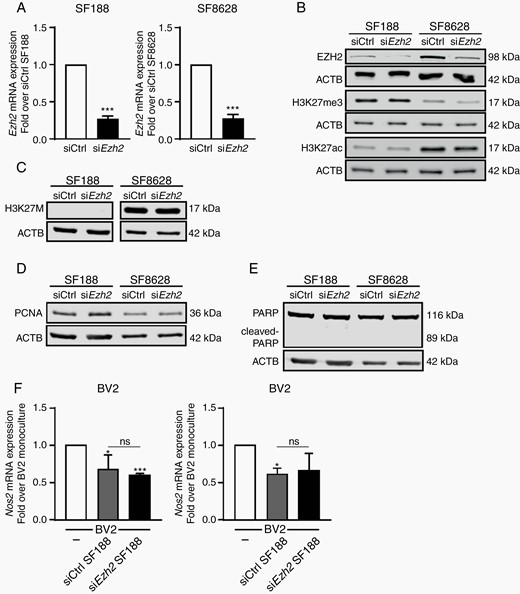
Repression of EZH2 gene expression in H3K-27M DIPG cancer cells does not impact on their survival or capability to activate microglia. (A) qPCR analysis of EZH2 in SF188 and SF8628 glioma cells transfected with either EZH2-specific siRNA (siEZH2) or scramble siRNA (siCtrl). Fold change is shown relative to siCtrl with siCtrl set as 1. Immunoblot analysis of siEZH2 and siCtrl transfected SF188 and SF8628 glioma cells for (B) EZH2, H3K27me3, H3K27ac and β-actin, (C) H3K-27M mutation and β-actin and (D) and (E) PCNA and cleaved PARP. (F) qPCR analysis of Nos2 expression in BV2 microglia grown as monocultures or with siEZH2 or siCtrl SF188 or siEZH2 or siCtrl SF8628 glioma cells as segregated co-cultures for 4 h, results are shown relative to BV2 monocultures, set as 1. P value: ***<.001.
Collectively, these data indicate that chemical or genetic inhibition of EZH2 and thereby functions, have limited if no effect, on the proliferative and survival capabilities of pediatric HGGs, regardless of H3-K27M mutational status. These observations would also argue against the concept that EZH2 inhibitors in animal models of DIPG would exert their beneficial effects by targeting exclusively the glioma cells. Thus, it suggests that EZH2 inhibitors might target other cells, in the DIPG tumor microenvironment that would be responsible for the beneficial effects seen in vivo.
Repression of Ezh2 Gene Expression in Microglia Promotes a Cell Phenotype Holding Anti-tumoral Properties
Thereafter, we decided to investigate whether targeting Ezh2 gene expression in microglia, instead of in the glioma cancer cells, could bring beneficial effects. Efficient knock-down of Ezh2 gene expression was achieved in BV2 microglia, as illustrated by the decrease in Ezh2 mRNA, as well as EZH2 protein expression after 24 h (Figure 4A and B). In contrast to what was observed in the pediatric HGG cells, Ezh2 knockdown was associated with a significant decrease in global H3K27me3 expression levels in the microglial cells after 24 h (Figure 4B). Similarly, to what was observed for EZH2 knockdown in pediatric HGG cells, we observed no increase in cell proliferation or cell death in siEzh2 BV2 microglia cells compared to control cells as measured by PCNA and cleaved PARP (Supplementary Figure 4A and B). In support of the acquisition of a microglial anti-tumoral phenotype, silencing of Ezh2 gene expression in microglia lead to a robust induction of Nos2 gene expression in these cells which is associated with an immune reactive activation state of these myeloid cells (Figure 4C). To further understand the mechanism behind the induction of Nos2 gene expression, we performed a chromatin immunoprecipitation (ChIP) analysis of H3K27me3 and acetylated H3K27 (H3K27ac) enrichment to the Nos2 promoter in siCtrl and siEzh2 BV2 cells. Actually, a significant decrease in the amount of H3K27me3 was observed on the Nos2 promoter that reflects a transcriptionally more active locus (Figure 4D). In parallel, an enrichment of H3K27ac, which is associated with higher activation of transcription, was observed in siEzh2 microglia compared to siCtrl one, although not significant (P = .09) (Figure 4D). Next, we investigated the expression of the potent anti-tumoral cytokine Il1b and found that this cytokine was not induced in microglia upon coculture with H3.3 wildtype SF188 tumor cells, H3-K27M SF8628, or a second patient derived DIPG cell line known as SU-DIPG-XIII (RRID:CVCL_IT41) (Figure 4E). However, we found that siEzh2 microglia expressed increased levels of Il1b that was maintained even when cocultured with SF188 tumor cells (Figure 4F). To further support that Il1b could be an important target of H3K27me3, we treated siEzh2 microglia with the inflammogen, lipopolysaccharide (LPS). Indeed, we found that this led to a robust and significant increase in Il1b expression in siEzh2 microglia compared to scramble control cells (Figure 4G). Recently it has been shown that the mechanistic target of rapamycin (mTOR) signaling pathway is a powerful regulator of the microglial inflammatory response, including LPS-induced Il1b expression.36 Therefore, mTOR kinase activation was examined in siCtrl and siEzh2 BV2 cells following coculture with H3-K27M SF8628 DIPG cells or wild-type H3 SF188 pHGG cells. Indeed, we found increased phosphorylation, of one of the main substrates of mTOR, S6 ribosomal protein, suggesting an activation of the mTOR pathway in siEzh2 BV2 cells consistent with an increased pro-inflammatory phenotype36 (Figure 4H). Overall, these observations suggested that siEzh2 microglia may exhibit anti-tumoral properties and therefore we decided to investigate this possibility using several functional assays to measure anti-tumoral capabilities.
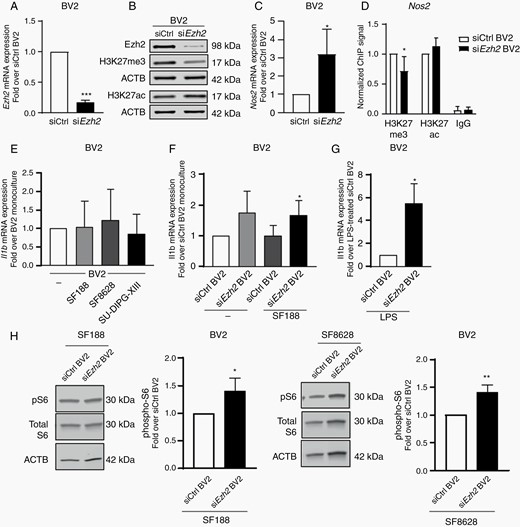
Repression of Ezh2 gene expression in microglia promotes the expression of anti-tumoral factors. (A) qPCR analysis of Ezh2 in BV2 microglia cells transfected with either Ezh2-specific siRNA (siEZH2) or scramble siRNA (siCtrl). Fold change is shown relative to siCtrl with siCtrl set as 1. (B) Immunoblot analysis of siEzh2 and siCtrl transfected microglia cells for EZH2, H3K27me3, H3K27ac and β-actin. qPCR analysis of (C) Nos2 expression in siEzh2 or siEzh2 BV2 microglia (D) Chromatin Immunoprecipitation (ChIP) analysis of H3K27me3 and H3K27ac on the Nos2 promoter in siCtrl or siEzh2 BV2 cells (E) Il1b expression of BV2 microglia grown as monocultures or with SF188, SF8628 or SU-DIPG-XIII glioma cells as segregated co-cultures for 4 h, results are shown relative to BV2 monocultures, set as 1. qPCR analysis of Il1b for (F) siEzh2 or siCtrl microglia grown as monocultures or with SF188 as segregated co-cultures for 4 h, results are shown relative to siCtrl BV2 monocultures, set as 1 and (G) LPS-treated siEzh2 or siCtrl BV2 4 h (H) Immunoblot analysis of siEzh2 and siCtrl transfected microglia cells following 4 h co-culture with SF188 or SF8628 glioma cells for phospho-S6, Total S6 and β-actin. P value: *<.05, and ***<.001.
Remarkably, transwell matrigel invasion assays revealed that Ezh2-siRNA transfected BV2 microglia, not only lost their ability to promote the invasion of the pediatric HGG cells, they even considerably repressed this capability, and this was independent of H3-K27M mutational status (Figure 5A), further suggesting the acquisition of an anti-tumoral microglial activation state. In order to further support that Ezh2 repression in microglia could exert anti-tumoral activities in the context of DIPG, we again used a second patient derived DIPG cell line, SU-DIPGX-III. Indeed, Ezh2 repression in microglia led to a drastic reduction in the migration capacity of SU-DIPG-XIII cells when cocultured with siEzh2 BV2 microglia for 24 h (Figure 5B). Finally, to validate that EZH2 repression in microglia could exert anti-tumoral activities in a human context, we carried out migration assays using the human microglial cell line, HMC3. We observed that siEZH2 HMC3 microglia, like siEzh2 BV2 microglia, were able to significantly reduce the migratory capacity of H3-K27M SF8628 DIPG cells or the wild-type H3 SF188 pHGG cells when compared to siCtrl HMC3 cells (Figure 5C). Next, as a further measure of anti-tumoral activity, we measured the capacity of siEzh2 microglia to phagocytose carboxyfluorecein succinimidyl ester (CFSE)-labelled tumor cells. We selected this assay as phagocytosis of live tumor cells is more physiologically relevant than measuring microglial phagocytosis of dextran or latex beads. Similarly, boosting phagocytosis in this way has been shown to be of benefit in preclinical animal models of DIPG and other pediatric brain tumors.37 Indeed, we found increased ability of siEzh2 microglia to phagocytose both SF188 H3.3 wildtype tumor cells as well as H3-K27 mutant DIPG cells, SF8628 and SU-DIPG-XIII compared to siCtrl microglial cells (Figure 5D and E). Finally, the ability of Ezh2-siRNA transfected microglia to promote the death of pediatric HGG cells, was examined by counting the number of dead microglia-exposed cancer cells. This analysis uncovered an increase in cell death for both wild-type H3 SF188 and H3-K27M SF8626 pediatric HGG cells exposed to Ezh2-siRNA transfected microglia, as compared to the same pediatric HGG cells exposed to microglia transfected with a scramble siRNA control (Figure 5F) with no effect on cell cycle progression being observed (Supplementary Figure 4C). Lastly, we pretreated BV2 cells with 3 µM of EZH2 inhibitors, GSK343 or EPZ-6438. Following this pretreatment, we removed the inhibitors and carried out migration assays with H3-K27M SF8628 DIPG cells or the wild-type H3 SF188 pHGG cells. Strikingly, we found that at 3 h pretreatment of BV2 microglia with GSK343 was enough to dramatically reduce the migratory capacity of both tumor cell lines (Supplementary Figure 5A). Similarly, we noticed that 24 h pretreatment with EPZ-6428 also reduced the migratory capacity of both cell lines albeit to a lesser extent than GSK343 (Supplementary Figure 5B). The anti-tumoral activity exerted by BV2 after short treatment with EZH2 inhibitors is in stark contrast to the lack of effects on tumor cell proliferation and cell death (Supplementary Figure 3). These findings provide further evidence that the effects of EZH2 inhibitors in the DIPG tumor microenvironment context are most probably mediated by targeting the TAMs rather than tumor cells per se. Altogether these data indicate that repression of Ezh2 expression, and the associated sustained decrease in global H3K27me3 in microglia, are linked to the acquisition of a cellular phenotype holding anti-tumoral properties (Figure 6).

Repression of Ezh2 gene expression in microglia promotes a cell phenotype holding anti-tumoral properties. (A) Quantification of the invasion capacity of SF188 and SF8628 glioma cells into matrigel coated transwells with siCtrl or siEzh2 BV2 microglia in the lower compartment; results are presented relative to siCtrl BV2 control, set as 1. (B) Quantification of the migration capacity of SU-DIPG-XIII glioma cells into transwells with siCtrl or siEzh2 BV2 microglia in the lower compartment; results are presented relative to siCtrl BV2 control, set as 1. (C) Quantification of the migration capacity of SF188 and SF8628 glioma cells into transwells with siCtrl or siEZH2 HMC3 human microglia in the lower compartment; results are presented relative to siCtrl HMC3 control, set as 1. (D, E) Capacity of siCtrl or siEzh2 BV2 to phagocytosis CFSE-labelled (D) SF188 and SF8628 glioma cells and (E) CFSE-labelled SU-DIPG-XIII when co-cultured for 4 h at a ratio of 2:1, results are presented relative to siCtrl BV2 set as 1. (F) Microglia-induced glioma cell death measured by counting the number of cell-tracker labelled SF188 or SF8628 glioma cells after co-culture with siCtrl or siEzh2 BV2 microglia for 4 h. results are presented relative to siCtrl BV2 set as 1. P value: *<.05, **<.01 and ***<.001.
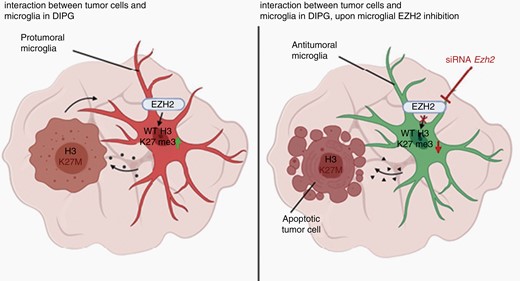
Schematic illustration showing proposed mechanism for how EZH2 inhibition can lead to apoptotic tumor cell death in the context of DIPG upon activation of a microglial anti-tumoral phenotype.
Discussion
Diffuse midline gliomas, including DIPG, are by definition in the most recent WHO classification defined by a H3-K27M mutation, and as a consequence of this mutation, by decreased global H3K27me3 expression levels.1 For some, it may appear counter-intuitive to attempt to inhibit the action of the enzyme responsible for placing this specific histone posttranslational modification, EZH2 within the PRC2 complex, as a therapeutic strategy to combat these tumors types that already exhibit low H3K27me3 expression level due to their H3-K27M mutation. The rationale behind this intervention has been placed on the observation that PRC2 activity is still detected in H3-K27M patient cells and that H3K27me3 peaks remain at hundreds of gene loci.11,12 It has been proposed that the paradoxical global reduction of H3K27me3, but its increase at specific genomic loci, could reprogram the epigenetic landscape and gene expression of H3-K27M expressing cells and drive DIPG tumorigenesis.9,11–13,28 One additional puzzling observation is that whereas the inhibition of the EZH2 H3K27 histone methyltransferase provided promising effects in preclinical animal models of the disease, in vitro, the effects of these EZH2 inhibitors on DIPG cells is far less convincing.16,17,34,35 Worth a note, early report suggested that p16INK4A-mediated senescence of neural stem cell-derived DIPG cancer cells could be responsible for the effects of EZH2 inhibition observed in the corresponding animal model of DIPG.17 However, it should be noted that those results have been found to be controversial.13 Taken as a whole, the existing literature argues for in vitro limited to no effect of EZH2 inhibitor on human DIPG cells13,17,28.34,35. Thus, these observations suggest that other cells within the DIPG tumor mass, beyond the glioma cancer cells themselves, potentially with a wild-type histone H3 and intact H3K27me3 expression levels, could contribute to the response to EZH2 inhibition.
In fact, solid tumors are heterogeneous with respect to the composition of bona fide tumors cells and with respect to a range of intermingling nonneoplastic cells which also play vital roles in controlling the course of pathology. DIPG should not be different in this respect; indeed, the cancer cells, of this diffusely infiltrative tumor, are embedded by the brain microenvironment, and the nonneoplastic cells surrounding them are thus expected to impact their growth and progression.18,21 In these pediatric HGG, immunohistochemistry analyses of animal models of the disease and patient tissue samples, as well as single-cell RNA sequencing analysis performed on fresh biopsies, demonstrate that myeloid cells, primarily microglia, are also found in significant numbers in the tumor mass.24–26 In contrast to adult glioblastoma, DIPG tumors do not show enhanced T-cell infiltration as compared to adjacent nontumor tissue.25,38 When direct comparison was made between adult glioblastoma and DIPG derived myeloid cells (likely representing microglia but in some cases infiltrating bone marrow derived macrophages), whereas both shared the expression of genes related to cell adhesion, angiogenesis, and extracellular matrix organization, which might be of benefit for a tumor-supportive phenotype, adult microglia expressed inflammatory properties which were not observed in cells collected from DIPG tumors.25 Furthermore, DIPG tumors appear to have neither a highly immunosuppressive nor inflammatory microenvironment.38 Thus, microglia appear to be present at the DIPG tumor site, and to exhibit an activation state that could promote tumor progression.
These observations also argue for a role for microglia, and possibly other cells in the tumor microenvironment, beyond the glioma cancer cells themselves, in the observed response of DIPG tumor to the inhibition of EZH2. Certainly, future in vivo studies would be beneficial for further understanding of the respective role of different cell types within the tumor microenvironment to the response to EZH2 inhibition in the context of DIPG tumors. In particular, experiments aiming at the conditional and inducible knockdown of microglial Ezh2 gene expression in the context of a DIPG animal model, could help further characterize the contribution of microglia to anti-tumoral effect reported in DIPG tumours upon EZH2 inhibition. Furthermore, understanding the detailed transcriptional program regulating the acquisition of the anti-tumoral microglial phenotype upon EZH2 inhibition, as well as the potential signaling pathways able to promote this microglial reprogramming would certainly be of interest in search a microglial-based therapeutic strategy to combat DIPG. Already, microglia and macrophages within the tumor microenvironment have been considered as both therapeutic tools and targets for the treatment of high-grade gliomas (recently reviewed in 39–41).
In fact, the targeting of the interaction between DIPG cancer cells and tumor-associated microglia/macrophages has already emerged as a promising strategy to combat this pediatric neoplasm. For example, blockade of the anti-phagocytic CD47 “don’t eat me” signal on DIPG cells using a humanized antibody has been reported to promote in vitro and in vivo their phagocytosis and dismiss by myeloid cells.37 However, it is important to note that other cells in the DIPG tumor microenvironment are also of therapeutic interest. For instance, recent studies revealed that neuronal activity-regulated secretion of the synaptic protein neuroligin-3 (NLGN3), which gene expression is reported to be regulated by PCR2-mediated H3K27me3, increased the proliferation of DIPG cells through the PI3K-mTOR pathway. Patient-derived orthotopic xenografts of DIPG fail to grow in Nlgn3 knockout mice and the expression of NLGN3 inversely correlated with survival in patients with high-grade gliomas.42–44 Therefore, when studying the potential acquisition of anti-tumoral properties by microglia, their crosstalk with other cell types, for example macrophages, astrocytes, and neurons, will have to be taken into account.45,46 Hence, these investigations should grant interest for further investigation of the contribution of microglia to the initiation, progression, and response of DIPG tumors to treatments.
In summary, we report that the activation of microglial cells towards a tumor-supportive phenotype by pediatric HGG, independently of their H3-K27M status, rely on the transient downregulation of H3K27me3 expression levels in these myeloid cells. Further, we demonstrate that targeting EZH2 gene expression in these pediatric HGG cells using an antisense RNA strategy, despite being effective in reducing the expression of the H3K27 histone methyltransferase, did not impact significantly on the expression level of H3K27me3 at the time point tested (up to 72 h). Silencing EZH2 in the pediatric HGG cells regardless of mutational status, did not impact on their proliferative or survival capabilities, neither on their ability to activate microglia. In contrast, targeting Ezh2 gene expression in microglia, led to a decrease in the global H3K27me3 expression levels, and promoted a microglial activation state that reduced glioma cell invasion capability as well as promoted their dismiss as well as exhibiting other antitumoral properties. Collectively, these findings uncover an epigenetic mechanism involved in pediatric HGG-induced microglia activation, involving the regulation of the methylation of H3K27. These results also argue for a role for microglia, and possibly other cells in the tumor microenvironment, beyond the glioma cancer cells themselves, in the observed response of DIPG tumor to the inhibition of EZH2. Finally, these investigations should grant interest for further investigation of the contribution of microglia to the initiation, progression, and response of DIPG tumors to treatments.
Funding
This work has been supported by grants from the Swedish Research Council, the Swedish Childhood Cancer Fund, Swedish Cancer Foundation, Swedish Cancer Society, Swedish Brain Foundation, Åke Wiberg Foundation, Petrus and Augusta Hedlund Foundation, Karolinska Institutet Foundations, Medical Research Council UK, UK Dementia Research Institute and Wellcome Trust.
Conflict of Interest. There are no conflicts of interest to disclose.
Authorship Statement. Study design: L.K., M.C., D.S., B.J., K.G., and B.W.M. Data collection: L.K., M.C., D.S., L.F., P.G.R., and M.E.G. Manuscript preparation: L.K., M.C., B.J., and D.S.
References
Author notes
These authors are co-first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}