






Abstract
Patients with rhabdoid tumor predisposition syndrome (RTPS) harbor germline alterations in the epigenetic regulator genes SMARCB1 or SMARCA4. Patients usually present with atypical teratoid/rhabdoid tumor (AT/RT) of the brain or malignant rhabdoid tumor (MRT) arising outside the central nervous system. Intensive treatment can lead to remissions, however tumors frequently recur or synchronous or metachronous tumors appear. A maintenance or secondary prevention regimen may prevent these aggressive tumors. Potential maintenance regimens may include low-dose traditional chemotherapy or different epigenetic therapies designed to target the epigenetic imbalance that drives RTs. We here review several potential maintenance regimens that may be useful in RTPS.
Rhabdoid tumor predisposition syndrome (RTPS) is an inherited genomic disorder characterized in most cases (85–95%) by heterozygous loss of SMARCB1 (also known as INI1/SNF5/BAF47) and rarely (5–15%) by SMARCA4.1–3 These genes encode proteins that are key components of the mammalian chromatin-remodeling SWI/SNF complex. Germline heterozygosity for SMARCB1 or SMARCA4 leads to increased risk of developing rhabdoid tumors (RTs) of the central nervous system (AT/RT) as well as soft tissues outside the central nervous system (malignant rhabdoid tumor [MRT]). AT/RT and MRT are the predominant tumor types in RTPS. MRT often occurs in the kidney, but can arise in virtually any soft tissue. SMARCA4 alterations may have decreased penetrance and be less lethal compared to SMARCB1.2 Most germline SMARCB1 mutant alleles are spontaneous, due to the high early mortality of SMARCB1-driven RTPS, however the SMARCA4 alteration may be inherited. Biallelic alteration of SMARCB1 or SMARCA4 leads to loss of expression of SMARCB1 or SMARCA4 protein, which is the diagnostic hallmark of RTs.
Most patients with RTPS present clinically with a primary tumor.1,2 Initial treatment involves best surgical resection followed by intensive chemotherapy and in many cases radiation therapy. Some AT/RT protocols involve maintenance chemotherapy regimens to prevent recurrence, and in this minireview we explore opportunities for regimens that may prevent the development of recurrent or additional tumors in patients with RTPS.
Traditional Chemotherapy Regimens
In the Dana Farber Cancer Institute AT/RT protocol (NCT00084838), patients received an additional 24 weeks of metronomic chemotherapy after completing intensive, sarcoma-like therapy with optional radiation therapy.4 Combining alternating low-dose oral chemotherapy regimens with intrathecal or intra-Ommaya chemotherapy infusions of agents such as methotrexate, cytarabine, or topotecan can effectively treat aggressive pediatric embryonal tumors, including AT/RT.5–7 These regimens however do include alkylator and other DNA-damaging agents. The extended time course of maintenance treatment needed to prevent additional tumors from forming in patients with RTPS might lead patients to exceed recommended lifetime doses of alkylators such as cyclophosphamide and temozolomide as well as topoisomerase inhibitors such as etoposide. High lifetime exposures to alkylator and topoisomerase inhibitor therapy can lead to increased risk of treatment related malignancies, including acute myeloid leukemia.8,9 The risks of secondary malignancies due to chemotherapy may limit the value of traditional chemotherapy maintenance regimens in RTPS.
Epigenetic Maintenance Therapy
Epigenetic therapy is particularly attractive in AT/RT and MRT due to the fundamentally unbalanced epigenome in these tumors (Figure 1). The decreased activity of SWI/SNF due to mutation in SMARCB1 or SMARCA4 leads to unopposed activity of the polycomb repressor complex 2 (PRC2), including the histone methyltransferase EZH2, raising the possibility that EZH2 inhibitors may be effective as part of initial therapy or as maintenance therapy. Tazemetostat is a first-in-class EZH2 inhibitor with activity against AT/RT tumor cells in preclinical models and early phase clinical trials.10 One caveat to extensive use of tazemetostat in young children is that the package insert for tazemetostat indicates that tazemetostat therapy is associated with increased risk of secondary malignancies, including AML, MDS, and T-cell lymphoblastic lymphoma.11
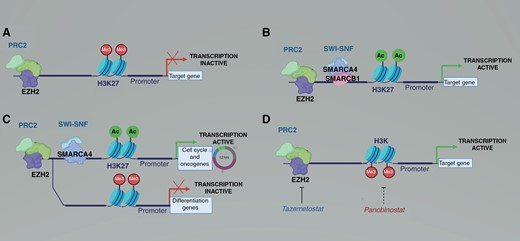
Epigenetic alterations in rhabdoid tumors prevent differentiation and drive proliferation. (A) EZH2 and the polycomb repressor complex 2 (PRC2) promote histone methylation and suppress transcription. (B) In normal development, the mammalian SWI/SNF complex including SMARCB1 and SMARCA4 antagonize PRC2, allowing transcription of pro-differentiation and cell cycle genes. (C) In rhabdoid tumors, loss of SMARCB1 is compensated at some promoters by SWI/SNF complexes containing SMARCA4, leading to transcription of cell cycle progression genes but not of pro-differentiation genes. (D) Tazemetostat inhibits EZH2, leading to decreased histone methylation. The HDAC inhibitor panobinostat leads to increased histone acetylation. Either decreased histone methylation or increased histone acetylation leads to increased transcription of pro-differentiation genes.
Panobinostat is a pan-histone deacetylase (HDAC) inhibitor.12 The inhibition of HDAC leads to increased histone acetylation, which is often a permissive epigenetic mark. The derepression of tumor suppressors and pro-differentiation genes should inhibit the growth and promote the differentiation of SMARCB1-deficient tumor cells. Preclinical studies show that panobinostat inhibits HDAC and promotes differentiation in AT/RT and MRT.12,13 The non-genotoxic mechanism of action is appealing to restore epigenetic balance in AT/RT cells. However, panobinostat can have severe side effects limiting the tolerability of the drug for long-term use, including life threatening hemorrhage, intractable diarrhea, electrolyte imbalance, and cardiac arrhythmia. In children, panobinostat administered 3 times per week led to thrombocytopenia, neutropenia, nausea, and diarrhea.14 A clinical trial studying maintenance panobinostat for patients with AT/RT (NCT04897880) closed because the drug maker withdrew panobinostat from the United States market. While panobinostat is no longer available in the USA, a number of other HDAC inhibitors, including vorinostat, romidepsin, and belinostat are FDA approved for hematologic malignancies. However, the brain penetration of panobinostat and other available HDAC inhibitors remains questionable, limiting their potential effectiveness against CNS AT/RT.15,16
Corin is a novel dual warhead molecule engineered to contain both an HDAC1/2 inhibitor and a LSD1 inhibitor connected by a linker.17 Corin selectively inhibits CoREST, a developmentally important epigenetic repressor. CoREST can antagonize residual SWI/SNF activity in AT/RT, potentially repressing neuronal differentiation programs (Figure 2). Treatment of AT/RT cells with corin suppresses growth and induces apoptosis. Corin also promotes differentiation in a number of cancer types, including melanoma, suggesting that corin removes the CoREST mediated block of differentiation.17 Because of the more tightly focused HDAC inhibition and the targeting of the inhibition to CoREST, corin may have a more favorable side effect profile than panobinostat or other more broadly targeted epigenetic agents.17
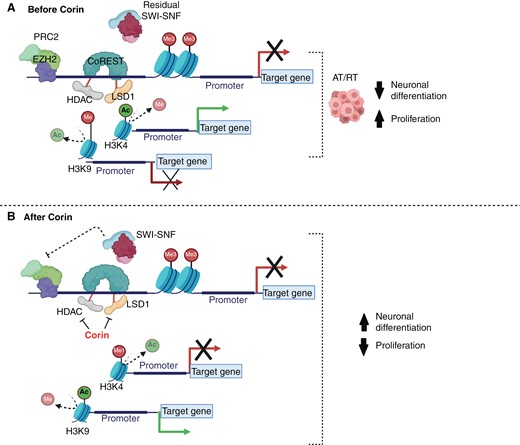
(A) CoREST may inhibit the residual mammalian SWI/SNF complex in rhabdoid tumors, leading to differentiation block due to decreased histone acetylation (H3K9Ac) and increased histone methylation (H3K9me), while increased H3K4Ac may drive pro-proliferation gene expression. (B) Inhibition of CoREST by corin may lead to increased H3K4 monomethylation (H3K4me), causing repression of pro-proliferation genes, as well as H3K9 aceylation (H3K9Ac), causing reactivation of tumor suppressors and rhabdoid tumor differentiation.
Summary
RTPS is a cancer predisposition syndrome that is highly likely to present with either a MRT or an AT/RT. These patients overall have a poor prognosis. Additional therapies to maintain tumor remission and to prevent additional tumor formation are needed. Because RTPS leads to AT/RT and/or MRT early in life, and it appears there is a critical period for formation of these aggressive tumors, a shorter period of preventative treatment may be needed compared to other tumor predisposition syndromes such as neurofibromatosis type 1 (NF1) and Li–Fraumeni syndrome. Identification and deployment of promising agents could significantly improve outcomes for patients with RTPS.
Funding
This work is supported by Allegheny Health Network-Johns Hopkins University research fund and by National Cancer Institute Core Grant to the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center (P30CA006973)
Conflict of interest statement
The authors report no conflicts of interest.

{kind=link}
{kind=link}