




Abstract
Adamantinomatous craniopharyngioma (ACP) is the most common benign tumor in the sellar region of children and originates from embryonic remnants. Owing to its unique location and frequent tight adhesion to and invasion of surrounding structures, the ACP poses significant challenges for neurosurgical treatment. Traditionally, the core treatment for ACPs has been surgical resection supplemented with radiotherapy in cases of residual or recurrent tumors. As a result, ACP classification has been based primarily on the tumor’s relationship with surrounding anatomical and histological structures, guiding the selection of surgical approaches and the prevention of complications. Moreover, efforts to explore pharmacological treatments for ACP have yielded varying results across different cases, creating confusion among researchers. This variability also suggests the possibility of different molecular subtypes within ACPs, despite being driven by a single-gene mutation. With advancements in molecular biological studies, such as ACP RNA sequencing, whole-exome sequencing, and methylation analysis, along with the discovery of interactions between different molecular pathways within ACP, researchers have been continuously exploring the molecular subtyping of ACP and predicting the efficacy of targeted therapies on the basis of these subtypes. This review focuses on summarizing and synthesizing the molecular mechanisms and potential subtypes of ACP, aiming to provide theoretical support for future research on the molecular subtyping of ACP.
Craniopharyngiomas are rare intracranial tumors originating from the craniopharyngeal duct at the base of the skull. It primarily occurs in children and adolescents, although adults can also be affected, with an incidence rate of approximately 0.5–2 cases per million person-years.1 Although craniopharyngiomas are histologically classified as benign epithelial tumors (World Health Organization [WHO] Grade I), their location, often near or adhered to critical neural and vascular structures, makes surgical treatment challenging, with high perioperative complication rates, high recurrence rates, and poor prognoses.2 As the pioneering neurosurgeon Professor Harvey Cushing once noted, craniopharyngioma represents one of the most formidable challenges faced by neurosurgeons.3 Previously, craniopharyngiomas were classified into adamantinomatous craniopharyngiomas (ACPs) and papillary craniopharyngiomas (PCPs) on the basis of their histological characteristics.4 However, owing to differences in their molecular biology and histopathology, these have now been recognized as distinct independent types of craniopharyngiomas.5 Among them, ACP is more common, accounting for approximately 90% of all craniopharyngioma cases. Therefore, an in-depth exploration of the detailed mechanisms of ACP is crucial for addressing craniopharyngiomas as a whole.6
Currently, there are 2 conventional treatment options for ACP: aggressive total resection or more conservative partial resection followed by radiotherapy.7 However, both options are associated with potential complications. Aggressive total resection may damage surrounding tissues, leading to cerebrovascular injury, cognitive decline, metabolic changes, and frequent panhypopituitarism.8–11 On the other hand, partial resection combined with radiotherapy may leave residual tumor tissue, increasing the risk of recurrence and repeat surgeries and increasing the risk of radiation-induced secondary malignancies.12–14 Consequently, there has been an ongoing exploration of anti-inflammatory treatments, immunotherapies, and other targeted therapies for ACP.15,16 Recent advances in the study of ACP molecular mechanisms have made significant progress, suggesting the potential for more effective targeted therapies that could reduce recurrence rates and treat related complications. Researchers have also explored the possibility of different molecular subtypes of ACP.17–20
This paper aims to systematically review the latest research advancements in ACP molecular mechanisms and the various molecular subtypes proposed on the basis of these advancements. This will further the development of ACP molecular research, aiding in the clinical prognosis assessment and precision treatment of patients.
Histopathological Features of ACP
Adamantinomatous craniopharyngioma is a nonneuroepithelial tumor located at the base of the third ventricle and the pia mater.21 It can occur at any age, with a peak incidence between the ages of 5–15 years and 45–60 years.7 In most cases, the tumor is tightly adhered to surrounding tissues, often infiltrating adjacent structures such as the pituitary gland, hypothalamus, and optic chiasm, making surgical separation challenging and the prognosis poor.22–25 Imaging of the ACP reveals different subtypes, including cystic, solid, or mixed cystic‒solid features (Figure 1A‒F). The cystic component may consist of one or more cysts filled with dark fluid, commonly referred to as “motor oil,” which is rich in lipids and inflammatory mediators.26,27 The solid part is primarily composed of epithelial tumor cells, which are highly heterogeneous and include rows of epithelium and stellate reticulum at the invasive front of the tumor. The epithelial cells form finger-like projections that often contain whorled cell structures23,28 (Figure 1G, H). The tumor is frequently surrounded by gliotic tissue, which consists mainly of astrocytes and immune cells,29 and this gliotic zone facilitates complete tumor resection. Additionally, ACPs often contain calcifications, which can be observed via computed tomography (CT), as well as wet keratin nodules (containing cells without visible nuclei), both of which are crucial for ACP diagnosis22 (Figure 1G). Notably, ACPs originate from precursor cells of the anterior pituitary gland, which are located outside the blood‒brain barrier, a histological feature that provides a natural advantage for ACP drug therapy.30
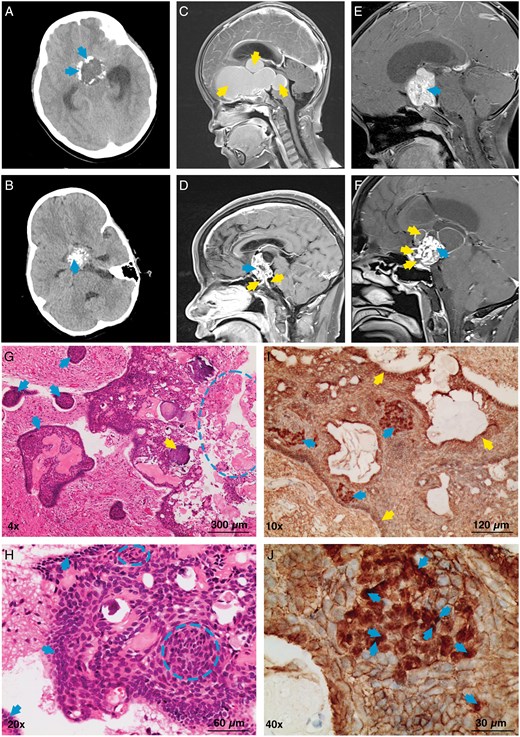
Imaging, HE, and IHC data related to ACPs. (A) CT scan showing egg-shell calcification of the ACP (arrows). (B) CT scan showing cluster-like calcification of the ACP (arrow). (C) MRI: sagittal T1 image with contrast enhancement showing a completely cystic lesion of the ACP (arrows). (D) MRI: sagittal T1 image with contrast enhancement showing a mixed cystic and solid lesion of the ACP. Solid lesion (upper left arrow), cystic lesion (two lower arrows). (E) MRI: sagittal T1 image with contrast enhancement showing a solid lesion of the ACP (arrow). (F) MRI: sagittal T1 image with contrast enhancement showing a mixed cystic and solid lesion of the ACP. Solid lesion (right arrow), cystic lesion (three left arrows). (G) HE staining: tumor papillary projection (four left arrows), wet keratin (circle), and calcification (arrow near the left side of the circle). (H) HE staining: tumor basal layer (arrows) and clusters of tumor cells (circles). (I) IHC: β-catenin expression in the nuclei and cytoplasm of some tumor cells within tumor cell clusters (three left arrows); the remaining tumor components show predominant membranous expression (top, right, and bottom arrows). (J) IHC: β-catenin nuclear expression (arrows).
Molecular Biology of ACP
On immunohistochemistry, ACPs typically exhibit abnormal accumulation of β-catenin (Figure 1I, J). Studies have shown that ACP is driven primarily by somatic mutations in exon 3 of the CTNNB1 gene, which encodes β-catenin. These mutations prevent the phosphorylation of β-catenin, increasing its resistance to degradation, thereby activating the Wnt/β-catenin signaling pathway, which induces the formation and development of ACPs28,31–33 (Figure 2A, B). The Wnt/β-catenin signaling pathway has been extensively studied in colorectal cancer, hepatocellular carcinoma, endometrial cancer, and medulloblastoma.34–36 These findings indicate that abnormal activation of this pathway can significantly promote tumorigenesis and progression, potentially disrupting cancer immunosurveillance, promoting immune evasion, and contributing to resistance to immunotherapies, including immune checkpoint inhibitors. In ACP studies, the reported incidence of CTNNB1 gene mutations varies significantly across different sources (16%~100%),37–40 and some researchers speculate that this variation may stem from differences in tumor site detection and the sensitivity of the techniques used. In ACPs, nuclear and cytoplasmic β-catenin accumulation is observed only in small clusters of cells, in areas with epithelial whorl-like structures, or a few cells near the invasive edge of the tumor. These regions influence ACP cell proliferation and differentiation through paracrine effects through the secretion of factors.22,41 No other somatic gene mutations associated with sporadic ACP have been identified, whereas somatic or germline mutations in the APC gene have been detected in some familial (nonsporadic) ACP patients, suggesting that familial ACP may involve a non-CTNNB1-dependent pathway.42
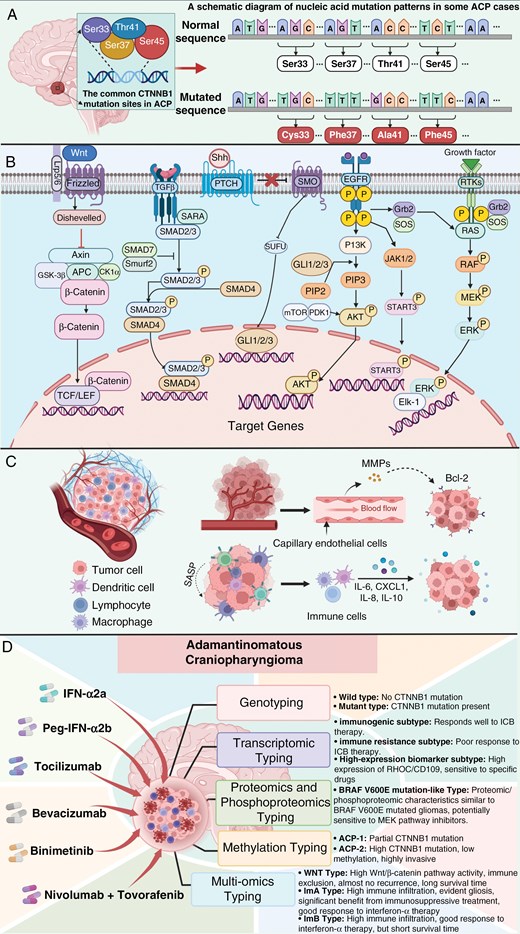
Schematic diagram of the molecular mechanisms and targeted therapies related to ACP development. (A) Common CTNNB1 mutation sites and nucleic acid mutations in ACPs. (B) Known intracellular signaling pathways in ACP tumor cells. (C) Schematic representation of MMPs; the cytokines IL-6, CXCL1, IL-8, and IL-10; and the role of the SASP-related paracrine pathway in the ACP microenvironment. (D) Schematic diagram of targeted therapies and molecular subtype classification of ACP. All panels were created with BioRender.com.
Recent research by Lu et al has shown that the TGF-β signaling pathway plays a significant role in promoting aggressive biological behavior and poor prognosis of ACP and is closely associated with recurrent ACP.43 The TGF-β pathway promotes epithelial‒mesenchymal transition (EMT) through various mechanisms, including autocrine and paracrine mechanisms, and coactivation with the WNT pathway, which is also common in other tumors.44–46 Additionally, through immunostaining and mouse model experiments, Apps et al reported that the MAPK/ERK pathway is highly active in certain areas of the ACP.27 Fibroblast growth factor (FGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and ERK1/2 colocalize with the proliferation marker Ki67 in the palisading epithelium around cluster structures and adjacent reactive tissues. In ACP treated with trametinib, pERK1/2 immunofluorescence is decreased, accompanied by dose-dependent apoptosis and significantly reduced proliferation,27 suggesting that the MAPK/ERK pathway could be a novel therapeutic target for ACP, providing preclinical evidence for the use of MEK inhibitors. In another concurrent study, researchers also reported that CD47—an immune checkpoint highly expressed in various tumor cells47–49—is overexpressed in ACPs, promoting ACP cell behavior through the activation of the MAPK/ERK pathway.50 Similarly, the sonic hedgehog (SHH) signaling pathway is also excessively activated in ACPs. Studies have shown that the mRNA expression of SHH, GLI1, and GLI3 is significantly greater in ACP samples than in normal pituitary tissues. These proteins belong to the SHH pathway, and analysis suggests that coactivation of the SHH and WNT pathways may play an important role in ACP pathogenesis.51–53 Moreover, the role of the epidermal growth factor receptor (EGFR) signaling pathway in ACP progression has garnered increasing attention.54 The EGFR pathway regulates cell migration by upregulating the expression of the low- and intermediate-molecular-weight keratins CK8 and CK18, a pattern of keratin expression also observed in ACP tumor cells.55 Hölsken reported that activation of the EGFR pathway significantly promotes ACP cell migration and invasion, a finding confirmed in a mouse ACP model experiment56,57 (Figure 2B).
In addition to the above signaling pathways, several other molecular mechanisms are involved in ACP development (Figure 2C), such as the role of matrix metalloproteinases (MMPs) in tumor invasion.18 Matrix metalloproteinases are highly expressed in both the stromal capillaries and epithelial components of ACPs, inducing the expression of the antiapoptotic protein B-cell leukemia/lymphoma 2 (Bcl-2) and participating in the regulation of tumor cell growth, thereby promoting ACP growth and recurrence through autocrine‒paracrine mechanisms.58,59 Immunomodulatory factors also play crucial roles in the tumor microenvironment.60 Donson et al reported high levels of cytokines and chemokines, especially IL-6, CXCL1, IL-8, IL-10, and their receptors, in ACP cyst fluid and tumor tissues.61 Grob et al reported significant shrinkage of ACP cysts upon treatment with monoclonal antibodies against VEGF and IL-6 receptor antagonists.62 In light of this evidence, Apps et al suggested that inflammation in ACPs is a key driver of ACP pathogenesis.2
The role of the senescence phenotype in ACP development is noteworthy, as recent studies have investigated how senescent cells induce tumorigenesis through paracrine pathways in ACPs.28,63–65 For a long time, cellular senescence has been widely regarded as a natural protective mechanism that limits cancer cell proliferation and tumor growth. However, this view is being increasingly challenged.66 Research suggests that the senescence-associated secretory phenotype (SASP) produced by senescent cells can trigger various protumorigenic cellular and molecular processes through paracrine pathways, leading to changes in the cellular microenvironment characterized by inflammation and immune response. This phenomenon has been confirmed in ACP studies.63,66 Single-cell and single-nucleus gene expression analyses further confirmed the presence of senescent cells with active SASP in ACPs, revealing their complex relationship with tumor progression.64
Historical Classification of Craniopharyngiomas
The classification of any disease is rooted in the evolving understanding of that disease, and craniopharyngiomas (CPs) are no exception. Since CP is considered a benign tumor (CNS WHO Grade I), it was historically believed that complete radical resection was the best treatment for craniopharyngiomas.67 However, radical resection increases the risk of intraoperative damage to the hypothalamus, significantly increasing surgical risk.68 This finding indicates that the extent of tumor adherence to the hypothalamus dictates the surgical treatment strategy for craniopharyngiomas. Consequently, classifying craniopharyngiomas on the basis of the degree of adhesion between the tumor and the hypothalamus has become natural. Currently, 3 main grading systems are mentioned in the literature to assess hypothalamic involvement or the adherence of the tumor to the hypothalamus: the Puget grading system,69 the Prieto grading system,70 and the Hori T grading system.71 It is indisputable that the adhesion and invasion of craniopharyngiomas to surrounding tissues significantly influence treatment decisions and patient prognosis.72 Therefore, a systematic and comprehensive preoperative analysis of the adhesion between the craniopharyngioma and surrounding structures, such as the hypothalamus, is crucial in determining the surgical approach.73
Once the surgical principles for craniopharyngiomas have been established, whether for aggressive or conservative treatment, selecting the correct and appropriate surgical approach becomes key to a successful operation. The goal of surgical resection of craniopharyngiomas is to remove as much of the tumor as possible while minimizing damage to the hypothalamus.74 To achieve this surgical goal, surgeons should choose the correct and appropriate surgical approach on the basis of the anatomical relationship between the tumor and surrounding neurovascular structures, as well as the surgeon’s personal experience. Over the past few decades, researchers have proposed a series of topographic classifications of craniopharyngiomas to facilitate precise assessment of the anatomical location of the tumor, thereby enabling the determination of the correct and appropriate surgical approach (Table 1).
The Topographic (Anatomical) Classification of CPs
| Author | Year | Classification basis | Classification system |
|---|---|---|---|
| Ciric IS et al75 | 1980 | Relationship of the tumor with the surrounding arachnoid and pia mater | -Intraventricular type -Subpial type -Subarachnoid extrameningeal type -Intraextraarachnoid type -Intra-arachnoid suprasellar type -Extra-arachnoid intrasellar type |
| Steno J76 | 1985 | Tumor location | -Intrasellar type -Suprasellar type (a) Extraventricular subtype (b) Intraextraventricular subtype (c) Intraventricular subtype |
| Yaşargil MG et al77 | 1990 | Relationship of the tumor with the diaphragm sellae | -Entirely intrasellar–subdiaphragmatic type -Intrasellar and suprasellar–sellar diaphragm type -Suprasellar para-chiasmatic–extraventricular type -Intraextraventricular type -Paraventricular type -Intraventricular type |
| Hoffman HJ78 | 1994 | Relationship of the tumor with the optic chiasm | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Giant type |
| Samii M et al79 | 1997 | The levels of tumor vertical invasion (5 grades) | -I: intrasellar type -II: suprasellar cistern type -III: inferior third ventricle type -IV: superior third ventricle type -V: lateral ventricle septum pellucidum type |
| Pascual JM et al80 | 2004 | Relationship of the tumor with the third ventricle | -I: suprasellar tumor compressing the third ventricle (third ventricle wall intact) -II: suprasellar tumor breaking through the floor of the third ventricle and invading inside -III: tumor inside the third ventricle, invading the floor of the third ventricle -IV: tumor inside the third ventricle, with the third ventricle floor intact |
| Wang KC et al81 | 2005 | Tumor origin and relationship with the diaphragma sellae | -Origin below the diaphragma sellae, diaphragma sellae intact -Origin below the diaphragma sellae, diaphragma sellae incomplete -Origin above the diaphragma sellae |
| Kassam AB et al82 Tang B et al83 | 2008 2019 | Relationship of the tumor with the pituitary stalk (endoscopic perspective) Subclassification of Type II (endoscopic perspective) | -I: preinfundibular type -II: trans-infundibular type -II1: tumor confined to the pituitary stalk -II2: (a) tumor extends upward into the third ventricle; (b) tumor extends downward into the sella -II3: tumor extends both upward into the third ventricle and downward into the sella -III: postinfundibular type -IIIa: tumor extends upward into the third ventricle -IIIb: tumor extends upward into the interpeduncular cistern -IIIa + b: tumor extends in both directions IV: completely intrathird ventricle type |
| Fatemi N et al84 | 2009 | Anatomical extension of the tumor | -Postchiasmatic type -Intrasellar-suprasellar type -Cavernous sinus involvement type -Far-lateral extension type |
| Yamada S et al85 | 2010 | Location of the tumor | -Below the sellar diaphragm type (a) Pure intrasellar type (b) Suprasellar extension type -Above the sellar diaphragm type |
| Qi S et al86,87 | 2011, 2021 | Growth pattern of the tumor around the arachnoid sleeve near the pituitary stalk | -Q type: tumor originating from the area below the sellar diaphragm -S type: tumor originating from the suprasellar pituitary stalk area -T type: tumor originating from the infundibular tubercle region |
| Matsuo T et al88 | 2014 | Anatomical relationships of the tumor with the diaphragma sellae, pituitary stalk, and optic nerves | -Relationship with the sellar diaphragm: below the sellar diaphragm type (complete, incomplete), above the sellar diaphragm type -Relationship with the pituitary stalk: preinfundibular, lateral-infundibular, postinfundibular, and Infiltrating infundibulum type -Relationship with optic nerve: prechiasmatic type, postchiasmatic type, and other (pure intrasellar type) -Tumor extension: third ventricle, interpeduncular cistern, prepontine cistern, frontal base, and cavernous sinus -Sphenoid sinus: intrasellar type, anterior sellar type, and external ear type |
| Morisako H et al89 | 2016 | Tumor origin | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Intrathird ventricle type |
| Jeswani S et al90 | 2016 | Relationship of the tumor with the pituitary stalk (endoscopic view) | -I: tumor mainly anterior to the pituitary stalk -II: tumor mainly within the pituitary stalk -III: tumor mainly posterior to the pituitary stalk |
| Tang B et al91 | 2018 | Relationship of the tumor with the pituitary stalk | -Central type -Lateral type (based on lesion origin): hypothalamic-pituitary stalk subtype Suprasellar pituitary stalk subtype Intrasellar pituitary stalk subtype |
| Prieto R et al92,93 | 2017, 2020 | Mammillary body angle (MBA) | -Intrasellar-suprasellar type -Suprasellar and third ventricle extraventricular type -Suprasellar–intraventricular invasive type -Infundibulum-tuber cinereum type -Pure intrathird ventricle type |
| Almeida JP et al94 | 2020 | Tumor extension | -A. Lower-middle type (intrasellar type) -B. Upper-middle type (suprasellar type) -C. Lateral type (lateral fissure type) -D. Intraventricular type -E. Posterior type (interpeduncular cistern and prepontine cistern type) |
| Lei C et al95 | 2021 | Location of the tumor (endoscopic perspective) | -Intrasellar type -Intrasellar-suprasellar type -Suprasellar type -Intrathird ventricle type |
| Author | Year | Classification basis | Classification system |
|---|---|---|---|
| Ciric IS et al75 | 1980 | Relationship of the tumor with the surrounding arachnoid and pia mater | -Intraventricular type -Subpial type -Subarachnoid extrameningeal type -Intraextraarachnoid type -Intra-arachnoid suprasellar type -Extra-arachnoid intrasellar type |
| Steno J76 | 1985 | Tumor location | -Intrasellar type -Suprasellar type (a) Extraventricular subtype (b) Intraextraventricular subtype (c) Intraventricular subtype |
| Yaşargil MG et al77 | 1990 | Relationship of the tumor with the diaphragm sellae | -Entirely intrasellar–subdiaphragmatic type -Intrasellar and suprasellar–sellar diaphragm type -Suprasellar para-chiasmatic–extraventricular type -Intraextraventricular type -Paraventricular type -Intraventricular type |
| Hoffman HJ78 | 1994 | Relationship of the tumor with the optic chiasm | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Giant type |
| Samii M et al79 | 1997 | The levels of tumor vertical invasion (5 grades) | -I: intrasellar type -II: suprasellar cistern type -III: inferior third ventricle type -IV: superior third ventricle type -V: lateral ventricle septum pellucidum type |
| Pascual JM et al80 | 2004 | Relationship of the tumor with the third ventricle | -I: suprasellar tumor compressing the third ventricle (third ventricle wall intact) -II: suprasellar tumor breaking through the floor of the third ventricle and invading inside -III: tumor inside the third ventricle, invading the floor of the third ventricle -IV: tumor inside the third ventricle, with the third ventricle floor intact |
| Wang KC et al81 | 2005 | Tumor origin and relationship with the diaphragma sellae | -Origin below the diaphragma sellae, diaphragma sellae intact -Origin below the diaphragma sellae, diaphragma sellae incomplete -Origin above the diaphragma sellae |
| Kassam AB et al82 Tang B et al83 | 2008 2019 | Relationship of the tumor with the pituitary stalk (endoscopic perspective) Subclassification of Type II (endoscopic perspective) | -I: preinfundibular type -II: trans-infundibular type -II1: tumor confined to the pituitary stalk -II2: (a) tumor extends upward into the third ventricle; (b) tumor extends downward into the sella -II3: tumor extends both upward into the third ventricle and downward into the sella -III: postinfundibular type -IIIa: tumor extends upward into the third ventricle -IIIb: tumor extends upward into the interpeduncular cistern -IIIa + b: tumor extends in both directions IV: completely intrathird ventricle type |
| Fatemi N et al84 | 2009 | Anatomical extension of the tumor | -Postchiasmatic type -Intrasellar-suprasellar type -Cavernous sinus involvement type -Far-lateral extension type |
| Yamada S et al85 | 2010 | Location of the tumor | -Below the sellar diaphragm type (a) Pure intrasellar type (b) Suprasellar extension type -Above the sellar diaphragm type |
| Qi S et al86,87 | 2011, 2021 | Growth pattern of the tumor around the arachnoid sleeve near the pituitary stalk | -Q type: tumor originating from the area below the sellar diaphragm -S type: tumor originating from the suprasellar pituitary stalk area -T type: tumor originating from the infundibular tubercle region |
| Matsuo T et al88 | 2014 | Anatomical relationships of the tumor with the diaphragma sellae, pituitary stalk, and optic nerves | -Relationship with the sellar diaphragm: below the sellar diaphragm type (complete, incomplete), above the sellar diaphragm type -Relationship with the pituitary stalk: preinfundibular, lateral-infundibular, postinfundibular, and Infiltrating infundibulum type -Relationship with optic nerve: prechiasmatic type, postchiasmatic type, and other (pure intrasellar type) -Tumor extension: third ventricle, interpeduncular cistern, prepontine cistern, frontal base, and cavernous sinus -Sphenoid sinus: intrasellar type, anterior sellar type, and external ear type |
| Morisako H et al89 | 2016 | Tumor origin | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Intrathird ventricle type |
| Jeswani S et al90 | 2016 | Relationship of the tumor with the pituitary stalk (endoscopic view) | -I: tumor mainly anterior to the pituitary stalk -II: tumor mainly within the pituitary stalk -III: tumor mainly posterior to the pituitary stalk |
| Tang B et al91 | 2018 | Relationship of the tumor with the pituitary stalk | -Central type -Lateral type (based on lesion origin): hypothalamic-pituitary stalk subtype Suprasellar pituitary stalk subtype Intrasellar pituitary stalk subtype |
| Prieto R et al92,93 | 2017, 2020 | Mammillary body angle (MBA) | -Intrasellar-suprasellar type -Suprasellar and third ventricle extraventricular type -Suprasellar–intraventricular invasive type -Infundibulum-tuber cinereum type -Pure intrathird ventricle type |
| Almeida JP et al94 | 2020 | Tumor extension | -A. Lower-middle type (intrasellar type) -B. Upper-middle type (suprasellar type) -C. Lateral type (lateral fissure type) -D. Intraventricular type -E. Posterior type (interpeduncular cistern and prepontine cistern type) |
| Lei C et al95 | 2021 | Location of the tumor (endoscopic perspective) | -Intrasellar type -Intrasellar-suprasellar type -Suprasellar type -Intrathird ventricle type |
The Topographic (Anatomical) Classification of CPs
| Author | Year | Classification basis | Classification system |
|---|---|---|---|
| Ciric IS et al75 | 1980 | Relationship of the tumor with the surrounding arachnoid and pia mater | -Intraventricular type -Subpial type -Subarachnoid extrameningeal type -Intraextraarachnoid type -Intra-arachnoid suprasellar type -Extra-arachnoid intrasellar type |
| Steno J76 | 1985 | Tumor location | -Intrasellar type -Suprasellar type (a) Extraventricular subtype (b) Intraextraventricular subtype (c) Intraventricular subtype |
| Yaşargil MG et al77 | 1990 | Relationship of the tumor with the diaphragm sellae | -Entirely intrasellar–subdiaphragmatic type -Intrasellar and suprasellar–sellar diaphragm type -Suprasellar para-chiasmatic–extraventricular type -Intraextraventricular type -Paraventricular type -Intraventricular type |
| Hoffman HJ78 | 1994 | Relationship of the tumor with the optic chiasm | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Giant type |
| Samii M et al79 | 1997 | The levels of tumor vertical invasion (5 grades) | -I: intrasellar type -II: suprasellar cistern type -III: inferior third ventricle type -IV: superior third ventricle type -V: lateral ventricle septum pellucidum type |
| Pascual JM et al80 | 2004 | Relationship of the tumor with the third ventricle | -I: suprasellar tumor compressing the third ventricle (third ventricle wall intact) -II: suprasellar tumor breaking through the floor of the third ventricle and invading inside -III: tumor inside the third ventricle, invading the floor of the third ventricle -IV: tumor inside the third ventricle, with the third ventricle floor intact |
| Wang KC et al81 | 2005 | Tumor origin and relationship with the diaphragma sellae | -Origin below the diaphragma sellae, diaphragma sellae intact -Origin below the diaphragma sellae, diaphragma sellae incomplete -Origin above the diaphragma sellae |
| Kassam AB et al82 Tang B et al83 | 2008 2019 | Relationship of the tumor with the pituitary stalk (endoscopic perspective) Subclassification of Type II (endoscopic perspective) | -I: preinfundibular type -II: trans-infundibular type -II1: tumor confined to the pituitary stalk -II2: (a) tumor extends upward into the third ventricle; (b) tumor extends downward into the sella -II3: tumor extends both upward into the third ventricle and downward into the sella -III: postinfundibular type -IIIa: tumor extends upward into the third ventricle -IIIb: tumor extends upward into the interpeduncular cistern -IIIa + b: tumor extends in both directions IV: completely intrathird ventricle type |
| Fatemi N et al84 | 2009 | Anatomical extension of the tumor | -Postchiasmatic type -Intrasellar-suprasellar type -Cavernous sinus involvement type -Far-lateral extension type |
| Yamada S et al85 | 2010 | Location of the tumor | -Below the sellar diaphragm type (a) Pure intrasellar type (b) Suprasellar extension type -Above the sellar diaphragm type |
| Qi S et al86,87 | 2011, 2021 | Growth pattern of the tumor around the arachnoid sleeve near the pituitary stalk | -Q type: tumor originating from the area below the sellar diaphragm -S type: tumor originating from the suprasellar pituitary stalk area -T type: tumor originating from the infundibular tubercle region |
| Matsuo T et al88 | 2014 | Anatomical relationships of the tumor with the diaphragma sellae, pituitary stalk, and optic nerves | -Relationship with the sellar diaphragm: below the sellar diaphragm type (complete, incomplete), above the sellar diaphragm type -Relationship with the pituitary stalk: preinfundibular, lateral-infundibular, postinfundibular, and Infiltrating infundibulum type -Relationship with optic nerve: prechiasmatic type, postchiasmatic type, and other (pure intrasellar type) -Tumor extension: third ventricle, interpeduncular cistern, prepontine cistern, frontal base, and cavernous sinus -Sphenoid sinus: intrasellar type, anterior sellar type, and external ear type |
| Morisako H et al89 | 2016 | Tumor origin | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Intrathird ventricle type |
| Jeswani S et al90 | 2016 | Relationship of the tumor with the pituitary stalk (endoscopic view) | -I: tumor mainly anterior to the pituitary stalk -II: tumor mainly within the pituitary stalk -III: tumor mainly posterior to the pituitary stalk |
| Tang B et al91 | 2018 | Relationship of the tumor with the pituitary stalk | -Central type -Lateral type (based on lesion origin): hypothalamic-pituitary stalk subtype Suprasellar pituitary stalk subtype Intrasellar pituitary stalk subtype |
| Prieto R et al92,93 | 2017, 2020 | Mammillary body angle (MBA) | -Intrasellar-suprasellar type -Suprasellar and third ventricle extraventricular type -Suprasellar–intraventricular invasive type -Infundibulum-tuber cinereum type -Pure intrathird ventricle type |
| Almeida JP et al94 | 2020 | Tumor extension | -A. Lower-middle type (intrasellar type) -B. Upper-middle type (suprasellar type) -C. Lateral type (lateral fissure type) -D. Intraventricular type -E. Posterior type (interpeduncular cistern and prepontine cistern type) |
| Lei C et al95 | 2021 | Location of the tumor (endoscopic perspective) | -Intrasellar type -Intrasellar-suprasellar type -Suprasellar type -Intrathird ventricle type |
| Author | Year | Classification basis | Classification system |
|---|---|---|---|
| Ciric IS et al75 | 1980 | Relationship of the tumor with the surrounding arachnoid and pia mater | -Intraventricular type -Subpial type -Subarachnoid extrameningeal type -Intraextraarachnoid type -Intra-arachnoid suprasellar type -Extra-arachnoid intrasellar type |
| Steno J76 | 1985 | Tumor location | -Intrasellar type -Suprasellar type (a) Extraventricular subtype (b) Intraextraventricular subtype (c) Intraventricular subtype |
| Yaşargil MG et al77 | 1990 | Relationship of the tumor with the diaphragm sellae | -Entirely intrasellar–subdiaphragmatic type -Intrasellar and suprasellar–sellar diaphragm type -Suprasellar para-chiasmatic–extraventricular type -Intraextraventricular type -Paraventricular type -Intraventricular type |
| Hoffman HJ78 | 1994 | Relationship of the tumor with the optic chiasm | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Giant type |
| Samii M et al79 | 1997 | The levels of tumor vertical invasion (5 grades) | -I: intrasellar type -II: suprasellar cistern type -III: inferior third ventricle type -IV: superior third ventricle type -V: lateral ventricle septum pellucidum type |
| Pascual JM et al80 | 2004 | Relationship of the tumor with the third ventricle | -I: suprasellar tumor compressing the third ventricle (third ventricle wall intact) -II: suprasellar tumor breaking through the floor of the third ventricle and invading inside -III: tumor inside the third ventricle, invading the floor of the third ventricle -IV: tumor inside the third ventricle, with the third ventricle floor intact |
| Wang KC et al81 | 2005 | Tumor origin and relationship with the diaphragma sellae | -Origin below the diaphragma sellae, diaphragma sellae intact -Origin below the diaphragma sellae, diaphragma sellae incomplete -Origin above the diaphragma sellae |
| Kassam AB et al82 Tang B et al83 | 2008 2019 | Relationship of the tumor with the pituitary stalk (endoscopic perspective) Subclassification of Type II (endoscopic perspective) | -I: preinfundibular type -II: trans-infundibular type -II1: tumor confined to the pituitary stalk -II2: (a) tumor extends upward into the third ventricle; (b) tumor extends downward into the sella -II3: tumor extends both upward into the third ventricle and downward into the sella -III: postinfundibular type -IIIa: tumor extends upward into the third ventricle -IIIb: tumor extends upward into the interpeduncular cistern -IIIa + b: tumor extends in both directions IV: completely intrathird ventricle type |
| Fatemi N et al84 | 2009 | Anatomical extension of the tumor | -Postchiasmatic type -Intrasellar-suprasellar type -Cavernous sinus involvement type -Far-lateral extension type |
| Yamada S et al85 | 2010 | Location of the tumor | -Below the sellar diaphragm type (a) Pure intrasellar type (b) Suprasellar extension type -Above the sellar diaphragm type |
| Qi S et al86,87 | 2011, 2021 | Growth pattern of the tumor around the arachnoid sleeve near the pituitary stalk | -Q type: tumor originating from the area below the sellar diaphragm -S type: tumor originating from the suprasellar pituitary stalk area -T type: tumor originating from the infundibular tubercle region |
| Matsuo T et al88 | 2014 | Anatomical relationships of the tumor with the diaphragma sellae, pituitary stalk, and optic nerves | -Relationship with the sellar diaphragm: below the sellar diaphragm type (complete, incomplete), above the sellar diaphragm type -Relationship with the pituitary stalk: preinfundibular, lateral-infundibular, postinfundibular, and Infiltrating infundibulum type -Relationship with optic nerve: prechiasmatic type, postchiasmatic type, and other (pure intrasellar type) -Tumor extension: third ventricle, interpeduncular cistern, prepontine cistern, frontal base, and cavernous sinus -Sphenoid sinus: intrasellar type, anterior sellar type, and external ear type |
| Morisako H et al89 | 2016 | Tumor origin | -Intrasellar type -Prechiasmatic type -Postchiasmatic type -Intrathird ventricle type |
| Jeswani S et al90 | 2016 | Relationship of the tumor with the pituitary stalk (endoscopic view) | -I: tumor mainly anterior to the pituitary stalk -II: tumor mainly within the pituitary stalk -III: tumor mainly posterior to the pituitary stalk |
| Tang B et al91 | 2018 | Relationship of the tumor with the pituitary stalk | -Central type -Lateral type (based on lesion origin): hypothalamic-pituitary stalk subtype Suprasellar pituitary stalk subtype Intrasellar pituitary stalk subtype |
| Prieto R et al92,93 | 2017, 2020 | Mammillary body angle (MBA) | -Intrasellar-suprasellar type -Suprasellar and third ventricle extraventricular type -Suprasellar–intraventricular invasive type -Infundibulum-tuber cinereum type -Pure intrathird ventricle type |
| Almeida JP et al94 | 2020 | Tumor extension | -A. Lower-middle type (intrasellar type) -B. Upper-middle type (suprasellar type) -C. Lateral type (lateral fissure type) -D. Intraventricular type -E. Posterior type (interpeduncular cistern and prepontine cistern type) |
| Lei C et al95 | 2021 | Location of the tumor (endoscopic perspective) | -Intrasellar type -Intrasellar-suprasellar type -Suprasellar type -Intrathird ventricle type |
Among all the anatomical classifications of craniopharyngiomas, the Yaşargil MG classification, Samii M classification, Morisako H classification, and Qi S classification are the most widely referenced. The Yaşargil MG classification is commonly used in clinical practice, primarily because of the widespread adoption of pterional surgical approaches and microsurgical techniques at the time. The Samii M and Morisako H classifications are, in fact, modifications of the Yaşargil MG classification. However, different surgical practices and the frequent need to adapt surgical approaches on the basis of the tumor’s location and morphological characteristics during surgery make it difficult to assess the strengths and weaknesses of each classification solely on the basis of surgical outcomes from different centers.
The QST classification, which is based on the tissue of origin, is particularly noteworthy for researchers. As individualized surgical plans become more common and the integration of endoscopic techniques with transcranial microsurgery advances,96–98 different anatomical classifications may increasingly focus on hypothalamic protection during resection and the decision of whether to pursue total tumor removal.99,100
Molecular Subtyping of ACP
As described in the previous discussion on molecular biology, the development of ACPs involves the interaction of multiple signaling pathways and molecular mechanisms, including Wnt/β-catenin, MAPK, SHH, and others, which play significant roles in the formation and progression of ACPs. In recent years, with the application of high-resolution technologies such as single-cell RNA sequencing, researchers have gradually revealed significant heterogeneity within ACP tumors.101 This internal heterogeneity also leads to considerable differences in the clinical presentations of patients with ACP.102 For example, the age of onset varies widely, affecting both children and adults; radiologically, ACP can present in various forms, including cystic, solid, or a combination of cystic and solid components.103 These diverse presentations contrast sharply with the traditional view of ACP as a tumor driven solely by single-gene mutations. More notably, different patients also exhibit significant variability in their responses to drug treatments.104 For example, some patients respond well to anti-inflammatory therapy and achieve disease control, whereas others may show no significant response to the same treatment.15 These findings further suggest that ACP might exist in different molecular subtypes, each with distinct mechanisms of onset, progression, and treatment response. Tumor classification has gradually shifted from histological to molecular subtyping, and new molecular subtyping is not only the basis for precision therapy but also crucial for elucidating tumor mechanisms, identifying prognostic factors, and developing personalized treatment strategies.105,106
Research on the molecular subtyping of ACP has evolved from the discovery of single-gene mutations to comprehensive multiomics analyses. Early studies focused primarily on mutations in the CTNNB1 gene (encoding β-catenin), revealing aberrant activation of the Wnt signaling pathway in ACP.107–109 Subsequently, specific epigenetic markers, such as DNA methylation and histone modifications, were identified, revealing different patterns among numerous ACP patients.110 With technological advancements, researchers have employed high-throughput sequencing techniques, including transcriptomics, proteomics, single-cell sequencing, and spatial transcriptomics, to perform comprehensive analyses of the ACP genome, discovering additional gene mutations and structural variations.4,101,111,112 Clinical studies indicate that the biological heterogeneity of ACP tumors results in significant variations in clinical presentation, pathological features, and prognosis. For example, ACPs with certain molecular characteristics may exhibit increased invasiveness and recurrence rates, whereas others demonstrate relatively benign behavior.39,113,114 These findings suggest the possibility of finer molecular subtypes within ACPs. Current research on the molecular subtypes of ACP has focused mainly on defining different tumor subtypes through gene mutation profiles, expression profiles, and epigenetic changes22,110,115 (Figure 2D).
In early studies on ACP, due to significant variations in the reported incidence of CTNNB1 gene mutations among different studies, classifying ACP into wild-type and mutant-type seemed to be a feasible approach.37–40 However, this classification currently lacks sufficient clinical data to support it.33,116 Moreover, the so-called wild-type ACP, IHC, and HE staining images have shown the presence of similar whorled cluster structures and nuclear accumulation of β-catenin in these samples.20 A recent study employed laser capture microdissection to isolate tumor cells and conducted deep sequencing of targeted amplified fragments. The results revealed that all ACP samples tested carried CTNNB1 mutations.40 The study by Zhang et al also confirmed that the detection of wild-type cases may be affected by the low proportion of tumor cells and the limited sensitivity of current sequencing technologies.117 Therefore, the existence of wild-type and mutant-type still requires further investigation.
Given the challenges of molecular genotyping, researchers have begun exploring other classification criteria. The cyst wall cells of craniopharyngiomas express programmed death-ligand 1 (PD-L1), while programmed death-1 (PD-1) is expressed in whorl-like cells with nuclear accumulation of β-catenin, suggesting that PD-1/PD-L1 immune checkpoint-targeted therapy for craniopharyngiomas might have potential clinical value.118 Nevertheless, in subsequent studies, we observed that the expression of PD-L1 in ACP tumor cells is challenging to quantify definitively, including determining the level of expression and how to classify it. It remains to be further determined which types of patients could benefit from this treatment.119–121 Yuan et al retrieved RNA data from 39 pediatric ACP patients from the public databases GSE60815 and GSE94349 and, through screening and analysis, classified 725 ACP-related immune genes into 2 types: immune resistance subtype and immunogenic subtype.115 These results indicate that these 2 types respond differently to immune checkpoint blockade (ICB) therapy. However, Yuan et al’s classification is merely a bioinformatics-based classification, lacking comparative analysis with clinical data such as imaging or histology, and it has not been validated in clinical practice. Moreover, this classification does not clarify the relationship between these 2 subtypes and the gene-driven pathways of ACP.118
Wu et al reported that ACPs are diverse at the genetic level, providing an important basis for the molecular subtyping of ACPs. They introduced the concept that ACP patients with high expression of RHOC and CD109 have different sensitivities to various drugs on the basis of sequencing databases.112 However, like previous studies, they did not obtain clinical research support. Many scholars pessimistically believe that it might not be possible to subtype ACPs on the basis of RNA omics.122 A recently published study proposed a new classification method that classifies certain tumor cells as the BRAF V600E mutation-like type on the basis of proteomics and phosphoproteomics theories.123 According to this new classification method, the proteomic changes in the MAPK/MEK pathway of these tumor cells resemble the previously observed BRAF V600E gene variation expression, suggesting that MEK pathway inhibitors might be effective for ACP treatment.27,124 Additionally, an epigenetic study by Gutiérrez et al identified 2 distinct ACP methylation clusters, with the ACP-2 cluster exhibiting significantly low methylation and being associated with CTNNB1 mutations, which are more aggressive.110 These findings suggest that DNA methylation features might be used for future ACP diagnosis and prognosis.
Recently, Professor Lin Zhixiong’s research team conducted whole-exome sequencing (WES) on 151 samples from 143 craniopharyngioma patients, along with RNA sequencing on 84 samples and DNA methylation analysis on 95 samples (the largest dataset globally).20 The results revealed completely distinct molecular subtypes of ACP at the RNA level, which was consistent with the findings of the methylation-based subtypes, indicating that ACP has 3 different molecular subtypes: the WNT type, ImA type, and ImB type. Among them, the WNT type can be overactivated through the Wnt/β-catenin pathway, whereas the ImA type and ImB type exhibit increased levels of immune infiltration, with the ImA type showing a significant gliosis reaction. These molecular characteristics of the 3 subtypes were confirmed through immunohistochemistry and proteomics analysis. Computed tomography and MRI observations indicate that the WNT type is predominantly solid, whereas the ImA type and ImB type are primarily cystic. Prognostic statistics show that patients with the WNT type almost never experience recurrence, and their survival time is significantly longer than that of patients with the ImB type. A multivariate Cox regression analysis suggested that this classification is the most significant prognostic predictor apart from the extent of tumor resection. According to the TIDE (tumor immune dysfunction and exclusion) model predictions, the ImA type is expected to benefit the most from immunosuppressive therapy among the 3 subtypes, whereas the WNT type shows an immune exclusion status. Single-sample gene set enrichment analysis (GSEA) revealed that only ImA type and ImB type samples were enriched for the interferon-α response pathway, which is consistent with a retrospective analysis of clinical trial results: interferon-α treatment responses were observed only in ACP cases with predominantly cystic components.125 These findings are highly important for the future precision diagnosis and treatment of ACP. However, the study did not clarify why tumors driven by single-gene mutations present different molecular subtypes. This may result from interactions between the molecular pathways driven by single-gene mutations and the tumor microenvironment.
Precision Therapy for ACP on the Basis of Molecular Characteristics
In recent years, there has been a gradual exploration of precision-targeted therapies for ACP126 (Figure 2D). Ros et al investigated the inflammatory environment of ACP and its therapeutic importance and reported that ACP displays numerous inflammatory and immune markers, suggesting the presence of numerous potential treatment targets.15 Nowadays, various related drugs are being tested for the treatment of ACP.127 Earlier, Jakacki et al studied the application of interferon-2α (IFN-2α) in ACP patients,128 followed by clinical studies evaluating the response of ACPs to locally and systemically administered pegylated IFN-α-2b.129 However, a recent phase II clinical trial indicated that pegylated IFN-α-2b had limited efficacy.130,131 Transcriptomic analyses have shown that both solid and cystic components of ACP exhibit inflammasome activation.61 In this context, Grob, Vos-Kerkhof, and others explored the use of the monoclonal antibody tocilizumab, which targets IL-6, for the treatment of ACP and reported good efficacy in cystic ACP patients.62,132 Currently, a phase II clinical trial (NCT05233397) is further evaluating the efficacy of tocilizumab (Actemra) for progressive or recurrent ACP.133 Additionally, De Rosa et al reported a case where ACP was treated with bevacizumab, resulting in a 66.1% reduction in tumor volume after 3 months, with partial remission achieved.134 In another case study by Patel et al, the use of MEK inhibitor binimetinib was shown to reduce the tumor volume and stabilize the condition of the patient.124 Related to this, a phase II clinical trial (NCT05286788) is also assessing the efficacy of binimetinib (Mektovi) in pediatric ACP patients.135 In addition to the aforementioned studies, another phase II trial (NCT05465174) is investigating the potential of the PD-1 inhibitor nivolumab in combination with the pan-RAF kinase inhibitor tovorafenib for the treatment of ACP in children and adolescents, which is also worth ongoing attention.136 While research on targeted therapies for ACP highlights the sustained efforts of scholars to develop effective interventions, current treatment options remain limited.126 Additionally, the efficacy of most treatments is limited, with relatively few cases and inconsistent outcomes. Given the heterogeneity of ACPs and the urgent need for personalized treatment strategies, further research is needed to enrich and validate targeted therapy options for ACPs.
Conclusion and Outlook
Adamantinomatous craniopharyngioma is a complex and challenging type of intracranial tumor. Although histologically classified as benign, its invasive growth and high recurrence rate make treatment and prognosis extremely difficult.137 Over the past few decades, significant progress has been made in understanding ACP, particularly in the field of molecular biology. Research has revealed the critical roles of several key signaling pathways, including the Wnt/β-catenin signaling pathway, the TGF-β signaling pathway, and the MAPK/ERK pathway, in the development and progression of ACP. Additionally, the senescence phenotype, immune modulators, and MMPs play significant roles in the tumor microenvironment. These discoveries have laid the groundwork for molecular subtyping and have provided potential targets for the development of new therapeutic strategies.
Currently, molecular subtyping is gradually emerging in ACP research. Through the analysis of mutation profiles, expression profiles, and epigenetic changes, researchers have identified multiple molecular subtypes of ACP. Recent studies have indicated that ACPs can be classified into the WNT type, ImA type, and ImB type. These subtypes differ significantly in their clinical presentation and prognosis, suggesting that future treatment strategies should fully consider the characteristics of these molecular subtypes to enable precise treatment, in line with the growing trend toward precision medicine in cancer research.138
Future research on the molecular biology of ACP should focus on understanding why tumors driven by single-gene mutations present different molecular subtypes. Previous studies have suggested a close relationship between inflammation and ACP development.41 This raises the following question: do the different molecular subtypes of ACP result from interactions between single-gene mutation-driven molecular pathways and the inflammatory microenvironment of the tumor? If this hypothesis is confirmed, future targeted therapies for ACP must inevitably involve a combination of both approaches.
Funding
The study was supported by the Sanbo Brain Hospital Management Group [SBJT-KY-2020-002 to Z.L.], the Capital Funds for Health Improvement and Research [2022-2-8013 to Z.L.], and the National Key R&D Program of China [2023YFC2411603 to Z.L.].
Conflict of interest statement. None declared.
Acknowledgments
Figure 2 was created with BioRender.com.
Author contributions
W.A., S.L., and Y.A. were responsible for the conception and design of the study. W.A. drafted and edited the manuscript. Z.L. reviewed and revised the manuscript. All the authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
This study was performed in accordance with the principles of the Declaration of Helsinki. Approval was granted by the Institutional Review Board/Ethics Committee of Sanbo Brain Hospital of Capital Medical University.
Consent for publication
All authors have read and approved the final manuscript.
Data availability
Not applicable.



{kind=link}
{kind=link}