





PLAIN ENGLISH SUMMARY
Endothelin-1 (ET-1) is a peptide that is involved in various chronic diseases including cardiovascular and kidney disease. ET-1 can bind to two receptors, endothelin A (ETA) and endothelin B (ETB), which are found in different organs and tissues. When ET-1 binds to the ETA receptor, it causes blood vessels to narrow, while binding of ET-1 to the ETB receptor causes blood vessels to widen. These receptors help regulate fluid and electrolyte balance in the kidneys, as well as the kidney's ability to filter various substances out of the body. Overactivation of ET-1 can occur in people with diabetes or obesity, which can damage the structure and function of the kidney.
Studies in mice and humans with kidney disease have shown that blocking the ETA receptor improves kidney health. As a result, medicines that specifically block the ETA receptor, known as endothelin receptor antagonists (ERAs), are a promising option for treating these kidney diseases. The first ERA (sparsentan) is now available for use in patients with immunoglobulin A (IgA) nephropathy, a specific type of kidney disease. It should be noted that ERAs can cause side effects. Fluid retention, which can increase the risk of heart failure, is a side effect that is particularly observed in patients with type 2 diabetes and severe kidney disease. This side effect is less often observed in patients with IgA nephropathy or patients without diabetes.
Treatment strategies to optimize safe and effective use of ETA blockers are being developed. Overall, these insights offer hope for better care of patients with kidney disease.
Endothelin-1 (ET-1) is a 21–amino acid peptide involved in numerous cardiovascular and renal processes. ET-1 can bind to endothelin receptor A (ETA) and endothelin receptor B (ETB), which are found in various organs and tissues. In general, binding of ET-1 to the ETA receptor causes vasoconstriction, whereas activation of the ETB receptor leads to vasodilation. In the kidney, endothelin receptors regulate fluid and electrolyte balance, regional blood flow and glomerular filtration rate. In pathological conditions, ET-1 promotes kidney injury through adverse effects on the endothelial glycocalyx, podocytes and mesangial cells, and stimulating inflammation and fibrosis in the tubules. In experimental and clinical studies, inhibition of the ETA receptor has been shown beneficial in a variety of kidney diseases. These include diabetic kidney disease, immunoglobulin A nephropathy, focal segmental glomerulosclerosis and Alport syndrome. Accordingly, selective ETA endothelin receptor antagonists (ERA) may prove a viable therapeutic option in these diseases. However, clinical application is challenged by the occurrence of fluid retention which can lead to heart failure, in particular in patients with severe CKD. Concomitant use of sodium-glucose cotransporter 2 inhibitors (SGLT2i) may mitigate these adverse effects through their diuretic actions. The development of highly selective ETA antagonists, such as atrasentan and zibotentan, and the opportunities of combining these with SGLT2i, holds promise to optimize efficacy and safety of ERAs in clinical practice.
INTRODUCTION
Endothelin-1 (ET-1) is a peptide consisting of 21 amino acids and is involved in regulation of basal vascular tone [1]. Soon after the discovery of ET-1, it became clear that endothelin may contribute to hypertension and vascular spasms. ET-1 is generated from an inactive precursor, big endothelin, through proteolytic processing by endothelin-converting enzyme. ET-1 can bind to two receptors, the endothelin receptor A (ETA) and endothelin B (ETB) receptor. Both receptors are present in many organs and tissues, although the ETA receptor is mainly located in the vasculature, heart, lungs, ovaries and uterus. ETB receptors are mainly found in the brain and lungs [2]. Binding of ET-1 to the ETA receptor causes vasoconstriction, hypertension, inflammation and fibrosis. In contrast, activation of ETB causes vasodilation via a nitric oxide–mediated pathway. In the kidney ETB activation causes natriuresis. ETB also serves as a clearance receptor for ET-1 thereby opposing the actions of ETA activation. However, ETB does not always oppose the actions of ETA, and therefore the situation is not as straightforward as it seems (Fig. 1). For example, activation of ETB on vascular smooth muscle cells can also promote vasoconstriction [3]. The objective of this review is to summarize the current knowledge on the role of ET-1 in the pathophysiology of chronic kidney disease (CKD) and the efficacy and safety of endothelin receptor antagonists in patients with CKD.
ENDOTHELIN AND THE KIDNEY
Endothelin activation and kidney injury
Multiple experimental studies have demonstrated that activation of the endothelin pathway leads to progressive kidney injury. ETA and ETB receptors are present in the kidney where they are critically involved in maintaining fluid and electrolyte homeostasis, regional blood flow and glomerular filtration rate (GFR) [4]. Increased ET-1 production causes pathological changes in many kidney cell types. ET-1 production is stimulated by multiple biochemical (growth factors, oxidative stress, cytokines), metabolic (dysglycaemia, obesity), vasoactive (angiotensin II, aldosterone) and pathological (proteinuria) factors which are typically dysregulated in people with CKD [5]. Binding of ET-1 to the ETA receptor in the glomeruli or tubuli causes damage to many different cell types or structures including the endothelial glycocalyx, podocytes and cytoskeleton, and mesangial cells [6].
ETA activation causes endothelial glycocalyx damage thereby affecting the charge selective properties of the glomeruli and resulting in increased albumin and protein leakage [7]. Increased tubular exposure to excessive amounts of filtered proteins activates a cascade of events including feedback loops to promote ET-1 release. This leads to recruitment of monocytes which secrete pro-inflammatory cytokines which in turn initiate and sustain a pro-inflammatory environment leading to tubulointerstitial remodelling and scarring [8]. There is also experimental evidence that ET-1-stimulated crosstalk between podocytes and endothelial cells results in endothelial glycocalyx damage and aggravates albuminuria [9]. Such processes are expected to lead to excessive proteinuria which feedback to enhance ET-1 release.
In experimental models of kidney disease there is abundant evidence that ETA activation leads to podocyte damage, slit diaphragm dysfunction and disruption of the cytoskeleton [10, 11].
Mesangial cells are also typically targeted by ET-1 via ETA receptor activation which lead to cell proliferation and fibrosis. It is thus beyond doubt that ET-1 and ETA receptor activation are critically involved in the pathogenesis of multiple kidney disease aetiologies.
It is therefore perhaps no surprise that pharmacological inhibition of the ETA receptor using selective endothelin receptor antagonist (ERA) have shown benefits in a range of experimental models of kidney disease including experimental models of diabetic kidney disease, immunoglobulin A (IgA) nephropathy, focal segmental glomerulosclerosis (FSGS) and Alport syndrome (Table 1) [12–19]. Some of these studies compared ETA with ETB inhibition or assessed effects of combined ETA/B inhibition. The majority of these studies demonstrated no kidney benefit of ETB inhibition and sometimes even harmful effects [20–36].
Experimental studies with selective ETA and ETB receptor antagonists.
| Animal model | ERA | Comparator | Endpoint | Result |
|---|---|---|---|---|
| Diabetic kidney disease | ||||
| Streptozotocin-induced diabetic rats [12] | Darusentan | No treatment | Daily urinary protein excretion | Treatment with darusentan reduced the daily protein excretion by >50% 36 weeks after diabetes induction, compared with non-treated rats |
| Streptozotocin-induced diabetic rats [13] | Atrasentan | Vehicle | Daily urinary protein excretion | Treatment with atrasentan for 6 weeks reduced the daily protein excretion by 40%, compared with rats treated with vehicle |
| IgA nephropathy | ||||
| gddY mice [14] | Sparsentan | Losartan | Development of renal injury | Sparsentan compared with losartan attenuated UACR more rapidly and to a greater extent, which correlated with increased podocyte and glycocalyx protection |
| gddY mice [15] | Atrasentan | None | Change in UACR after 4 days of treatment | Atrasentan reduced UACR from baseline by 28 ± 44% (P = ns), 62 ± 8% (P = .0498) and 63 ± 6% (P = .029) at 10, 20 and 30 mg/kg/day, respectively |
| Focal segmental glomerulosclerosis | ||||
| FSGS mice (TRPC6 overexpression) [16] | Sparsentan | Losartan | Glomerular endothelial glycocalyx thickness | The increase in glomerular endothelial glycocalyx was fully restored by sparsentan compared with intermediate effects of losartan |
| Adriamycin-induced nephropathy rats [17] | Sparsentan | Vechicle | Development of GSS | In groups dosed with sparsentan 60 mg/kg or 180 mg/kg, development of GSS was significantly reduced (P < .0001; Spar60, 1.14 ± 0.21 or Spar180, 1.23 ± 0.21 vs vehicle, 2.70 ± 0.14) |
| Alport syndrome | ||||
| 129 Sv autosomal Alport mice [18] | Sitaxentan | None | Fibrosis score and % sclerotic glomeruli | Fibrosis scores and % sclerotic glomeruli were reduced to near wild-type levels in mice treated with sitaxentan compared with untreated Alport mice |
| Col4a3 KO mice [19] | Sparsentan | Losartan/vehicle | Development of glomerulosclerosis, fibrosis and proteinuria, and decline in GFR | Sparsentan significantly delayed onset of glomerulosclerosis, interstitial fibrosis and proteinuria compared with losartan, and significantly attenuated GFR decline compared with vehicle |
| Animal model | ERA | Comparator | Endpoint | Result |
|---|---|---|---|---|
| Diabetic kidney disease | ||||
| Streptozotocin-induced diabetic rats [12] | Darusentan | No treatment | Daily urinary protein excretion | Treatment with darusentan reduced the daily protein excretion by >50% 36 weeks after diabetes induction, compared with non-treated rats |
| Streptozotocin-induced diabetic rats [13] | Atrasentan | Vehicle | Daily urinary protein excretion | Treatment with atrasentan for 6 weeks reduced the daily protein excretion by 40%, compared with rats treated with vehicle |
| IgA nephropathy | ||||
| gddY mice [14] | Sparsentan | Losartan | Development of renal injury | Sparsentan compared with losartan attenuated UACR more rapidly and to a greater extent, which correlated with increased podocyte and glycocalyx protection |
| gddY mice [15] | Atrasentan | None | Change in UACR after 4 days of treatment | Atrasentan reduced UACR from baseline by 28 ± 44% (P = ns), 62 ± 8% (P = .0498) and 63 ± 6% (P = .029) at 10, 20 and 30 mg/kg/day, respectively |
| Focal segmental glomerulosclerosis | ||||
| FSGS mice (TRPC6 overexpression) [16] | Sparsentan | Losartan | Glomerular endothelial glycocalyx thickness | The increase in glomerular endothelial glycocalyx was fully restored by sparsentan compared with intermediate effects of losartan |
| Adriamycin-induced nephropathy rats [17] | Sparsentan | Vechicle | Development of GSS | In groups dosed with sparsentan 60 mg/kg or 180 mg/kg, development of GSS was significantly reduced (P < .0001; Spar60, 1.14 ± 0.21 or Spar180, 1.23 ± 0.21 vs vehicle, 2.70 ± 0.14) |
| Alport syndrome | ||||
| 129 Sv autosomal Alport mice [18] | Sitaxentan | None | Fibrosis score and % sclerotic glomeruli | Fibrosis scores and % sclerotic glomeruli were reduced to near wild-type levels in mice treated with sitaxentan compared with untreated Alport mice |
| Col4a3 KO mice [19] | Sparsentan | Losartan/vehicle | Development of glomerulosclerosis, fibrosis and proteinuria, and decline in GFR | Sparsentan significantly delayed onset of glomerulosclerosis, interstitial fibrosis and proteinuria compared with losartan, and significantly attenuated GFR decline compared with vehicle |
GSS, glomerulosclerosis score; KO, knock-out.
Experimental studies with selective ETA and ETB receptor antagonists.
| Animal model | ERA | Comparator | Endpoint | Result |
|---|---|---|---|---|
| Diabetic kidney disease | ||||
| Streptozotocin-induced diabetic rats [12] | Darusentan | No treatment | Daily urinary protein excretion | Treatment with darusentan reduced the daily protein excretion by >50% 36 weeks after diabetes induction, compared with non-treated rats |
| Streptozotocin-induced diabetic rats [13] | Atrasentan | Vehicle | Daily urinary protein excretion | Treatment with atrasentan for 6 weeks reduced the daily protein excretion by 40%, compared with rats treated with vehicle |
| IgA nephropathy | ||||
| gddY mice [14] | Sparsentan | Losartan | Development of renal injury | Sparsentan compared with losartan attenuated UACR more rapidly and to a greater extent, which correlated with increased podocyte and glycocalyx protection |
| gddY mice [15] | Atrasentan | None | Change in UACR after 4 days of treatment | Atrasentan reduced UACR from baseline by 28 ± 44% (P = ns), 62 ± 8% (P = .0498) and 63 ± 6% (P = .029) at 10, 20 and 30 mg/kg/day, respectively |
| Focal segmental glomerulosclerosis | ||||
| FSGS mice (TRPC6 overexpression) [16] | Sparsentan | Losartan | Glomerular endothelial glycocalyx thickness | The increase in glomerular endothelial glycocalyx was fully restored by sparsentan compared with intermediate effects of losartan |
| Adriamycin-induced nephropathy rats [17] | Sparsentan | Vechicle | Development of GSS | In groups dosed with sparsentan 60 mg/kg or 180 mg/kg, development of GSS was significantly reduced (P < .0001; Spar60, 1.14 ± 0.21 or Spar180, 1.23 ± 0.21 vs vehicle, 2.70 ± 0.14) |
| Alport syndrome | ||||
| 129 Sv autosomal Alport mice [18] | Sitaxentan | None | Fibrosis score and % sclerotic glomeruli | Fibrosis scores and % sclerotic glomeruli were reduced to near wild-type levels in mice treated with sitaxentan compared with untreated Alport mice |
| Col4a3 KO mice [19] | Sparsentan | Losartan/vehicle | Development of glomerulosclerosis, fibrosis and proteinuria, and decline in GFR | Sparsentan significantly delayed onset of glomerulosclerosis, interstitial fibrosis and proteinuria compared with losartan, and significantly attenuated GFR decline compared with vehicle |
| Animal model | ERA | Comparator | Endpoint | Result |
|---|---|---|---|---|
| Diabetic kidney disease | ||||
| Streptozotocin-induced diabetic rats [12] | Darusentan | No treatment | Daily urinary protein excretion | Treatment with darusentan reduced the daily protein excretion by >50% 36 weeks after diabetes induction, compared with non-treated rats |
| Streptozotocin-induced diabetic rats [13] | Atrasentan | Vehicle | Daily urinary protein excretion | Treatment with atrasentan for 6 weeks reduced the daily protein excretion by 40%, compared with rats treated with vehicle |
| IgA nephropathy | ||||
| gddY mice [14] | Sparsentan | Losartan | Development of renal injury | Sparsentan compared with losartan attenuated UACR more rapidly and to a greater extent, which correlated with increased podocyte and glycocalyx protection |
| gddY mice [15] | Atrasentan | None | Change in UACR after 4 days of treatment | Atrasentan reduced UACR from baseline by 28 ± 44% (P = ns), 62 ± 8% (P = .0498) and 63 ± 6% (P = .029) at 10, 20 and 30 mg/kg/day, respectively |
| Focal segmental glomerulosclerosis | ||||
| FSGS mice (TRPC6 overexpression) [16] | Sparsentan | Losartan | Glomerular endothelial glycocalyx thickness | The increase in glomerular endothelial glycocalyx was fully restored by sparsentan compared with intermediate effects of losartan |
| Adriamycin-induced nephropathy rats [17] | Sparsentan | Vechicle | Development of GSS | In groups dosed with sparsentan 60 mg/kg or 180 mg/kg, development of GSS was significantly reduced (P < .0001; Spar60, 1.14 ± 0.21 or Spar180, 1.23 ± 0.21 vs vehicle, 2.70 ± 0.14) |
| Alport syndrome | ||||
| 129 Sv autosomal Alport mice [18] | Sitaxentan | None | Fibrosis score and % sclerotic glomeruli | Fibrosis scores and % sclerotic glomeruli were reduced to near wild-type levels in mice treated with sitaxentan compared with untreated Alport mice |
| Col4a3 KO mice [19] | Sparsentan | Losartan/vehicle | Development of glomerulosclerosis, fibrosis and proteinuria, and decline in GFR | Sparsentan significantly delayed onset of glomerulosclerosis, interstitial fibrosis and proteinuria compared with losartan, and significantly attenuated GFR decline compared with vehicle |
GSS, glomerulosclerosis score; KO, knock-out.
Endothelin and fluid retention
The promising experimental data encouraged clinical studies to assess the efficacy and safety of ERAs in patients with CKD. Although ERAs consistently show marked reductions in proteinuria in multiple aetiologies of kidney disease [37–39], their clinical use for the treatment of CKD is still limited. This is mainly due to the fact that ERAs cause fluid retention, a side effect that occurs more frequently in patients with reduced kidney function in whom fluid and sodium homeostasis is impaired.
The mechanisms of fluid retention have been an area of intensive research. Since ETB receptor activation causes sodium excretion and promotes vasodilatation, initial studies mainly focused on ETB receptors and demonstrated that ETB knock-out animals or specific ETB antagonism cause fluid retention which, clinically, can lead to oedema and heart failure. It is nowadays clear that ETB receptors in the thick ascending limb and collecting duct inhibit sodium reabsorption and their inhibition enhance sodium excretion [40]. Subsequent studies showed that non-selective ETA antagonist may also promote fluid retention when administered at doses sufficient to inhibit ETB receptors as well [41]. Based on these initial studies one can thus conclude that for safe and appropriate use of ERAs in clinical practice it is critical to use highly selective ETA antagonists at concentrations low enough to avoid ETB activation. However, the fluid retention cannot only be attributed to ETB inhibition. More recent studies also suggest a role for nephron ETA receptors. Nephron ETA knock-out animals showed a mild and variable degree of fluid retention suggesting that ETA activation in the nephron exert natriuretic effects [42]. That natriuretic and diuretic actions of ET-1 are at least partly mediated through ETA receptors is also evident from experimental studies demonstrating that ET-1 administration to ETB knock-out mice causes natriuresis and this effect is inhibited by ETA antagonism. Moreover, combined ETA and ETB knock-out mice are significant more volume overloaded and hypertensive compared with ETB knock-out mice. Thus, diuretic effects of ET-1 are to a large extent mediated by ETB receptors, but the role of ETA receptors should not be excluded.
It is of interest that ETB receptor activation is involved in the renal actions of other diuretic or vasoactive hormones. ETB activation inhibits the anti-diuretic actions of vasopressin in the collecting duct [43, 44]. In addition, the effects of relaxin, a naturally occurring hormone with vasoactive and haemodynamic effects, are mediated through ETB receptors and nitric oxide synthase 1. Specifically, matrix metalloproteinases convert big endothelin to ET-1 which is required for relaxin-induced vasodilation through ETB receptor activation [45]. These data are relevant in the context of new long-acting relaxin analogues that are currently being evaluated for their effects on kidney function [46].
CLINICAL STUDIES
Clinical trials in participants with type 2 diabetes and CKD
Multiple small randomized controlled clinical studies with short follow-up have been performed which demonstrated that different ERA reduce albuminuria in patients with CKD with and without diabetes. To date, four phase 3 randomized controlled clinical trials designed to characterize the efficacy and safety of ERAs have been completed and two others are ongoing (Table 2, Fig. 2). The ASCEND (Avosentan on doubling of Serum Creatinine, ENd-stage renal disease and death in Diabetic nephropathy) trial was the first phase 3 randomized controlled clinical trial with the ERA avosentan in patients with type 2 diabetes and CKD. Participants had a mean estimated GFR (eGFR) of 33 mL/min/1.73 m2 and median urinary albumin:creatinine ratio (UACR) of 1.4 g/g creatinine [47]. The trial tested high doses (25 and 50 mg/day) of a rather unselective ERA and was halted early due to excess heart failure and mortality in the avosentan treatment groups. The high avosentan doses used in the ASCEND trial were also associated with fluid retention in a phase 2 dose-finding clinical trial in patients with relatively preserved kidney function, eGFR ∼80 mL/min/1.73 m2. Since the presence of type 2 diabetes and a low eGFR are independent risk markers of heart failure, the high rates of heart failure in the ASCEND trial are perhaps no surprise [39, 47, 56, 57]. The subsequent phase 3 SONAR (Study Of diabetic Nephropathy with AtRasentan) trial incorporated lessons from the ASCEND trial in the design of the trial by selecting a very low dose (0.75 mg/day) of a highly selective ETA antagonist atrasentan. In addition, patients at risk of heart failure were excluded, and importantly, the trial design incorporated an active 6-week single blind run-in period during which all patients were treated with atrasentan 0.75 mg/day [58]. Only patients who tolerated atrasentan and did not develop signs of fluid retention were eligible to proceed to the randomization visit during which they were randomly allocated to continue atrasentan or to transition to placebo treatment. In addition, the main stratum of the trial to test the efficacy of atrasentan in reducing the risk of the primary kidney outcome included patients who also responded to atrasentan defined as a reduction in albuminuria of more than 30% during the active run-in period. This was done to select a population with maximal benefit and minimal side effects. The trial was stopped prematurely due to a decision of the sponsor to discontinue the development program. At the termination of the trial atrasentan compared with placebo significantly reduced the primary kidney outcome by 35% in the main efficacy stratum, an effect size similar to sodium-glucose cotransporter 2 inhibitors (SGLT2i) [48, 59]. In addition, the heart failure rates were markedly lower compared with the ASCEND trial, illustrating that the mitigation strategy included in the design of the trial was, at least partly, successful (Fig. 3). However, despite the precautionary measures, adverse events related to fluid retention occurred more frequently in the atrasentan group.
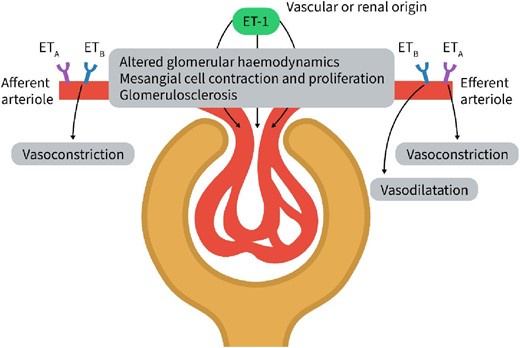
ETA and ETB receptors are present in the afferent and efferent arteriole. ETA and ETB antagonism is required to diminish the vasoconstrictive effects of ET-1 in the afferent arteriole indicating that both ETA and ETB activation modulate the vasoconstrictive effects of ET-1. In the efferent arteriole, ETA inhibition causes vasodilatation. Inhibition of the ETB receptor counteracts this effects and results in vasoconstriction. This indicates that in the efferent arteriole binding of ET-1 to ETA results in vasoconstriction and binding to ETB in vasodilatation. Renal blood flow is thus regulated by ET-1 through its effects on ETA and ETB in the afferent and efferent arteriole. The figure was adapted from Dhaun et al., Nature Reviews Cardiology 2019:16:491–502.
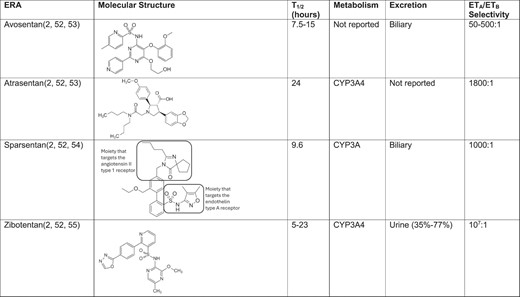
Molecular structure and pharmacokinetic/dynamic parameters of ERAs being investigated in phase 3 clinical trials of CKD progression.
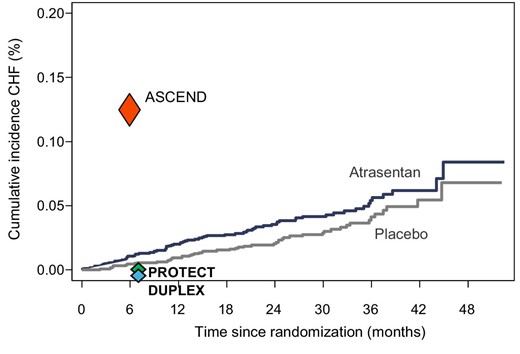
Incidence of heart failure in clinical trials with ERAs. Kaplan–Meier curve of heart failure hospitalizations in the SONAR trial is shown. The incidence of heart failure in the SONAR trial at 6 months (the median follow-up time in the ASCEND trial) was markedly lower compared with the ASCEND trial (red triangle), suggesting that the careful patient selection and the use of low-dose atrasentan in the SONAR trial were successful. In the PROTECT trial in patients with IgA Nephropathy (PROTECT) only one heart failure event occurred in each treatment arm (green triangle). In the DUPLEX trial in patients with FSGS, heart failure did not occur (blue triangle).
Completed and ongoing ERA phase 3 trials.
| Trial acronym | Patient population | No. of patients | Drug and dose | Follow-up | Endpoint | Result |
|---|---|---|---|---|---|---|
| Diabetic kidney disease | ||||||
| ASCEND [47] | eGFR >15 mL/min; UACR >309 mg/g | 1392 | Avosentan 25 and 50 mg | 26 weeks | Time to doubling of serum creatinine, ESRD or death | Trial was discontinued early due to excessive heart failure and mortality in the avosentan treatment group |
| SONAR [48] | eGFR 25–75 mL/min; UACR 300–5000 mg/g | 2648 | Atrasentan 0.75 mg | 2.2 years | Doubling of serum creatinine (sustained for ≥30 days) or ESRD | Trial was discontinued early due to a sponsor decision. Atrasentan reduced the primary kidney outcome by 35% (95% CI 12 to 51) |
| IgA nephropathy | ||||||
| PROTECT [49] | Biopsy-proven IgAN eGFR >30 mL/min Proteinuria >1 g/day | 404 | Sparsentan 400 mg | 110 weeks | Proteinuria change at 36 weeks and rate of eGFR slope | Sparsentan compared with irbesartan reduced proteinuria by 41% (95% CI 31 to 49); the difference in eGFR slope over 2 years was 1.0 mL/min (95% CI –0.03 to 1.94; P = .058) |
| ALIGN [50] | Biopsy-proven IgAN; eGFR >30 mL/min; proteinuria >1 g/day | 404 | Atrasentan 0.75 mg | 132 weeks | Proteinuria change at 36 weeks and eGFR change at Week 136 | Ongoing |
| FSGS | ||||||
| DUPLEX [51] | Biopsy-proven FSGS; eGFR >30 mL/min; UPCR >1.5 g/g | 371 | Sparsentan 800 mg | 108 weeks | FSGS partial remission of proteinuria (UPCR <1.5 g/g and >40% reduction in ratio from baseline) at 36 weeks and eGFR slope at the time of final analysis | Remission in proteinuria was achieved in 42% and 26% in the sparsentan and irbesartan groups, respectively (difference 16%; 95% CI 4.0 to 28.0). The between-group difference in total eGFR slope was 0.3 mL/min (95% –1.7 to 2.4; P = .75) |
| CKD | ||||||
| ZENITH-HP (NCT06087835) | CKD of different aetiologies; eGFR 20–90 mL/min; UACR >700 mg/g or UPCR >1000 mg/g | Target 1500 | Zibotentan 0.25/0.75 mg | 108 weeks | Change in eGFR from baseline to Week 24 | Ongoing |
| Trial acronym | Patient population | No. of patients | Drug and dose | Follow-up | Endpoint | Result |
|---|---|---|---|---|---|---|
| Diabetic kidney disease | ||||||
| ASCEND [47] | eGFR >15 mL/min; UACR >309 mg/g | 1392 | Avosentan 25 and 50 mg | 26 weeks | Time to doubling of serum creatinine, ESRD or death | Trial was discontinued early due to excessive heart failure and mortality in the avosentan treatment group |
| SONAR [48] | eGFR 25–75 mL/min; UACR 300–5000 mg/g | 2648 | Atrasentan 0.75 mg | 2.2 years | Doubling of serum creatinine (sustained for ≥30 days) or ESRD | Trial was discontinued early due to a sponsor decision. Atrasentan reduced the primary kidney outcome by 35% (95% CI 12 to 51) |
| IgA nephropathy | ||||||
| PROTECT [49] | Biopsy-proven IgAN eGFR >30 mL/min Proteinuria >1 g/day | 404 | Sparsentan 400 mg | 110 weeks | Proteinuria change at 36 weeks and rate of eGFR slope | Sparsentan compared with irbesartan reduced proteinuria by 41% (95% CI 31 to 49); the difference in eGFR slope over 2 years was 1.0 mL/min (95% CI –0.03 to 1.94; P = .058) |
| ALIGN [50] | Biopsy-proven IgAN; eGFR >30 mL/min; proteinuria >1 g/day | 404 | Atrasentan 0.75 mg | 132 weeks | Proteinuria change at 36 weeks and eGFR change at Week 136 | Ongoing |
| FSGS | ||||||
| DUPLEX [51] | Biopsy-proven FSGS; eGFR >30 mL/min; UPCR >1.5 g/g | 371 | Sparsentan 800 mg | 108 weeks | FSGS partial remission of proteinuria (UPCR <1.5 g/g and >40% reduction in ratio from baseline) at 36 weeks and eGFR slope at the time of final analysis | Remission in proteinuria was achieved in 42% and 26% in the sparsentan and irbesartan groups, respectively (difference 16%; 95% CI 4.0 to 28.0). The between-group difference in total eGFR slope was 0.3 mL/min (95% –1.7 to 2.4; P = .75) |
| CKD | ||||||
| ZENITH-HP (NCT06087835) | CKD of different aetiologies; eGFR 20–90 mL/min; UACR >700 mg/g or UPCR >1000 mg/g | Target 1500 | Zibotentan 0.25/0.75 mg | 108 weeks | Change in eGFR from baseline to Week 24 | Ongoing |
ESRD, end-stage renal disease.
Completed and ongoing ERA phase 3 trials.
| Trial acronym | Patient population | No. of patients | Drug and dose | Follow-up | Endpoint | Result |
|---|---|---|---|---|---|---|
| Diabetic kidney disease | ||||||
| ASCEND [47] | eGFR >15 mL/min; UACR >309 mg/g | 1392 | Avosentan 25 and 50 mg | 26 weeks | Time to doubling of serum creatinine, ESRD or death | Trial was discontinued early due to excessive heart failure and mortality in the avosentan treatment group |
| SONAR [48] | eGFR 25–75 mL/min; UACR 300–5000 mg/g | 2648 | Atrasentan 0.75 mg | 2.2 years | Doubling of serum creatinine (sustained for ≥30 days) or ESRD | Trial was discontinued early due to a sponsor decision. Atrasentan reduced the primary kidney outcome by 35% (95% CI 12 to 51) |
| IgA nephropathy | ||||||
| PROTECT [49] | Biopsy-proven IgAN eGFR >30 mL/min Proteinuria >1 g/day | 404 | Sparsentan 400 mg | 110 weeks | Proteinuria change at 36 weeks and rate of eGFR slope | Sparsentan compared with irbesartan reduced proteinuria by 41% (95% CI 31 to 49); the difference in eGFR slope over 2 years was 1.0 mL/min (95% CI –0.03 to 1.94; P = .058) |
| ALIGN [50] | Biopsy-proven IgAN; eGFR >30 mL/min; proteinuria >1 g/day | 404 | Atrasentan 0.75 mg | 132 weeks | Proteinuria change at 36 weeks and eGFR change at Week 136 | Ongoing |
| FSGS | ||||||
| DUPLEX [51] | Biopsy-proven FSGS; eGFR >30 mL/min; UPCR >1.5 g/g | 371 | Sparsentan 800 mg | 108 weeks | FSGS partial remission of proteinuria (UPCR <1.5 g/g and >40% reduction in ratio from baseline) at 36 weeks and eGFR slope at the time of final analysis | Remission in proteinuria was achieved in 42% and 26% in the sparsentan and irbesartan groups, respectively (difference 16%; 95% CI 4.0 to 28.0). The between-group difference in total eGFR slope was 0.3 mL/min (95% –1.7 to 2.4; P = .75) |
| CKD | ||||||
| ZENITH-HP (NCT06087835) | CKD of different aetiologies; eGFR 20–90 mL/min; UACR >700 mg/g or UPCR >1000 mg/g | Target 1500 | Zibotentan 0.25/0.75 mg | 108 weeks | Change in eGFR from baseline to Week 24 | Ongoing |
| Trial acronym | Patient population | No. of patients | Drug and dose | Follow-up | Endpoint | Result |
|---|---|---|---|---|---|---|
| Diabetic kidney disease | ||||||
| ASCEND [47] | eGFR >15 mL/min; UACR >309 mg/g | 1392 | Avosentan 25 and 50 mg | 26 weeks | Time to doubling of serum creatinine, ESRD or death | Trial was discontinued early due to excessive heart failure and mortality in the avosentan treatment group |
| SONAR [48] | eGFR 25–75 mL/min; UACR 300–5000 mg/g | 2648 | Atrasentan 0.75 mg | 2.2 years | Doubling of serum creatinine (sustained for ≥30 days) or ESRD | Trial was discontinued early due to a sponsor decision. Atrasentan reduced the primary kidney outcome by 35% (95% CI 12 to 51) |
| IgA nephropathy | ||||||
| PROTECT [49] | Biopsy-proven IgAN eGFR >30 mL/min Proteinuria >1 g/day | 404 | Sparsentan 400 mg | 110 weeks | Proteinuria change at 36 weeks and rate of eGFR slope | Sparsentan compared with irbesartan reduced proteinuria by 41% (95% CI 31 to 49); the difference in eGFR slope over 2 years was 1.0 mL/min (95% CI –0.03 to 1.94; P = .058) |
| ALIGN [50] | Biopsy-proven IgAN; eGFR >30 mL/min; proteinuria >1 g/day | 404 | Atrasentan 0.75 mg | 132 weeks | Proteinuria change at 36 weeks and eGFR change at Week 136 | Ongoing |
| FSGS | ||||||
| DUPLEX [51] | Biopsy-proven FSGS; eGFR >30 mL/min; UPCR >1.5 g/g | 371 | Sparsentan 800 mg | 108 weeks | FSGS partial remission of proteinuria (UPCR <1.5 g/g and >40% reduction in ratio from baseline) at 36 weeks and eGFR slope at the time of final analysis | Remission in proteinuria was achieved in 42% and 26% in the sparsentan and irbesartan groups, respectively (difference 16%; 95% CI 4.0 to 28.0). The between-group difference in total eGFR slope was 0.3 mL/min (95% –1.7 to 2.4; P = .75) |
| CKD | ||||||
| ZENITH-HP (NCT06087835) | CKD of different aetiologies; eGFR 20–90 mL/min; UACR >700 mg/g or UPCR >1000 mg/g | Target 1500 | Zibotentan 0.25/0.75 mg | 108 weeks | Change in eGFR from baseline to Week 24 | Ongoing |
ESRD, end-stage renal disease.
Clinical trials in participants with CKD without diabetes
The efficacy and safety of ERAs are also assessed in phase 3 trials in patients with CKD but without diabetes. The PROTECT (A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy) trial assessed the effect of the dual ET-1 angiotensin receptor (AT1) antagonist (DEARA) sparsentan [49, 60]. Sparsentan is a single molecule that can inhibit both the AT1 and ETA receptor [61]. An experimental study demonstrated enhanced blood pressure lowering effects of sparsentan compared with equimolar administration of irbesartan, supporting the idea that the inhibitory ETA effects of sparsentan contribute to its clinical efficacy [62]. Two phase 3 trials with sparsentan have been completed. The first trial was a 2-year clinical trial of sparsentan compared with irbesartan in 404 participants with biopsy-proven IgA nephropathy who had proteinuria >1 g/day despite optimal guideline recommended treatment. A prespecified interim analysis after the first 263 participants were followed for 9 months demonstrated that sparsentan compared with placebo reduced proteinuria by 41%. The final analysis after 110 weeks follow-up showed that this effect was sustained throughout the study. At Week 110, sparsentan compared with irbesartan reduced eGFR decline. In the sparsentan group the annual rate of eGFR decline was –2.9 mL/min/1.73 m2 compared with –3.9 mL/min/1.73 m2 in the irbesartan group. Based on these findings sparsentan is now approved to slow CKD progression for adult patients with IgA nephropathy.
Another trial assessed the effect of sparsentan in patients aged between 8 and 75 years with biopsy- proven FSGS or documented pathogenic variant in a podocyte protein associated with FSGS and a urinary protein:creatinine ratio (UPCR) of >1.5 g/g. At the pre-specified interim analysis after 36 weeks, 42.0% of the participants achieved the surrogate endpoint of partial remission (defined as a UPCR of ≤1.5 g/g and a >40% reduction in the ratio from baseline) in the sparsentan group compared with 26.0% in the irbesartan group [between group difference 16%; 95% confidence interval (CI) 4.0% to 28%]. Sparsentan compared with irbesartan also reduced UPCR as early as 6 weeks of follow-up and this effect was sustained through the end of the study after 108 weeks of treatment. Although the proteinuria endpoint was met, the difference in the confirmatory eGFR decline endpoint did not reach statistical significance. The between-group difference in favour of sparsentan in the total slope (calculated from randomization to Week 108) and chronic eGFR slope (calculated from Week 6 to Week 108) was 0.3 (95% CI –1.7 to 2.4; P = .75) and 0.9 (95% CI –1.3 to 3.0; P = .42). Numerically fewer patients in the sparsentan compared with irbesartan group reached kidney failure (6.5% vs 11.2%). The investigators of the trial speculate that the heterogeneity of the clinical trial population, the initial larger decline in eGFR with sparsentan compared with sparsentan, and the higher than anticipated proteinuria response with irbesartan may explain why the rates of eGFR decline did not statistically significantly differ between the two treatment groups [51].
After the completion of the SONAR trial, another phase 3 trial with atrasentan was initiated. In this trial [ALIGN (Atrasentan in Patients With IgA Nephropathy); ClinicalTrials.gov identifier: NCT04573478] 404 participants with biopsy-proven IgA nephropathy and urinary protein excretion >1 g/day were enrolled and assigned to atrasentan or placebo. The pre-specified primary proteinuria endpoint was change in proteinuria after 36 weeks of follow-up in the first 270 participants showed that atrasentan compared with placebo reduced UPCR [50]. The reduction in UPCR was present at the first study visit after 6 weeks follow-up and was sustained through Week 36. The trial is still ongoing to assess the effect of atrasentan on change in eGFR from baseline to Week 136.
In clinical trials in patients with IgA nephropathy or FSGS, both sparsentan and atrasentan were well tolerated. Importantly, fluid retention or heart failure did not emerge as a safety concern. In these trials very few patients were hospitalized due to heart failure, as could be expected because the incidence of heart failure is markedly lower in glomerulonephritis compared with type 2 diabetes (Fig. 3). Anaemia, which most likely reflect dilutional anaemia, occurred more frequently in the ERA compared with the control treatment group.
Combination treatment of ERA and SGLT2i
SGLT2i reduce the risk of kidney failure and the rate of kidney function decline in adults with CKD with and without type 2 diabetes. Clinical practice guidelines from endocrinology, nephrology and cardiology associations now recommend SGLT2i as a vital component of optimal kidney and heart failure protective treatment. SGLT2i exert diuretic effects and reduce extracellular volume. This effect is of particular interest in the context of ERA treatment since the diuretic effects of SGLT2i could theoretically mitigate ERA induced fluid retention. Indeed, a small nested case–control study from the SONAR trial demonstrated that combined initiation of SGLT2i and atrasentan reduced body weight while it increased with atrasentan alone. Moreover, the combination of the two drugs reduced albuminuria more than atrasentan suggesting that combination may augment kidney protection while abrogating fluid retention and heart failure [63]. This small study was followed by a phase 2b trial to test the hypothesis that combined dapagliflozin and low-dose zibotentan, a highly selective ETA antagonist, reduces albuminuria compared with dapagliflozin alone with an acceptable safety profile. The results showed that in participants with CKD combined treatment with a very low dose of zibotentan 0.25 mg/day and dapagliflozin 10 mg/day vs dapagliflozin 10 m/day reduced albuminuria by 27.0% after 12-week treatment and did not increase fluid retention (proportion of patients with fluid retention event 9% vs 8%) [64]. Other studies with other SGLT2i/ERA combinations, such as the ASSIST (Randomized, Double-blind, Placebo-controlled, Crossover Study of Atrasentan in Subjects With IgA Nephropathy) (ClinicalTrials.gov identifier: NCT05834738) and AFFINITY (Atrasentan in Patients With Proteinuric Glomerular Diseases) (ClinicalTrials.gov identifier: NCT04573920) basket trial, are ongoing to test this combination in adults with IgA nephropathy or type 2 diabetes and CKD.
Although concomitant treatment of ERAs with thiazide or loop diuretics have also been shown to reduce body weight, SGLT2i may have specific effects on intravascular and extravascular volume that are different from these conventional diuretics. ERAs may also increase vascular leak by decreasing venous constriction, which in turn can increase extravascular volume [41]. Since SGLT2i preferentially reduce extravascular volume, they theoretically provide better fluid mitigation of ERA-mediated fluid retention, independent of serum sodium concentrations. The ZENITH-High (Study to Investigate Efficacy, Safety, and Tolerability of Zibotentan/Dapagliflozin Compared to Dapagliflozin in Participants With Chronic Kidney Disease and High Proteinuria) Proteinuria study is an ongoing phase 3 clinical trial designed to determine the long-term benefit and safety of a fixed-dose combination of the SGLT2i dapagliflozin and the ERA zibotentan. The results of this trial will provide more definitive evidence about the long-term efficacy and safety of this interesting combination.
The completed clinical trials in patients with CKD have focused primarily on patients with diabetes or IgA nephropathy. Whether ERAs have clinical benefits in other CKD aetiologies, such as patients with Alport syndrome or patients already receiving dialysis, remains to be investigated. Combining ERAs with SGLT2i to optimize kidney protection and minimize side effects seems to be a promising therapeutic approach. It is however unknown whether ERAs have to be initiated simultaneously with SGLT2i or whether the two drug classes can be initiated sequentially to minimize fluid retention. The ongoing ZODIAC (Zibotentan and Dapagliflozin in Patients With Type 2 Diabetes and Elevated Albuminuria) trial is designed to address this mechanistic question (ClinicalTrials.gov identifier: NCT05570305). Finally, other drugs including the non-steroidal mineralocorticoid receptor antagonist finerenone and the glucagon-like peptide receptor agonist semaglutide slow the progression of CKD and reduce the risk of kidney failure. With multiple new interventions available to slow CKD progression, future studies should investigate which drug or combination of drugs can be used best for which patient or patient subgroup according the concept of personalized medicine.
In conclusion, experimental and clinical studies have univocally demonstrated the kidney protective effects of selective ETA ERA. The narrow therapeutic window along with the risk of fluid retention and heart failure in patients with severe CKD has been the biggest challenge in the past and has limited clinical application. However, the development of low doses of highly selective ETA antagonists, such as atrasentan and zibotentan, in combination with concomitant use of SGLT2i, will aid in translating the use of these agents from clinical research to daily practical use. The first dual ETA and AT1 antagonist, sparsentan, has become available for use in patients with IgA nephropathy. Other ERAs may follow in the future.
FUNDING
This paper was published as part of a supplement financially supported by Bayer AG and the scientific content has not been influenced in any way by the sponsor.
DATA AVAILABILITY STATEMENT
The data underlying this article will be shared on reasonable request to the corresponding author.
CONFLICT OF INTEREST STATEMENT
E.M. and V.S.W. report no conflicts. H.J.L.H. has received a grant/contract from AstraZeneca, Boehringer Ingelheim, Janssen and Novo Nordisk, and has received consulting fees from AstraZeneca, Abbvie, Boehringer Ingelheim, CSL Behring, Bayer, Chinook, Dimerix, EliLilly, Gilead, Goldfinch, Merck, Novartis, NovoNordisk, Janssen and Travere Pharmaceuticals. He has received honoraria from AstraZeneca and Novo Nordisk, and a travel grant from Eli-Lilly.


{kind=link}
{kind=link}
{kind=link}
Comments