





PLAIN ENGLISH SUMMARY
The kidneys produce daily about 180 liters of urine but only about 2 liters are excreted. The proximal tubule plays an important role in reabsorbing the majority of filtered urine and many metabolites such as sugars, amino acids, salts or phosphate that are contained in this large volume. Reabsorption of these important metabolites is mediated by a diverse group of highly specialized transport proteins. Another group of transport proteins in the proximal tubule is responsible for the active secretion of metabolic waste products or toxins and drugs into urine. All these transporters have in common that they are directly linked to kidney metabolism and indirectly to whole-body metabolism and functions. In recent years, it has become evident that modulation of these transporters may influence the onset, progression and consequences of kidney disease. This review summarizes recent developments in this field and discusses some examples of drugs already in clinical use or in development. The examples include inhibitors of sugar transporters (SGLT2 inhibitors) that are successfully used in patients with kidney disease, diabetes or heart failure. Likewise, indirect inhibitors (acetazolamide) of an transporter absorbing sodium in exchange for protons (NHE3) are used mostly in patients with heart failure or for prevention of high altitude disease, while direct inhibitors show promise in preclinical studies to reduce damage in episodes of acute kidney disease or high blood pressure. Modulators of transporters mediating the excretion of urate have been used in patients with gout and are also discussed to prevent kidney disease. Novel drugs in development target transporters for phosphate, amino acids, or toxin and drug excretion and may be helpful for specific conditions associated with kidney disease. The advantages and challenges associated with these (novel) drugs targeting proximal tubule transport are discussed.
The proximal tubule is responsible for reabsorbing about 60% of filtered solutes and water and is critical for the secretion of metabolic waste products, drugs and toxins. A large number of highly specialized ion channels and transport proteins belonging to the SLC and ABC transporter families are involved. Their activity is directly or indirectly linked to ATP consumption and requires large quantities of energy and oxygen supply. Moreover, the activity of these transporters is often coupled to the movement of Na+ ions thus influencing also salt and water balance, as well as transport and regulatory processes in downstream segments. Because of their relevance for systemic ion balance, for renal metabolism or for affecting regulatory processes, proximal tubule transporters are attractive targets for existing drug and for novel strategies to reduce kidney disease progression or to alleviate the consequences of decreased kidney function. In this review, the relevance of some major proximal tubule transport systems as drug targets in individuals with chronic kidney disease (CKD) is discussed. Inhibitors of the sodium-glucose cotransporter 2, SGLT2, are now part of standard therapy in patients with CKD and/or heart failure. Also, indirect inhibition of Na+/H+-exchangers by carbonic anhydrase inhibitors and uricosuric drugs have been used for decades. Inhibition of phosphate and amino acid transporters have recently been proposed as novel principles to remove excess phosphate or to protect the proximal tubule metabolically, respectively. In addition, organic cation and anion transporters involved in drug and toxin excretion may serve as targets of new drugs. The advantages and challenges associated with (novel) drugs targeting proximal tubule transport are discussed.
INTRODUCTION
The proximal tubule is the nephron segment responsible for the reabsorption of approximately 60% of the filtered amount of water and salt. Moreover, it reabsorbs nearly all filtered glucose and amino acids, and a major part of filtered phosphate, bicarbonate and low molecular weight proteins. Reabsorption of these important solutes is achieved by the concerted action of various plasma membrane transport proteins located in the luminal and basolateral membranes of the cells lining the proximal tubule.
Morphologically, the proximal tubule is optimized to maximize transport. Both, luminal and basolateral membranes have a highly enlarged surface to interact with primary urine or interstitial fluids/blood [1]. The luminal membrane is formed by microvilli, while the basolateral membrane is largely invaginated. Most transport processes are driven by the activity of the basolaterally located Na+/K+-ATPase, a pump that hydrolyses ATP to pump 3 Na+ to the basolateral interstitial space in exchange for 2 K+ ions that are pumped into cells (see Fig. 1). Na+/K+-ATPase activity creates a chemical gradient for sodium (which is around 140 mmol/L in primary urine and about 10 mmol/L intracellularly) and an electrical gradient (with a transmembrane potential of around –60 mV) that together favour the inward flux of Na+ and cotransport of other substrates even against their chemical gradient (e.g. for amino acids, glucose or phosphate). This transcellular transport of substrates then also energizes paracellular movement of further ions including also calcium, bicarbonate, sodium and chloride. The enormous transport activity of the proximal tubule thus depends mostly on Na+/K+-ATPase activity and the provision of sufficient amounts of ATP. Proximal tubule cells are very rich in mitochondria that mostly use aerobic fatty acid oxidation to generate ATP, while there is only very little glycolytic activity. Mitochondrial activity depends on sufficient oxygen supply and this strong dependency explains also the high vulnerability of proximal tubule cells to hypoxic insults such as in acute kidney injury. On the other hand, conditions that reduce or increase the workload of the proximal tubule, e.g. because of changes in filtered solute load, affect overall energy metabolism and requirements of proximal tubule cells.
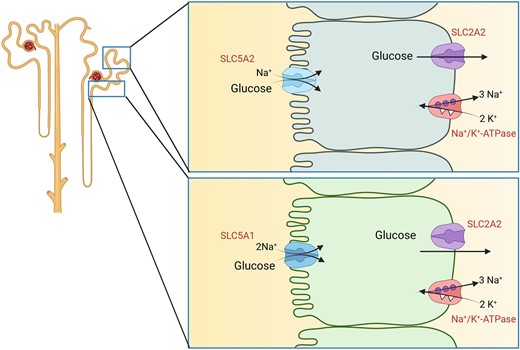
Renal glucose transport. Reabsorption of glucose by SGLT2 (SLC5A2) transporters in early proximal tubule (upper cell model) and SGLT1 (SLC5A1) in late proximal tubule (lower cell model). In both cell types, transport is energized by Na+/K+-ATPases. Glucose exits cells via GLUT2 (SLC2A2) transporters.
This review will focus on inhibitors of proximal tubule transport processes and their established or possible use in the context of kidney disease (Table 1). These inhibitors either increase clearance of metabolites or modulate proximal tubule metabolism, or both, and thereby exert beneficial effects. Open issues and challenges will be discussed.
SGLT2 INHIBITION
Inhibition of the renal sodium-glucose cotransporter 2 (SGLT2) has been a major success story and exemplifies the principle of targeting a proximal tubule transport protein to address a major metabolic disease with benefits for kidney and the cardiovascular system. Multiple recent reviews have covered underlying mechanisms and clinical use of SGLT2 inhibitors [2–6]. Nevertheless, this review will briefly highlight some of the major mechanisms of this class of drugs illustrating how studying basic physiologic principles of kidney function can lead to the development of new and powerful drugs.
About 180 g of glucose are daily filtered by the glomeruli but final urine is usually devoid of glucose except during postpranial periods or in individuals with (poorly controlled) diabetes. Glucose is reabsorbed by the proximal tubule by members of the SLC2 and SLC5 families of glucose transporters (Fig. 1). The threshold for the reabsorption of glucose is around 180–200 mg/dL (approx. 10 mmol/L) corresponding to the plasma glucose concentration above which glucose can be found in urine [7]. In the early proximal tubule, glucose is taken up from urine by the luminally located low affinity, high capacity Na+-driven glucose transporter SGLT2 (SLC5A2), while in the late proximal tubule the remaining glucose is reabsorbed by the high affinity, low capacity Na+-dependent glucose transporter SGLT1 (SLC5A1). It has been estimated that under normal conditions, SGLT2 reabsorbs up to 97% of filtered glucose, but if SGLT2 is absent or blocked, SGLT1 can reabsorb up to 80 g of glucose per day. In both segments, glucose reabsorption is completed by the release of glucose from proximal tubule cells into blood by the basolateral GLUT2 (SLC2A2) uniporter that allows for efflux of glucose along its concentration gradient. Since SGLT2 transporters have the capacity to accumulate very high intracellular glucose concentrations, this gradient is direct from intracellular to extracellular (blood side). The importance of these transporters is highlighted in individuals with loss of function variants in the SLC5A2 or SLC2A2 genes causing benign renal glucosuria and Fanconi–Bickel syndrome. Loss-of-function variants in SLC5A1 cause intestinal glucose-galactose malabsorption, a severe condition with massive diarrhoea and dehydration upon ingestion of glucose- or galactose-containing carbohydrates. The benign phenotype of patients with SLC5A2 variants has contributed to the development of SGLT2-specific inhibitors suggesting a potential good safety profile of such drugs, while inhibition of SGLT1 is expected to cause intestinal problems.
The class of gliflozines is derived from phlorizin, a natural product extracted from apple tree bark and leaves, had been known for a long time as a glucosuric substance and as inhibitor of Na+-dependent glucose transporters. Gliflozines affect kidney function and metabolism through various mechanisms. Reabsorption of glucose is linked to the reabsorption of sodium leading to lower downstream sodium concentrations which modulates the tubule-glomerular feedback to enhance glomerular filtration. In individuals with diabetes this contributes to glomerular hyperfiltration and a higher workload. SGLT2 inhibitors reduce hyperfiltration which may lead to an apparent fall in estimated glomerular filtration rate (eGFR) when drugs are initiated [2]. Inhibition of SGLT2 increases urinary salt, water and urate excretion. These effects are not only mediated by the direct inhibition of sodium/glucose cotransport but by the interaction of SGLT2 with other proximal tubular transporters including NHE3 and URAT1 (see below for their functions). The latter may be beneficial in patients with hyperuricemia and gout [5].
Lowering of blood glucose levels may contribute to reduced lipotoxicity in kidney and liver, which in turn may be linked to reduced systemic and local inflammation and fibrosis. Lower blood glucose levels may also stimulate energy production from beta-oxidation of lipids and synthesis of ketone bodies that serve as energy substrates for heart and brain. However, the glucose-lowering effect may not be necessary for kidney protection as indicated by the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD) trial [8]. Positive effects, however, were seen predominantly in those patients that had proteinuria [9].
Local redistribution of tubular workload, e.g. shifting Na-reabsorption to nephron segments downstream of the proximal tubule, may contribute to restoring the oxygen-sensitivity of erythropoietin producing cells and recovery of erythropoietin secretion [3].
Several of the beneficial effects of SGLT2 inhibitors are not easily explained by direct consequences of SGLT2 inhibition in kidney. Particularly the cardioprotective effects of SGLT2 inhibitors remain to be better understood even though lowering of circulating blood volume, increased availability of ketone bodies and less hypergylcemia as well as reduction of systemic inflammation can also positively affect cardiovascular functions [4, 6, 10]. However, the positive effects of SGLT2 inhibitors were also seen in normoglycemic patients. Several lines of explanation have been proposed and supported by data. There may be direct off-target effects of SGLT2 inhibitors on the cardiac Na+/H+-exchanger NHE1 that may improve mitochondrial function and energy balance of cardiomyocytes [10]. Alternatively, a recent study provided evidence in mouse models, including Sglt2 knockout (KO) mice, that SGLT2 inhibitors retained their beneficial effects on metabolic profiles, the kidney and cardiovascular system and related this to direct effects of these drugs on the gut microbiome and the production of uraemic toxins such as p-cresol [11]. SGLT2 inhibitors reduced p-cresol production, which in turn can damage cardiomyocytes.
Another important property of SGLT2 inhibitors may be their modulation of inflammatory processes. Examples are the endothelial cell protection and dampening of inflammation contributing to artherosclerosis, reducing oxidative stress, metabolic reprogramming with activation of sirtuins, reduced neuroinflammation and protection from cognitive decline, and less activation of the NLRP3 inflammasome. Ketone bodies such as β-hydroxybutyrate have been shown to block NLRP3 inflammaosme activation [12]. Again, these mechanisms may be independent of the anti-diabetic action of SGLT2 inhibitors as they have been observed also in non-diabetic humans and animal models [13–15].
DIRECT AND INDIRECT INHIBITION OF NHE3
The sodium-proton exchanger NHE3 (SLC9A3) is expressed in the kidney in the proximal tubule and thick ascending limb of the loop of Henle, where it mediates reabsorption of Na+ and bicarbonate (Fig. 2). NHE3 [and also NHE8 (SLC8A8) and H+-ATPases] secretes H+ into urine where it reacts with filtered HCO3– catalysed by membrane-bound carbonic anhydrase IV (CAIV) to form carbonic acid and eventually H2O and CO2. The latter diffuses into the cells of the proximal tubule where the reaction is reversed by cytosolic carbonic anhydrase II (CAII) to yield H+ and HCO3–. H+ is recycled again across the luminal membrane while HCO3– is further deprotonated to CO32– which is secreted into blood by the basolateral electrogenic Na+/CO32– cotransporter NBCe1A (SLC4A4-A) [16]. The activity of NHE3 is driven by proton and sodium gradients where basolateral Na+/K+-ATPases energize Na+ import and mediate basolateral Na+ efflux. NHE3 activity is regulated by a variety of endocrine and metabolic factors among them angiotensin II as a stimulator [17]. NHE3 is involved in mediating the rise of blood pressure by angiotensin II [18].
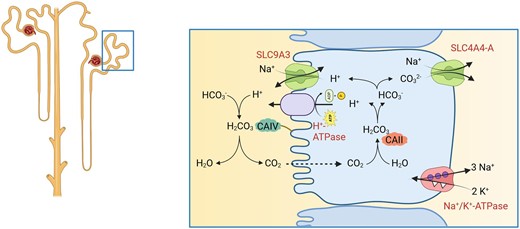
Function of NHE3. NHE3 (SLC9A3) together with other NHE isoforms mediates Na+/H+-exchange and thereby contributes to Na+ and HCO3– reabsorption.
Inhibition of NHE3 can be achieved directly and indirectly. Indirect inhibition can be induced by inhibitors of carbonic anhydrases such as acetazolamide that has been mostly used as a diuretic. However, acetazolamide and similar compounds are now used very little as stand-alone diuretics as they induce renal tubular acidosis and their efficacy as a diuretic is limited by down-stream compensatory upregulation of salt transport pathways. Also, acetazolamide may be associated with a higher risk of kidney injury in critically ill children [19]. However, in combination with other diuretics, mostly loop diuretics or thiazides, acetazolamide can be very effective in adult patients. In patients with decompensated heart failure, acetazolamide combined with a loop diuretic was more effective than loop diuretics alone and this effect persisted over a wide range of kidney function [20, 21].
Urinary alkalinization is a standard therapy to lower the risk for acute kidney injury in patients receiving nephrotoxic methotrexate at high doses. It has been suggested that acetazolamide may provide similar protection. However, a recent meta-analysis found no evidence supporting the use of acetazolamide in this context [22].
Direct inhibition of NHE3 as new strategy to prevent the angiotensin II–induced rise in blood pressure has been tested both by the use of proximal tubule specific Nhe3 KO mice and by compounds specifically blocking NHE3. The NHE3 inhibitor was given at a dose where no intestinal salt losses were measured in stool, suggesting that inhibition of renal salt reabsorption mediated by NHE3 may be critical [18].
NHE3 inhibition was also tested in the setting of ischaemia-induced acute kidney injury where it attenuated the fall in creatinine clearance, reduced excessive salt excretion, and also decreased tubular necrosis and dilatation, protein casts and cellular infiltrates [23]. As whole-organ pH was falling during ischaemia, NHE3 inhibition may reduce an overstimulation of this transporter and increased work load of proximal tubule cells.
A major limitation of direct and indirect inhibitors of NHE3 activity is the lack of organ specificity as NHE3 is expressed in various tissues beyond the proximal tubule. As evident from mouse models lacking globally NHE3 activity, salt wasting, electrolyte disturbances and acid–base dysregulation occur most likely due to inhibition of intestinal fluid, salt and bicarbonate absorption, and the lack of renal compensation.
TARGETING URATE TRANSPORT
Urate handling by the kidney follows a complex sequence of filtration, initial reabsorption of nearly all filtered urate in the early proximal tubule (S1 segment), followed by active secretion of urate (equal to about 50% of the filtered urate) in the S2 segment, and again reabsorption of most urate secreted. Final excretion of urate with urine is in the range of about 10% of filtered urate (Fig. 3, left panel).
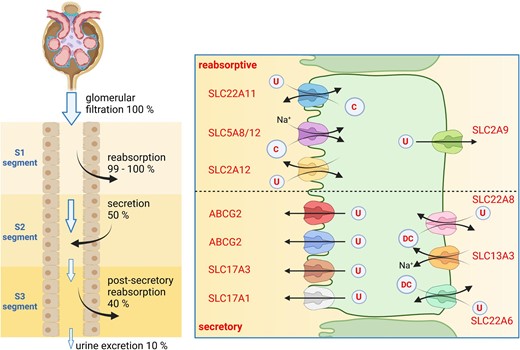
Renal urate handling. Left panel: tubular handling of urate with segmental reabsorption (S1 segment), secretion (S2 segment) and partial reabsorption (S3 segment). Percentages refer to the amount of filtered urate. Right panel: transport processes involved in reabsorption of urate (upper part of figure) or secretion (lower part). U, urate; C, carboxylates; DC, dicarboxylates.
Summary of proximal tubule transporters and expected effects of drugs modulating their activity. For details see text.
| Target | Expected effect | Evidence |
|---|---|---|
| SGLT2 | (i) Kidney protection, reduced progression of kidney disease | (i), (ii), (iv) animal studies and multiple RCTs |
| (ii) Reduced progression of cardiovascular disease | (iii) retrospective analysis of pats data, RCT ongoing | |
| (iii) Protection from kidney stones | ||
| (iv) Reduction in systemic inflammation | ||
| NHE3 | (i) Diuretic action in pats with decompensated heart failure | (i) RCTs |
| (ii) Reduce AKI | (ii) Animal studies | |
| (iii) Reduce salt-sensitive hypertension | (iii) Animal studies | |
| URAT1 | (i) Reduce serum urate levels in gout | (i) Animal studies and clinical trials |
| (ii) Prevent CKD progression | (ii) Animal studies, not supported by clinical trials | |
| NaPi-IIa | Reduce phosphate overload in CKD, reduce cardiovascular morbidity | Mouse and rat studies |
| OCT1/2, OATs | Reduce accumulation of nephrotoxic drugs/metabolites | Animal studies |
| B0AT1 | (i) Lowering of phenylalanine levels in phenylketonuria | Mouse studies, entering phase 1 and 2 clinical trials |
| (ii) Reduce severity in AKI/Balkan nephropathy |
| Target | Expected effect | Evidence |
|---|---|---|
| SGLT2 | (i) Kidney protection, reduced progression of kidney disease | (i), (ii), (iv) animal studies and multiple RCTs |
| (ii) Reduced progression of cardiovascular disease | (iii) retrospective analysis of pats data, RCT ongoing | |
| (iii) Protection from kidney stones | ||
| (iv) Reduction in systemic inflammation | ||
| NHE3 | (i) Diuretic action in pats with decompensated heart failure | (i) RCTs |
| (ii) Reduce AKI | (ii) Animal studies | |
| (iii) Reduce salt-sensitive hypertension | (iii) Animal studies | |
| URAT1 | (i) Reduce serum urate levels in gout | (i) Animal studies and clinical trials |
| (ii) Prevent CKD progression | (ii) Animal studies, not supported by clinical trials | |
| NaPi-IIa | Reduce phosphate overload in CKD, reduce cardiovascular morbidity | Mouse and rat studies |
| OCT1/2, OATs | Reduce accumulation of nephrotoxic drugs/metabolites | Animal studies |
| B0AT1 | (i) Lowering of phenylalanine levels in phenylketonuria | Mouse studies, entering phase 1 and 2 clinical trials |
| (ii) Reduce severity in AKI/Balkan nephropathy |
RCT, randomized controlled trial; AKI, acute kidney injury.
Summary of proximal tubule transporters and expected effects of drugs modulating their activity. For details see text.
| Target | Expected effect | Evidence |
|---|---|---|
| SGLT2 | (i) Kidney protection, reduced progression of kidney disease | (i), (ii), (iv) animal studies and multiple RCTs |
| (ii) Reduced progression of cardiovascular disease | (iii) retrospective analysis of pats data, RCT ongoing | |
| (iii) Protection from kidney stones | ||
| (iv) Reduction in systemic inflammation | ||
| NHE3 | (i) Diuretic action in pats with decompensated heart failure | (i) RCTs |
| (ii) Reduce AKI | (ii) Animal studies | |
| (iii) Reduce salt-sensitive hypertension | (iii) Animal studies | |
| URAT1 | (i) Reduce serum urate levels in gout | (i) Animal studies and clinical trials |
| (ii) Prevent CKD progression | (ii) Animal studies, not supported by clinical trials | |
| NaPi-IIa | Reduce phosphate overload in CKD, reduce cardiovascular morbidity | Mouse and rat studies |
| OCT1/2, OATs | Reduce accumulation of nephrotoxic drugs/metabolites | Animal studies |
| B0AT1 | (i) Lowering of phenylalanine levels in phenylketonuria | Mouse studies, entering phase 1 and 2 clinical trials |
| (ii) Reduce severity in AKI/Balkan nephropathy |
| Target | Expected effect | Evidence |
|---|---|---|
| SGLT2 | (i) Kidney protection, reduced progression of kidney disease | (i), (ii), (iv) animal studies and multiple RCTs |
| (ii) Reduced progression of cardiovascular disease | (iii) retrospective analysis of pats data, RCT ongoing | |
| (iii) Protection from kidney stones | ||
| (iv) Reduction in systemic inflammation | ||
| NHE3 | (i) Diuretic action in pats with decompensated heart failure | (i) RCTs |
| (ii) Reduce AKI | (ii) Animal studies | |
| (iii) Reduce salt-sensitive hypertension | (iii) Animal studies | |
| URAT1 | (i) Reduce serum urate levels in gout | (i) Animal studies and clinical trials |
| (ii) Prevent CKD progression | (ii) Animal studies, not supported by clinical trials | |
| NaPi-IIa | Reduce phosphate overload in CKD, reduce cardiovascular morbidity | Mouse and rat studies |
| OCT1/2, OATs | Reduce accumulation of nephrotoxic drugs/metabolites | Animal studies |
| B0AT1 | (i) Lowering of phenylalanine levels in phenylketonuria | Mouse studies, entering phase 1 and 2 clinical trials |
| (ii) Reduce severity in AKI/Balkan nephropathy |
RCT, randomized controlled trial; AKI, acute kidney injury.
On a cellular level, a series of urate transporters has been identified that mediate either net secretion or net reabsorption. It should also be noted that these transporters mediate urate but not uric acid transport. Reabsorption of urate is mediated on the luminal side by at least two members of the SLC22 transporter family, SLC22A11 (OAT4) and SLC22A12 (URAT1) which both act as obligatory exchangers that secrete carboxylates such as lactate into urine in exchange for urate. Transport is energized by Na+-driven lactate/carboxylate transporters SLC5A8 (SMCT1) and SLC5A12 (SMCT2) importing lactate from urine, which in turn are driven by basolateral Na+/K+-ATPases. Urate exits cells via SLC2A9 (GLUT9). In contrast, urate excretion into urine is initiated by importing urate from blood via the SLC22A6 (OAT1) and SLC22A8 (OAT3) exchangers at the basolateral side that take up urate in exchange for intracellular dicarboxylates such as citrate, succinate or α-ketoglutarate. Dicarboxylates are reimported into cells by the Na+/dicarboxylate cotransporters SLC13A3 (NaDC3). Secretion into urine is completed by transporters located at the luminal membrane: SLC17A1 (NaPi-I or NPT1), SLC17A3 (NPT4), ABCG2 and ABCC4 (MRP4) (Fig. 3, right panel). The relevance of these transporters is partly supported by human genetics showing that loss-of-function variants in SLC22A12 (URAT1) cause hypouricemia and urate kidney stones while inactivating variants and single nucleotide polymorphisms in ABCG2 associate with higher serum uric acid levels and gout [24]. Dysregulation of urate transporters in metabolic disease such as diabetes causes an imbalance between reabsorptive and secretory mechanisms, and contributes to hyperuricemia and elevated risk for gout [25].
Treatment of gout and hyperuricemia includes also inhibition of renal urate reabsorption with uricosuric drugs such as benzbromarone or probenecid that competitively inhibit urate reabsorption. However, more recently, sale and use of benzbromarone has been stopped in various countries due to its liver toxicity [25]. Also, some renin–angiotensin–aldosterone system inhibitors such as losartan act as unspecific inhibitors of urate transporters and can increase urate excretion. A specific inhibitor of URAT1 (SLC22A12), lesinurad was developed but taken from market after an increasing incidence of acute kidney injury linked to this drug. Lower dose of lesinurad had also no clear clinical benefits for hyperuricema and gout [25]. The alternative URAT1 inhibitor dotinurad has been shown to reduce serum urate levels but is currently approved for the treatment of hyperuricemia in only some countries [25].
CKD is often associated with hyperuricemia and it has been assumed based on animal experiments that serum urate-lowering therapies may provide a benefit to patients with CKD. However, two larger placebo-controlled trials, the Preventing Early Renal Loss in Diabetes (PERL) and Slowing of Kidney Disease Progression from the Inhibition of Xanthine Oxidase (CKD-FIX) trials, showed no benefit of adding allopurinol to standard treatment [26]. The randomized placebo controlled Study of verinurAd and alloPurinol in Patients with cHronic kIdney disease and hyperuRicaEmia (SAPPHIRE) trial tested the combination of allopurinol and the URAT1 inhibitor verinurad to reduce albuminuria and preserve eGFR in hyperuricemic patients with eGFR ≥25 mL/min/1.73 m2, and a urinary albumin–creatinine ratio 30–5000 mg/g over 34 weeks, and found no evidence for a positive effect except for lowering serum urate levels [27].
Retrospective analysis of patients with diabetes receiving SGLT2 inhibitors demonstrate also strong uricosuric effects that are likely indirect due to the effect of high urinary glucose levels of GLUT9 [25]. However, SGLT2 inhibitors may not increase the risk for urate stones as urine pH tends to become more alkaline and diluted thereby reducing the propensity to form urate crystals.
Thus, current evidence does not support the concept that inhibition of renal urate transporters may provide a benefit in patients with CKD to halt disease progression. Reduction of serum urate levels can be achieved even in patients with reduced eGFR but care should be taken to avoid high levels of intratubular urate levels which at low urinary pH can precipitate as uric acid triggering the development of uric acid stones or uric acid nephropathy.
INHIBITION OF PROXIMAL TUBULE Pi-TRANSPORTERS TO PREVENT OR REDUCE HYPERPHOSPHATEMIA IN CKD
Renal handling of inorganic phosphate (Pi) is controls and maintains systemic Pi balance. Pi undergoes almost free glomerular filtration and about 80% of the filtered Pi load is reabsorbed, mainly at the level of the proximal tubule. Some Pi reabsorption may also occur in parts of the distal nephron. We recently identified expression of NaPi-IIb in the loop of Henle but its function there has remained elusive [28]. Reabsorption of Pi in the proximal tubule is mediated by at least three different Pi transporters present in the apical brush border membrane, namely NaPi-IIa (SLC34A1), NaPi-IIc (SLC34A3) and PiT2 (SLC20A2) (Fig. 4) [29–31]. Their relative contribution to overall renal Pi handling may differ between species. NaPi-IIa is the major transporter mediating about 70% of tubular Pi reabsorption in mice [32, 33], while the genetic ablation of NaPi-IIc did not alter renal Pi handling [34, 35]. The relevance of PiT2 for renal Pi handling is unclear but currently believed to be very low. However, in humans loss-of-function variants in SLC34A1 (NaPi-IIa) cause hypophosphatemia and hyperphosphaturia without rickets, while loss of SLC34A3 (NaPi-IIc) causes hypophosphatemia, hyperphosphaturia and rickets, which often persists into adulthood. The relative contribution of NaPi-IIa and NaPi-IIc to renal Pi handling may differ between rodents and humans and requires additional clarification.
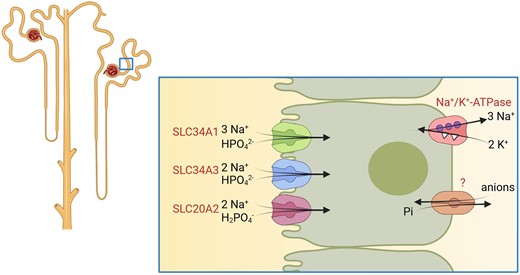
Proximal tubular phosphate transport. Transporters required for proximal tubular phosphate reabsorption. The basolateral exit pathway for phosphate has not been identified on a molecular basis to date.
Individuals with reduced renal clearance due to chronic kidney disease (CKD) commonly develop phosphate overloads with and without overt hyperphosphatemia. Hyperphosphatemia in individuals with CKD or end-stage renal disease (ESRD) causes an increased risk of cardiovascular morbidity and mortality [36, 37]. Likewise, in patients with dialysis the risk for cardiovascular mortality is highly elevated in those with higher Pi levels [38–40]. Current recommended therapies to lower phosphate levels in patients with CKD or ESRD include dietary counselling and/or Pi binders which both suffer from various limitations [41].
At least two distinct inhibitors of proximal tubular Pi transporters have been developed with the aim to increase renal clearance of Pi in individuals with risk for phosphate overload. Both inhibitors have been shown in animal models to increase renal Pi excretion in intact animals as well as in animals with induced reduction of kidney function.
Pfizer developed a series of substances with inhibitory effects on SLC34 Pi transporters. Among them, PF-06869206 is an orally active, selective inhibitor of NaPi-IIa with an IC50 of around 0.4 μmol/L, while the IC50 values for NaPi-IIb and NaPi-IIc as well as for PiT1 and PiT2 are >25 μmol/L [42]. The compound was used by two different groups in mice and rats [43–45]. The compound has a short half-life time in vivo of approximately 45 min [44]. PF-06869206 causes acute phosphaturia, calciuria and increased NaCl losses, while excretion of other solutes such as glucose or amino acids remained normal. Enhanced phosphaturia lowers blood Pi levels in intact mice. In one mouse model but not another, parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF23) levels were also reduced [43, 44]. Owing to its short half-life time, the effects of a single dose of PF-06869206 vanish 4–6 h after application [43]. In mice and rats with 5/6 nephrectomy, PF-06869206 was less phosphaturic, possibly reflecting lower nephron mass and a possible downregulation of NaPi-IIa in CKD [43, 44]. PF-06869206 did not affect Pi excretion in NaPi-IIa KO mice but was effective in mice deficient for either FGF23 or Galnt-3 [43, 45]. The compound may have off-target effects on the epithelial sodium channel ENaC [45]. The substance has not been tested in humans to date and its short half-life time may limit its efficacy in humans.
Another compound was reported as a poster by Bayer Pharmaceuticals. BAY 767 blocked both rat and human NaPi-IIa with a Ki ∼3–6 nmol/L being selective for NaPi-IIa over NaPi-IIb, NaPi-IIc and PiT1 [46]. Acute application to intact rats raised urinary Pi excretion. In a rat model with induced vascular calcification, BAY 767 reduced hyperphosphatemia and vascular calcifications together with PTH and FGF23. Data have been presented only as poster and no trials in humans have been reported to date.
Thus, inhibition of proximal tubular phosphate absorption is feasible and two compounds targeting NaPi-IIa are available. However, there are a number of important unresolved issues: is NaPi-IIa the best target or should NaPi-IIc be inhibited, or both? The therapy will obviously depend on sufficient residual renal function to clear enough phosphate, limiting its use in individuals with advanced stages of CKD. Last, animal data suggest that NaPi-IIa and NaPi-IIc may be downregulated in CKD, further limiting the efficacy of NaPi-IIa inhibitors. These questions need to be addressed to validate phosphate transport inhibitors for therapy.
EXCRETION OF TOXINS AND DRUGS
Excretion of drugs, toxins and waste metabolites by the kidney occurs either by direct filtration or by active secretion. The latter is most important for substances that are bound to proteins in blood and are not readily filtered. Important examples of substrates that are excreted a para-amino-hippuric acid, loop and thiazide diuretics, many uraemic toxins including p-cresol, metabolites of steroid hormones, and cytotoxic drugs such as methotrexate or cisplatin.
The proximal tubule expresses a complex set of transporters mediating the uptake of drugs and metabolites from blood across the basolateral membrane and their secretion into urine over the luminal membrane (Fig. 5). Some transporters also mediate (re)uptake of drugs and metabolites. According to the substrate preferences these transporters may be grouped into transporters transporting cationic, neutral or anionic substrates. Cationic substrates are excreted into urine by the basolateral organic cation transporters OCT1 and OCT2 (SLC22A1 and SLC22A2) and the apical MATE1 and -2 (multidrug and toxin extrusion 1 and 2, SLC47A1 and SLC47A2) transporters [47]. OCT1 and OCT2 function as uniporters, while MATE1 and 2 mediate the exchange of intracellular cationic substrates against extracellular H+. The main substrates of the OCT and MATE transporters are metformin, cimetidine, procainamide, fexofenadine, fluoroquinolone antibiotics (cephalexin, ciprofloxacin), acyclovir and cisplatin. Also, the ATP-driven P-glycoprotein (Pgp) mediates drug excretion such as steroids, HIV protease inhibitors, antifungals, rapamycin, methotrexate, zoledronic acid or metformin. Transport of cisplatin by OCT2 transporters is involved in cisplatin nephrotoxicity [48]. The OCTN1 and OCTN2 transporters (SLC22A4 and SLC22A5) mediate reabsorption of carnitine and may also transport emetine or verapamil [49]. The PEPT2 (SLC15A2) transporter is an H+-driven transporter for tri- and dipeptides but may mediate also reabsorption of beta-lactam and aminocephalosporin antibiotics [50]. The contribution to the reabsorption of angiotensin-converting enzyme inhibitors has been discussed, but is likely low. Anionic substrates are secreted into urine by the cooperation of the basolateral anion transporters OAT1 (SLC22A6), OAT2 (SLC22A7), OAT3 (SLC22A8) and OATP4C1 (SLCO4C1) which all exchange anionic substrates against intracellular dicarboxylates such as α-keto-glutarate. Secretion into urine is completed by anion exchangers and transporters located in the luminal membrane and include the ATP-driven MRP2 and MRP4 efflux pumps, and the OAT4 (SLC22A11), URAT1 (SLC22A12) and OAT10 (SLC22A13) exchangers that secrete anionic drugs and toxins in exchange for organic anions taken up from urine.
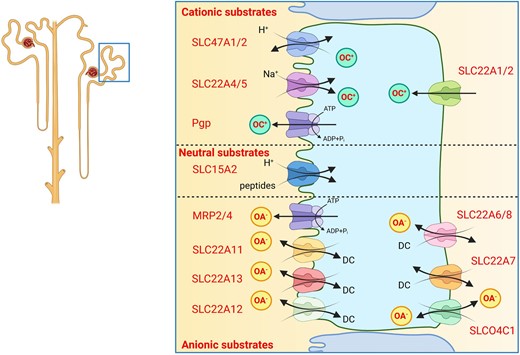
Excretion of organic cations and anions. Transport proteins mediating excretion of various types of drugs, waste metabolites and toxins. OC+, organic cations; OA–, organic anions; DC, dicarboxylates.
The function of these transporters in patients with CKD has several-fold implications. First, these transporters are involved in the excretion of uraemic toxins and their expression and activity is reduced in CKD. Second, several uraemic toxins act as inhibitors of these transporters and reduce thereby the excretion of endogenous waste products and affect the pharmacokinetics of drugs that are obligatorily excreted by these transport pathways [51]. Thus, dosing of drugs has to be adapted to the degree by which the injured kidney can excrete them. Third, drugs that interfere with the luminal transporters reduce their capacity to excrete drugs into urine leading to their intracellular accumulation. In case of metformin or cisplatin this inhibition of secretion can enhance their nephrotoxicity. Indeed, drugs such as ondansetron or topisetron, 5-HT3-antagonists, inhibit MATE1 and can aggravate nephrotoxicity of other drugs [52]. Likewise, multiple drug–drug interactions have been identified whereby some drugs can delay clearance of other drugs by competing for secretion by OCT, MATE or OAT transporters [53–55].
In addition, some of these transporters are also involved in excreting creatinine into urine and inhibition of creatinine excretion by drugs such as some of the anti-retroviral HIV drugs can cause a small rise in serum creatinine levels that should not be misinterpreted as a fall in GFR [56].
Inhibition of drug transporters may be viewed as a potential strategy to reduce nephrotoxicity of drugs that are excreted by these transporters. However, this will require selective inhibition of basolateral transporters avoiding the intracellular accumulation of nephrotoxic substances and needs also to ensure that drugs can be cleared from the organism to prevent other organ toxicities.
TARGETING NEUTRAL AMINO ACID TRANSPORTER B0AT1/SLC6A19
The proximal tubule absorbs filtered amino acids through a variety of amino acid transporters that are partly functionally coupled. Large neutral amino acid are mostly reabsorbed by the Na+-dependent B0AT1 amino acid transporter that forms a heterodimer with TMEM27 (collectrin) for stabilizing at the plasma membrane [57] (Fig. 6). B0AT1 is encoded by the SLC6A19 gene, and biallelic inactivating variants in SLC6A19 cause Hartnup disorder that leads to losses of neutral amino acids into urine. The transporter is also expressed in the small intestine and a number of other epithelia. B0AT1 is not the only amino acid transporter in the proximal tubule brush border membrane but functions along with other amino acid transporters such as PAT2 (SLC36A2) and IMINO (SLC6A20) which particularly transport proline and glycine, EAAT3 (SLC1A1) that accepts anionic amino acids (aspargate and glutamate), and the heterodimeric b0,+AT/rBAT (SLC3A1/SLC7A9) transporter [58]. The latter dimer is responsible for the reabsorption of cationic amino acids (arginine, histidine, ornithine and lysine) and cystine, and mutations in either subunit can cause cystinuria. This transporter functions as an obligatory exchanger of intracellular neutral amino acids against luminal cationic amino acids and cystine. Thus, reabsorption of cystine and cationic amino acids is functionally coupled to the import of neutral amino acids by B0AT1/TMEM27.
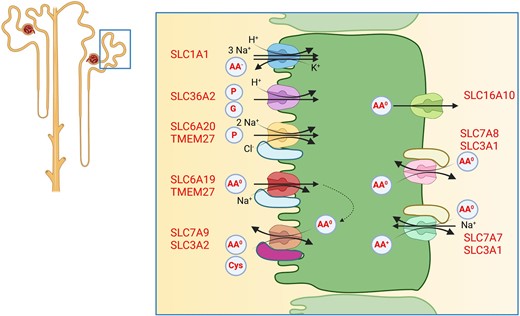
Amino acid reabsorption by the proximal tubule. Amino acid transporters located in the luminal brush border membrane and the basolateral membrane mediate transcellular amino acid reabsorption. Additional amino acid transporters, not shown, are involved in providing amino acids for cellular metabolism. AA–, anionic amino acids; AA0, neutral amino acids; AA+, cationic amino acids; P, proline; G, glycine; Cys, cystine.
At the basolateral side, amino acids are released into blood by another series of transporters that include TAT1 (SLC16A10) transporting mostly aromatic amino acids. These amino acids are then again taken up by the heteromeric LAT2/4F2hc (SLC7A8/4F2hc) transporter which exchanges intracellular neutral amino acids against extracellular large amino acids including aromatic amino acids. Cationic amino acids are released by the heteromeric y+LAT1/4F2hc (SLC7A7/SLC3A2) transporter in exchange for neutral amino acids and sodium. Loss-of-function variants in SLC7A7 cause lysinuric protein intolerance.
High-affinity transport inhibitors for B0AT1 in the nanomolar range have been developed by several groups and companies [59, 60]. Inhibition of intestinal B0AT1 increases Glucagon-like peptide 1 and FGF21, which may be beneficial in the setting of diabetes.
In the context of inhibiting kidney amino acid transport two specific applications are currently being developed: to enhance excretion of phenylalanine to reduce its blood levels in patients with phenylketonuria [61], to induce nitrogen excretion in ornithine transcarbamylase deficiency [62] and reduction of kidney damage in acute toxic nephropathy as evident from aristolochic acid induced nephropathy (a model for the Balkan nephropathy) [63]. The mechanisms mediating this protective effect in aristolochic acid–induced kidney injury remain to be established. B0AT1 inhibition may affect SGLT2 activity as well as tubular workload.
The use of B0AT1 inhibitors for phenylketonuria has already progressed to early clinical studies, which may also provide safety data.
SUMMARY AND OUTLOOK
Proximal tubule transporters are attractive drug targets and the increasing awareness of the importance of proximal tubular metabolism to the pathogenesis of CKD has further spurred interest. Potential problems of drugs targeting transporters in the proximal tubule are that many of these transporters are found also in other organs including small intestine, lung or brain, and side effects may arise from their role outside the kidney. The example of SGLT2, however, also suggests that targeting transporters with high kidney selectivity in their expression profile may reduce side effects. Ideally, drugs targeting proximal tubule transporters have high oral bioavailability and low hepatic metabolism with a sufficient half-life time.
Beyond the transport systems discussed in this review, additional transport systems may be taken into consideration for future drug development and may include specific amino acid transporters, transporters for metals and citrate, and potentially also transporters operating mostly in intracellular organelles such as mitochondria (e.g. SLC25 family) or lysosomes. While most drugs currently inhibit transporters, the identification of substances that can enhance transporter activities may be beneficial, e.g. drugs enhancing drug and uraemic toxin clearance in CKD. Surprisingly, there is a substantial number of orphan transporters expressed in the proximal tubule as well as numerous transport pathways that lack the identification of the underlying molecular mechanisms. Advances in our understanding the basic physiology of these molecules and pathways will aid in the development of new drugs targeting the proximal tubule.
ACKNOWLEDGEMENTS
Studies in the laboratory of the author have been supported by the Swiss National Science Foundation. Figures were created using BioRender.
FUNDING
This paper was published as part of a supplement financially supported by Bayer AG and the scientific content has not been influenced in any way by the sponsor.
DATA AVAILABILITY STATEMENT
No new data were generated or analysed in support of this research.
CONFLICT OF INTEREST STATEMENT
C.A.W. reports honoraria from Kyowa Kirin and Medice, and collaborations with Chugai and Bayer AG.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments