





ABSTRACT
The management of immunoglobulin A nephropathy, membranous nephropathy, lupus nephritis, anti-neutrophil cytoplasmic antibody–associated vasculitis, C3 glomerulonephritis, autoimmune podocytopathies and other immune-mediated glomerular disorders is focused on two major treatment goals, preventing overall mortality and the loss of kidney function. Since minimizing irreversible kidney damage best serves both goals, the management of immune-mediated kidney disorders must focus on the two central pathomechanisms of kidney function decline, i.e., controlling the underlying immune disease process (e.g. with immunotherapies) and controlling the non-immune mechanisms of chronic kidney disease (CKD) progression. Here we review the pathophysiology of these non-immune mechanisms of CKD progression and discuss non-drug and drug interventions to attenuate CKD progression in immune-mediated kidney disorders. Non-pharmacological interventions include reducing salt intake, normalizing body weight, avoiding superimposed kidney injuries, smoking cessation and regular physical activity. Approved drug interventions include inhibitors of the renin–angiotensin–aldosterone system and sodium–glucose cotransporter-2. Numerous additional drugs to improve CKD care are currently being tested in clinical trials. Here we discuss how and when to use these drugs in the different clinical scenarios of immune-mediated kidney diseases.
INTRODUCTION
Immune-mediated glomerular disorders, namely glomerulonephritis (GN) and podocytopathies are a heterogeneous group of immune-mediated forms of glomerular inflammation triggered either by infections, autoinflammation, monoclonal gammopathies, alloimmunity or autoimmunity (Table 1) [1]. The latter subgroup shares a loss of tolerance to a broad spectrum of different self-antigens. Autoimmunity against kidney-specific antigens can cause membranous nephropathy (MN; antigenic podocyte proteins) or anti-glomerular basement membrane (GBM) GN (antigenic NC1 domain of collagen IV). Autoimmunity against ubiquitous antigens occurs in lupus nephritis (LN; antigenic chromatin particles), anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (neutrophil antigens), immunoglobulin A (IgA) nephropathy (IgAN; antigenic IgA), cryoglobulinaemic GN (antigenic IgG) or autoimmune variants of complement factor 3 GN (C3GN; antigenic complement regulators).
Forms of immune-mediated glomerular disorders and immunological activity.
| Type | Antigen | Lesion pattern | Activity | Prevalence |
|---|---|---|---|---|
| Podocytopathies | ||||
| Idiopathic NS | Nephrin, ? | MCD | Variable | +++ |
| Nephrin, ? | FSGS | Variable | ++ | |
| Glomerulonephritis | ||||
| Infection-related GN | Pathogens | Diverse | Variable | +++ |
| Autoimmune GN | IgA | Crescentic GN | High | + |
| Chronic GN | Low | +++ | ||
| IgG | Cryoglobulinaemic GN | High | + | |
| Neutrophils | Crescentic GN | High | ++ | |
| Complement | Crescentic C3GN | Variable | (+) | |
| C3GN | Variable | + | ||
| Chromatin | Proliferative LN | Variable | ++ | |
| LN V | Variable | + | ||
| GBM | Crescentic GN | High | + | |
| Podocytes | Membranous GN | Variable | ++ | |
| Alloimmune GN | Endothelium | Transplant glomerulopathy | Variable | + |
| Autoinflammation-related GN | −, (IEI) | Diverse | Variable | + |
| Monoclonal gammopathy-related GN | −, MGRS | Diverse | Variable | ++ |
| Type | Antigen | Lesion pattern | Activity | Prevalence |
|---|---|---|---|---|
| Podocytopathies | ||||
| Idiopathic NS | Nephrin, ? | MCD | Variable | +++ |
| Nephrin, ? | FSGS | Variable | ++ | |
| Glomerulonephritis | ||||
| Infection-related GN | Pathogens | Diverse | Variable | +++ |
| Autoimmune GN | IgA | Crescentic GN | High | + |
| Chronic GN | Low | +++ | ||
| IgG | Cryoglobulinaemic GN | High | + | |
| Neutrophils | Crescentic GN | High | ++ | |
| Complement | Crescentic C3GN | Variable | (+) | |
| C3GN | Variable | + | ||
| Chromatin | Proliferative LN | Variable | ++ | |
| LN V | Variable | + | ||
| GBM | Crescentic GN | High | + | |
| Podocytes | Membranous GN | Variable | ++ | |
| Alloimmune GN | Endothelium | Transplant glomerulopathy | Variable | + |
| Autoinflammation-related GN | −, (IEI) | Diverse | Variable | + |
| Monoclonal gammopathy-related GN | −, MGRS | Diverse | Variable | ++ |
C3GN: Complement 3 glomerulopathy; FSGS: focal segmental glomerulosclerosis; IEI: inborn error of immunity; MCD: minimal change disease; MGRS: monoclonal gammopathy of renal significance.
Forms of immune-mediated glomerular disorders and immunological activity.
| Type | Antigen | Lesion pattern | Activity | Prevalence |
|---|---|---|---|---|
| Podocytopathies | ||||
| Idiopathic NS | Nephrin, ? | MCD | Variable | +++ |
| Nephrin, ? | FSGS | Variable | ++ | |
| Glomerulonephritis | ||||
| Infection-related GN | Pathogens | Diverse | Variable | +++ |
| Autoimmune GN | IgA | Crescentic GN | High | + |
| Chronic GN | Low | +++ | ||
| IgG | Cryoglobulinaemic GN | High | + | |
| Neutrophils | Crescentic GN | High | ++ | |
| Complement | Crescentic C3GN | Variable | (+) | |
| C3GN | Variable | + | ||
| Chromatin | Proliferative LN | Variable | ++ | |
| LN V | Variable | + | ||
| GBM | Crescentic GN | High | + | |
| Podocytes | Membranous GN | Variable | ++ | |
| Alloimmune GN | Endothelium | Transplant glomerulopathy | Variable | + |
| Autoinflammation-related GN | −, (IEI) | Diverse | Variable | + |
| Monoclonal gammopathy-related GN | −, MGRS | Diverse | Variable | ++ |
| Type | Antigen | Lesion pattern | Activity | Prevalence |
|---|---|---|---|---|
| Podocytopathies | ||||
| Idiopathic NS | Nephrin, ? | MCD | Variable | +++ |
| Nephrin, ? | FSGS | Variable | ++ | |
| Glomerulonephritis | ||||
| Infection-related GN | Pathogens | Diverse | Variable | +++ |
| Autoimmune GN | IgA | Crescentic GN | High | + |
| Chronic GN | Low | +++ | ||
| IgG | Cryoglobulinaemic GN | High | + | |
| Neutrophils | Crescentic GN | High | ++ | |
| Complement | Crescentic C3GN | Variable | (+) | |
| C3GN | Variable | + | ||
| Chromatin | Proliferative LN | Variable | ++ | |
| LN V | Variable | + | ||
| GBM | Crescentic GN | High | + | |
| Podocytes | Membranous GN | Variable | ++ | |
| Alloimmune GN | Endothelium | Transplant glomerulopathy | Variable | + |
| Autoinflammation-related GN | −, (IEI) | Diverse | Variable | + |
| Monoclonal gammopathy-related GN | −, MGRS | Diverse | Variable | ++ |
C3GN: Complement 3 glomerulopathy; FSGS: focal segmental glomerulosclerosis; IEI: inborn error of immunity; MCD: minimal change disease; MGRS: monoclonal gammopathy of renal significance.
The management of immune-mediated GN focusses either on extrarenal autoimmunity or on the mechanisms of glomerular inflammation to minimize irreversible kidney damage and dysfunction or even kidney failure [2]. In this context, proteinuria is used as a biomarker of immunological activity, e.g. when defining treatment response [2]. Indeed, this approach has its strengths during the initial management phase of acute GN characterized by high immunological activity (Table 1). However, the management of GN also features other scenarios where immunological disease activity is less obvious, smouldering or completely absent (Table 1) [3], and this can occur in the presence or absence of proteinuria because the presence of proteinuria has a wide differential diagnosis, also in patients with GN (Table 2). Given that proteinuria per se is viewed as injurious to the kidney, controlling immunological activity is not the only treatment goal in GN [2].
Factors contributing to the presence and level of proteinuria in immune-mediated glomerular disorders.
| Type of factor | Immunity-related factors | Non-immune factors |
|---|---|---|
| Genetic | Thresholds for cytokine release, signalling, leukocyte activity, complement activation | Poor nephron endowment (hyperfiltration) |
| Weaknesses of endothelial, GBM or podocytes leading to more injury at a given filtration pressure | ||
| Other intrinsic factors | Persistence and exposure of antigen | Hyperfiltration due to obesity, diabetes, pregnancy or nephron loss/CKD |
| Affinity and activity of autoreactive lymphocyte clones | ||
| Drug metabolism | ||
| Environmental factors | Unspecific immune activity triggered by, e.g. infections | Hyperfiltration due to salt content in diet, protein-rich diet or vasodilator therapy |
| Adherence to immunotherapy |
| Type of factor | Immunity-related factors | Non-immune factors |
|---|---|---|
| Genetic | Thresholds for cytokine release, signalling, leukocyte activity, complement activation | Poor nephron endowment (hyperfiltration) |
| Weaknesses of endothelial, GBM or podocytes leading to more injury at a given filtration pressure | ||
| Other intrinsic factors | Persistence and exposure of antigen | Hyperfiltration due to obesity, diabetes, pregnancy or nephron loss/CKD |
| Affinity and activity of autoreactive lymphocyte clones | ||
| Drug metabolism | ||
| Environmental factors | Unspecific immune activity triggered by, e.g. infections | Hyperfiltration due to salt content in diet, protein-rich diet or vasodilator therapy |
| Adherence to immunotherapy |
Factors contributing to the presence and level of proteinuria in immune-mediated glomerular disorders.
| Type of factor | Immunity-related factors | Non-immune factors |
|---|---|---|
| Genetic | Thresholds for cytokine release, signalling, leukocyte activity, complement activation | Poor nephron endowment (hyperfiltration) |
| Weaknesses of endothelial, GBM or podocytes leading to more injury at a given filtration pressure | ||
| Other intrinsic factors | Persistence and exposure of antigen | Hyperfiltration due to obesity, diabetes, pregnancy or nephron loss/CKD |
| Affinity and activity of autoreactive lymphocyte clones | ||
| Drug metabolism | ||
| Environmental factors | Unspecific immune activity triggered by, e.g. infections | Hyperfiltration due to salt content in diet, protein-rich diet or vasodilator therapy |
| Adherence to immunotherapy |
| Type of factor | Immunity-related factors | Non-immune factors |
|---|---|---|
| Genetic | Thresholds for cytokine release, signalling, leukocyte activity, complement activation | Poor nephron endowment (hyperfiltration) |
| Weaknesses of endothelial, GBM or podocytes leading to more injury at a given filtration pressure | ||
| Other intrinsic factors | Persistence and exposure of antigen | Hyperfiltration due to obesity, diabetes, pregnancy or nephron loss/CKD |
| Affinity and activity of autoreactive lymphocyte clones | ||
| Drug metabolism | ||
| Environmental factors | Unspecific immune activity triggered by, e.g. infections | Hyperfiltration due to salt content in diet, protein-rich diet or vasodilator therapy |
| Adherence to immunotherapy |
Kidney Disease: Improving Global Outcomes (KDIGO) defines chronic kidney disease (CKD) as every kidney abnormality that persists for >3 months, including proteinuria, haematuria and histopathological abnormalities [4]; hence most patients with GN fulfil these criteria, except for uncomplicated cases of steroid-sensitive nephrotic syndrome. This raises the question whether general CKD care should be a ubiquitous element of GN management and, more specifically, what type of CKD care should be adopted in which phase of GN. There is a consensus to treat all patients with GN with an inhibitor of the renin–angiotensin system (RAS) [2] but, for example, the recent approval of inhibitors of sodium–glucose cotransporter-2 (SGLT-2) for the treatment of CKD raises the question whether all patients with GN should now also receive an SGLT2 inhibitor [5–7].
This review addresses the above question in a conceptual manner not only by defining patients with GN as CKD patients, but also by explaining the pathophysiological consequences of immunopathology on the remaining nephrons that are to be protected from injury. We discuss the rationale for non-drug interventions as well as approved drugs and the drug pipeline to attenuate CKD progression and its significance for patients with GN. Finally, we address practical questions that arise during CKD management in these patients and define the known and the unknown as a starting point for fruitful research.
Treatment goals and markers of disease activity in glomerulonephritis
The first treatment goal is to minimize mortality related to systemic autoimmunity, kidney disease, cardiovascular disease and infections. Most of these are due, at least in part, to CKD or kidney failure, e.g. cardiovascular risk and secondary immunodeficiency correlate with the progressive decline of kidney function [8]. In addition, high immune disease activity and intense immunosuppressive therapy, namely corticosteroids, contribute to the risk of cardiovascular disease and infections [9]. Thus any decline of glomerular filtration rate (GFR) beyond age-related GFR loss contributes to a decreased kidney lifespan and the risk for a premature GN-related death. In this context, rapidly minimizing GN-related GFR loss, as determined by the annual slope of GFR, is now considered the most relevant predictor of GN outcome and is increasingly used as an endpoint in clinical trials [10]. GFR slope is gradually replacing proteinuria as a clinically more relevant endpoint [10, 11]. Regarding GN, one of the factors that most determines the loss of glomerular filtration is control of the underlying immune disease. The remission criteria are not uniformly defined across immune-mediated GN but still adhere mostly to proteinuria. Proteinuria is a biomarker of treatment response in acute GN, but in smouldering or immunologically inactive forms of GN, proteinuria may represent kidney scaring and/or glomerular hyperfiltration, which are prevalent and benefit from other treatments [12].
Two complementary approaches to improve kidney and cardiovascular outcomes in GN
To improve long-term kidney and cardiovascular outcomes in immune-mediated GN implies two main avenues of treatment (Fig. 1).
Immunotherapy to control immunological disease activity
Immunological activity is generally assumed to be present in GN but is not always easily identifiable, especially when active extrarenal manifestations of systemic autoimmunity are absent. The presence of high levels of a nephrotoxic autoantibody can serve as an indicator of size and activity of causative autoreactive lymphocyte clone(s) [13], but not all GN comes with such a biomarker or offers a good correlation between antibody titres and GN activity. The other gold standard remains (repeated) kidney biopsy, especially when applying activity and chronicity indices for histological assessment [2, 14]. Currently these indices are in use only for LN (Table 3), although they could be similarly informative in other forms of GN whenever immunological disease activity cannot be deducted from the standard laboratory evaluation and physical exam. This applies for all primary GNs lacking a reliable serum autoantibody or biomarker. Assessing immunological disease activity is particularly relevant to identify GN relapses versus other causes of increasing proteinuria or whether to ultimately stop immunosuppressive treatment after years of complete clinical remission.
How to assess immunological activity versus chronicity in GN.
| Diagnostic test | Criteria |
|---|---|
| Kidney biopsy | Active lesions (TMA, cellular crescents, loop necrosis, immune cell infiltrates, endothelial activation, mesangial proliferation) |
| Chronic lesions (glomerulosclerosis, fibrous crescents, interstitial fibrosis, tubular atrophy) | |
| Urinary sediment | Acanthocyturia |
| Autoantibody levels | Titre level |
| Complement | C3, C4 levels low, C5b-9 high |
| Proteinuria response | Proteinuria responding quickly and profoundly to immunotherapy as an ex post proof of immunological activity |
| Proteinuria responding quickly and profoundly to either RAS inhibitor or SGLT2 inhibitor as an ex post proof for the contribution of the RAS and SGLT2 to proteinuria | |
| Other urinary biomarkers | Immunity-related biomarkers validated against repeat biopsy occurring before a clinical disease flare and quickly responding to immunotherapy |
| Diagnostic test | Criteria |
|---|---|
| Kidney biopsy | Active lesions (TMA, cellular crescents, loop necrosis, immune cell infiltrates, endothelial activation, mesangial proliferation) |
| Chronic lesions (glomerulosclerosis, fibrous crescents, interstitial fibrosis, tubular atrophy) | |
| Urinary sediment | Acanthocyturia |
| Autoantibody levels | Titre level |
| Complement | C3, C4 levels low, C5b-9 high |
| Proteinuria response | Proteinuria responding quickly and profoundly to immunotherapy as an ex post proof of immunological activity |
| Proteinuria responding quickly and profoundly to either RAS inhibitor or SGLT2 inhibitor as an ex post proof for the contribution of the RAS and SGLT2 to proteinuria | |
| Other urinary biomarkers | Immunity-related biomarkers validated against repeat biopsy occurring before a clinical disease flare and quickly responding to immunotherapy |
TMA: thrombotic microangiopathy.
How to assess immunological activity versus chronicity in GN.
| Diagnostic test | Criteria |
|---|---|
| Kidney biopsy | Active lesions (TMA, cellular crescents, loop necrosis, immune cell infiltrates, endothelial activation, mesangial proliferation) |
| Chronic lesions (glomerulosclerosis, fibrous crescents, interstitial fibrosis, tubular atrophy) | |
| Urinary sediment | Acanthocyturia |
| Autoantibody levels | Titre level |
| Complement | C3, C4 levels low, C5b-9 high |
| Proteinuria response | Proteinuria responding quickly and profoundly to immunotherapy as an ex post proof of immunological activity |
| Proteinuria responding quickly and profoundly to either RAS inhibitor or SGLT2 inhibitor as an ex post proof for the contribution of the RAS and SGLT2 to proteinuria | |
| Other urinary biomarkers | Immunity-related biomarkers validated against repeat biopsy occurring before a clinical disease flare and quickly responding to immunotherapy |
| Diagnostic test | Criteria |
|---|---|
| Kidney biopsy | Active lesions (TMA, cellular crescents, loop necrosis, immune cell infiltrates, endothelial activation, mesangial proliferation) |
| Chronic lesions (glomerulosclerosis, fibrous crescents, interstitial fibrosis, tubular atrophy) | |
| Urinary sediment | Acanthocyturia |
| Autoantibody levels | Titre level |
| Complement | C3, C4 levels low, C5b-9 high |
| Proteinuria response | Proteinuria responding quickly and profoundly to immunotherapy as an ex post proof of immunological activity |
| Proteinuria responding quickly and profoundly to either RAS inhibitor or SGLT2 inhibitor as an ex post proof for the contribution of the RAS and SGLT2 to proteinuria | |
| Other urinary biomarkers | Immunity-related biomarkers validated against repeat biopsy occurring before a clinical disease flare and quickly responding to immunotherapy |
TMA: thrombotic microangiopathy.
CKD therapy for kidney disease resulting from GN
Severe acute GN or ongoing injury along the chronic phase both lead to irreversible damage accrual, i.e. CKD, and non-immune mechanisms of CKD progression contribute to the overall prognosis [15]. These non-immune mechanisms cannot be controlled by immunotherapy and require general CKD care. In the following paragraphs we will focus on the latter aspect by recapitulating the consequences of kidney injury, key elements of the pathophysiology of CKD progression as well as approved and upcoming options to attenuate progressive loss of kidney function by targeting the non-immune drivers of CKD progression.
Visible and non-visible consequences of GN-related CKD
GN-related loss of nephrons reduces the total glomerular filtration surface and the total tubular reabsorption capacity. This implies an increasing workload of the remaining nephrons, in sum leading to intraglomerular hypertension, hyperfiltration and tubular hyperreabsorption [16]. Increased workload initiates adaptive hypertrophy of the remaining nephrons, which for some time compensates for nephron loss and allows the preservation of a normal total GFR. In these situations, biomarkers of kidney function such as serum creatinine and eGFR commonly underestimate the extent of irreversible kidney injury, interstitial fibrosis and tubular atrophy [17]. In GN, podocyte loss is a driving force of nephron atrophy [18]. Besides direct immune-mediated podocyte injury, hyperfiltration-related podocyte shear stress (haemodynamic overload) ultimately leads to podocyte detachment, proteinuria and glomerulosclerosis followed by tubular atrophy [19]. The remaining tubules also suffer from the increased amounts of solutes and albumin processed for reabsorption, which increases oxygen demand in the proximal tubule, leading to a state of relative hypoxia and lysosomal stress (metabolic overload) [16]. The process of nephron loss persistently activates humoral stress pathways such as the RAS and mineralocorticoid receptor signalling, which also contribute to interstitial fibrosis and diminish the peritubular vascular network [20]. A decreasing number of nephrons, functional tubular epithelial cells and intact peritubular capillaries has several consequences:
Retention of sodium: hypertension and oedema
In active GN and early CKD, activation of the RAS is sufficient to cause sodium retention, hypertension and oedema. In nephrotic syndrome, additional mechanisms of sodium retentions, such as activation of the epithelial sodium channel by proteolytic enzymes that enter the tubular lumen in heavy proteinuria [21]. Hypertension can persist as a sign of ongoing activation of the RAS, but also of nephron loss and CKD due to a declining capacity to excrete sodium.
Impaired phosphate clearance and bone disease
Decreasing phosphate clearance activates numerous phosphaturic mechanisms, including the release of fibroblast growth factor-23 (FGF-23) and parathyroid hormone (PTH), with all the well-known consequences for bone health. Control of phosphate and PTH levels is necessary to avoid excessive bone loss and soft tissue calcifications, namely in the cardiovascular system.
Complex mechanisms of accelerated cardiovascular disease
Cardiovascular risk starts to increase once GFR drops below 60 ml/min and becomes a major determinant of morbidity and mortality in patients with advanced CKD. The high cardiovascular risk in certain autoimmune diseases may indeed relate to the rate of kidney damage as well as to systemic inflammation. Numerous mechanisms contribute to this phenomenon, including hypertension, renal anaemia, FGF-23-related cardiac hypertrophy, accelerated vascular calcification and shifts in the secretome of the intestinal microbiota, among others [22].
Complex mechanisms of secondary immunodeficiency
The metabolic shifts that occur during declining kidney function also affect the immune system [23]. Indeed, infections are the second most common cause of death in patients with CKD [11]. In patients with GN, the use of immunosuppressive drugs is obviously a major determinant of immunodeficiency. Thus, preventing CKD progression is important to improve various outcomes in patients with GN.
Targeting non-immune CKD progression in immune-mediated GN
Non-drug interventions are an important component of CKD care to attenuate CKD progression and to endorse patient self-responsibility, shared decision making and drug adherence.
Reducing salt intake
Western diets are notoriously rich in salt, especially in processed foods. Healthy but not diseased kidneys can handle salty diets, because persistent activation of the RAS and mineralocorticoid receptor signalling promote chronic sodium retention. Sodium is a natural antagonist of RAS inhibitors and diuretics. The result is hypervolaemia presenting as arterial hypertension, interstitial oedema, glomerular hyperfiltration/proteinuria and tubular hyperreabsorption [24]. Patients susceptible to dietary interventions can significantly benefit from reducing salt intake, e.g. better blood pressure control, less oedema, less proteinuria and possibly a slower progression of GFR decline [25]. Salt and fluid tonicity can also regulate immune functions in vitro and in animal models, e.g. T helper 17 polarization of T lymphocytes [26]. Whether reducing salt intake also modifies autoimmune disease activity and kidney inflammation in humans is unknown.
Normalization of body weight
Overweight and obesity are risk factors for CKD progression because additional body mass implies more fluid to filter and metabolites to reabsorb in tubules [19]. Thus obesity contributes to the haemodynamic and metabolic overload of remaining nephrons [16]. In addition, excess adipose tissue produces adipokines shown to enhance kidney inflammation. Significant weight reduction can reduce proteinuria and potentially improve long -erm outcomes of GN [27]. Glucagon-like peptide-1 receptor agonist (GLP-1) and a novel glucose-dependent insulinotropic polypeptide (tirzepatide) could become useful in this context [28].
Avoiding superimposing kidney injury/drug nephrotoxicity
To maximize the remaining kidney lifespan, any other kidney injury should be avoided. This implies patient education regarding possible other causes of kidney injury and common nephrotoxins. We advocate to limit the use of proton pump inhibitors, nephrotoxic antibiotics, high volumes of radiocontrast agents and non-steroidal anti-inflammatory drugs to situations with a clear benefit over the potential nephrotoxic risk. Dihidropyridine calcium channel blockers can exert a vasodilatory effect on the afferent arteriole and increase glomerular hyperfiltration and proteinuria [29]. These effects may be misinterpreted as immunological GN activity and lead to unjustified immunosuppressive treatment. Alternative antihypertensive drugs are available.
Smoking cessation
Smoking is associated with poor kidney and cardiovascular outcomes in all forms of CKD [30, 31]. Although high-level trial evidence is lacking in GN, there is international consensus to advise all CKD patients to stop smoking.
Physical activity
Regular sport activity is advised in all forms of CKD to normalize body weight and to reduce blood pressure. Patients should be educated, however, to avoid particularly strenuous physical activity, such as heavy weightlifting, which is associated with massive short blood pressure peaks. In this context, bodybuilding and anabolic use place the kidneys at risk for added damage.
Low protein diet
High protein intake increases glomerular hyperfiltration and proteinuria, hence, high protein (fitness) diets should generally be avoided in CKD patients. Improving kidney outcomes with low protein diets was a CKD management concept for decades to reduce haemodynamic and metabolic overload. However, carefully conducted randomized controlled trials could not verify this hypothesis [32]. Current CKD management guidelines recommend to consider dietary protein restriction in adults to 0.8 g/kg/day in patients with proteinuria >3.5 g/24 hours and eGFR <60 ml/min/1.73 m2 and replacement of nephrotic losses to attenuate CKD progression.
Approved drugs that control non-immune drivers of CKD progression and reduce cardiovascular morbidity and mortality
RAS blockade with angiotensin-converting enzyme inhibitors (ACEis) or angiotensin receptor blockers (ARBs) has been the cornerstone therapy to slow CKD progression and cardiovascular risk in GN, e.g. IgAN [33, 34]. Therefore their use is recommended as a baseline therapy for forms of chronic GN [2]. These benefits appear to be comparable between ACEis and ARBs when they are used in equivalent doses and carry comparable adverse effects besides cough, which is exclusive to ACEis. Our target should be to reach the maximum tolerated RAS blockade dose. RAS inhibitors are thought to attenuate CKD progression for several reasons, including a reduction in intraglomerular pressure and glomerular hyperfiltration, the related antiproteinuric effect, anti-inflammatory and antifibrotic effects as well as supporting regenerative mechanisms [35].
SGLT2 inhibitors have shown remarkable additional benefits in delaying CKD progression and cardiovascular protection on top of the standard RAS blockade in non-diabetic CKD patients [11, 36]. Post hoc studies from large clinical trials have demonstrated this effect in IgAN and podocytopathies presenting as focal segmental glomerulosclerosis [37–39]. Patients with LN and ANCA-associated vasculitis were excluded from the DAPA-CKD trial but not from the EMPA-Kidney trial, but dedicated subgroup analyses are still pending [11, 36]. The findings of these trials have been projected into 7.4 more years of survival free of kidney failure, the ultimate meaningful patient-oriented outcome [40]. An additional and beneficial diuretic effect on nephrotic syndrome might be expected with the use of these drugs. In case other diuretics are used, careful monitoring for potential hypovolaemia should be performed.
Pipeline of drugs that control non-immune drivers of CKD progression
Other new approaches with potential additional synergistic non-immune mechanisms of action have been developed but have not yet been approved by regulatory authorities for the treatment of GN. Drugs that block endothelin receptors (sparsentan, atrasentan and avosentan) and the non-steroidal MRA finerenone belong to this category.
Finerenone differs from established steroidal MRA by its different structure, volume of distribution and half-life that, when added to RAS inhibitors, slows the progression of CKD and reduces cardiovascular events in patients with diabetic kidney disease [41]. Results of subgroup analyses suggest that the benefits of finerenone are similar with or without concomitant SGLT2 inhibitors or GLP-1 receptor agonist treatment, so a synergic effect could be expected [42]. A small crossover study testing combinations of dapagliflozin and the MRA eplerenone found synergistic effects on proteinuria reduction [43]. Moreover, the risk of hyperkalaemia was significantly reduced by the presence of a SGLT2 inhibitor. Recently a clinical trial has been launched testing finerenone in patients with non-diabetic kidney disease [FIND-CKD (NCT05047263)].
As endothelin-1 (ET-1), through activation of endothelin receptor type A (ET-A), promotes vasoconstriction, kidney cell injury, inflammation and fibrosis, and hence proteinuria. Therefore it is not surprising that ET-A receptor antagonists have renoprotective effects in both experimental and clinical studies in proteinuric CKD patients, especially when they are combined with RAS inhibitors [44]. In the latter trial, fluid retention appeared as a critical drawback in diabetic patients with a significant cardiovascular risk profile, but atrasentan and sparsentan are currently being tested in patients with other glomerular disorders, including IgAN and podocytopathies presenting as focal segmental glomerulosclerosis [45]. The effects of an ET-A receptor antagonist could be complementary to dual RAS–SGLT2 inhibition or even finerenone.
Considerations on the time of initiation and handling of CKD therapy in GN
Starting CKD care when GFR decreases to <60 ml/min/1.73 m2
Many consider CKD only after the GFR decreases to <60 ml/min/1.73 m2, i.e. CKD G3 (Fig. 2). This makes sense in terms of the CKD-related cardiovascular risk, which starts to increase not before CKD G3. In this regard, CKD care should definitely start at least at this point, especially in patients with fast CKD progression as evidenced by a linear annual GFR decline predicted to continue to decline in the future [2, 4]. If CKD care can improve the negative GFR slope [38], this can translate into months or years longer kidney lifespan. But data from other forms of progressive CKD suggest that the effect is limited compared with an earlier onset of CKD therapy (Fig. 1). Of note, a GFR >60 ml/min/1.73 m2 in the absence of urinary abnormalities and hypertension in elderly patients likely represents normal kidney aging and does not require CKD care.
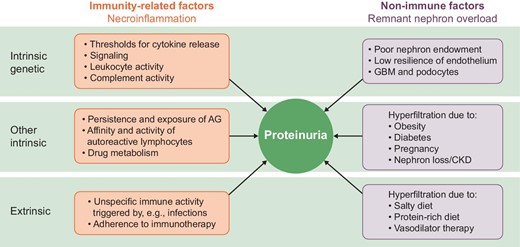
Immune and non-immune factors contribute to proteinuria in GN. Both immune as well as non-immune mechanisms of proteinuria can be grouped according to genetic and other intrinsic and extrinsic factors. AG: antigen.
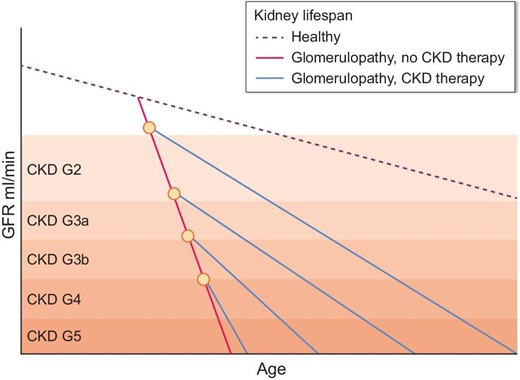
How time of onset of CKD therapy impacts kidney lifespan. In healthy individuals, GFR decreases with aging. Chronic or relapsing disease activity of GN can significantly shorten kidney lifespan despite immunotherapy. Therapy of CKD with renoprotective drugs can attenuate the slope of GFR decline and hence prolong (dialysis-free) kidney lifespan. Inhibition of the RAS and potentially of SGLT2 as well as other renoprotective drugs elicit significantly stronger renoprotective effects when started at an early CKD stage. This ‘the earlier, the better’ concept of renoprotective therapy implies an early awareness for CKD in GN and the perspective of maximizing (dialysis-free) kidney lifespan as a critical patient-oriented treatment goal.
Starting after induction of a successful treatment response in highly active GN
Data from other progressive forms of CKD suggest that earlier initiation of RAS blockade is associated with a larger effect size, i.e. longer kidney lifespan [46]. This finding fits the ‘the earlier, the better’ paradigm. However, whether good control of GN still puts the patient at risk for progressive CKD is not always clear. Risk factors for CKD progression may be concomitant diabetes, hypertension, age, persistent proteinuria, male sex and a history of repetitive episodes of active GN [15].
Starting whenever immunotherapy is unable to achieve proteinuria goals
Residual proteinuria may or may not represent persistent immunological activity and only immunophenotyping can clarify this point [47]. However, residual proteinuria is likely due to glomerular hyperfiltration in patients with a low nephron number (low birthweight or advanced CKD), concomitant obesity, hypertension, diabetes, pregnancy and/or exposure to a high salt or protein diet [19]. Non-drug (and drug) interventions as part of CKD care should be the standard of care in these settings to limit kidney overload [16].
Starting when rebiopsy shows signs of chronicity
Irreversible chronic lesions such as glomerulosclerosis, interstitial fibrosis and tubular atrophy are not susceptible to currently used immunomodulatory drugs and their presence implies a more advanced stage of CKD and a risk for further kidney function decline. Thus, initiating CKD care is strongly recommended in patients where any evidence of irreversible kidney injury is documented by kidney biopsy [2].
Starting during the highly active phase of GN
RAS inhibitors are frequently initiated during the active phase of GN to treat arterial hypertension [14]. At the same time, they reduce glomerular hyperfiltration and hence proteinuria in the phase of glomerular inflammation when the filtration barrier is most vulnerable to injury [33], i.e. glomerular barotrauma due to high filtration pressure. Theoretically, SGLT2 inhibitors should elicit additive effects in this context, but clinical evidence is still lacking [7]. In addition, unlike RAS inhibitors, SGLT2 inhibitor therapy is associated with infectious complications, e.g. mucosal candida infections [11], which may raise concerns for more severe infectious complications in the context of concomitant treatment with (high-dose) corticosteroids and sometimes other immunosuppressants. However, the use of SGLT2 inhibitors in kidney allograft recipients all exposed to combination immunosuppressive therapies was not associated with more or more severe infectious complications [48], which adds confidence in a potential use of SGLT2 inhibitors during highly active GN. Nevertheless, we feel that clinical studies are needed before this can be broadly recommended.
Do antiproteinuric drugs mask disease activity of GN?
Proteinuria is frequently used as a biomarker of immunological disease activity in GN, hence antiproteinuric drugs may interfere with such an interpretation. However, the presence of proteinuria always implies a differential diagnosis that includes other hypotheses, e.g. glomerular hyperfiltration, which is another important pathomechanism of CKD progression [19]. In view of the complex treatment goals, it is important not to withhold the best possible CKD therapy for this reason but to assess immunological activity by specific diagnostic tests such as immunophenotyping and repeat biopsy unless specific urinary biomarkers become available.
Balancing pill burden and potential drug non-adherence against treatment benefits
Pill burden and concerns about drug non-adherence are sometimes used as an argument against CKD therapy beyond immunotherapy of GN. Dual RAS–SGLT2 inhibition implies the additional intake of two pills per day, which together elicit profound effects in terms of the treatment goals of cardiovascular mortality, organ protection and even proteinuria. In the future, polypill-like combinations may help to sustain drug adherence and to avoid unnecessary immunotherapies considered for persistent proteinuria that instead is hyperfiltration related. Nevertheless, the use or non-use of additional drugs follows a benefit–risk assessment in each individual patient.
When not to use supportive care in CKD
Early RAS and SGLT2 inhibition is to be avoided in patients with nephrotic syndrome–related intravascular volume depletion, as this may further impair kidney perfusion, e.g. in children with an abrupt onset of nephrotic syndrome. No data support CKD therapy in acute GN with good prospects for full recovery, e.g. mild forms of infection-related GN. There has been concern regarding the use of RAS inhibitor in late-stage CKD, but a recent randomized trial could not find any difference in terms of onset of kidney replacement therapy [49].
Conclusions and outlook
Most paediatric and adult patients with immune-mediated glomerular disorders are CKD patients and may benefit from CKD therapy independent from the control of the immune-mediated process. Non-immune mechanisms contribute to the progression of CKD and can be controlled by numerous non-drug and drug interventions with a strong effect size in terms of the treatment goals of mortality, organ failure, organ dysfunction and proteinuria. It is likely that early treatment can maximize the renoprotective effects (Fig. 2), but more clinical data and randomized trials are needed to confirm this concept at a high level of scientific evidence for patients with the various forms of GN.
FUNDING
H.-J.A. received support from the Deutsche Forschungsgemeinschaft (AN372/29-1 and 30-1). G.M.F.J. received support from the Instituto de Salud Carlos II (PI19/01695) and RICORS (RD21/0005/0030). J.F. received support from the Deutsche Forschungsgemeinschaft (Clinical Research Unit 5011, project number 445703531) and ERK-NET.
AUTHORS’ CONTRIBUTIONS
All authors contributed equally to this review and approved the final version.
DATA AVAILABILITY STATEMENT
No new data were generated or analysed in support of this research.
CONFLICT OF INTEREST STATEMENT
H.-J.A. received honoraria or lecture fees from AstraZeneca, Bayer, Eleva, GlaxoSmithKline, Kezar, Eli Lilly, Novartis, Otsuka, PreviPharma and Vifor. G.M.F.J. received honoraria or lecture fees from AstraZeneca, GlaxoSmithKline, Otsuka and Vifor. J.F. received honoraria or lecture fees from AstraZeneca, Bayer, Boehringer, Calliditas, Novartis, Omeros, Travere and Vifor and serves on data safety monitoring boards for Novo Nordisk and Visterra. A.V. and P.R. have nothing to disclose.
REFERENCES


{kind=link}
{kind=link}
Comments