





Chronic kidney disease (CKD) is a public health problem. In a large national registry, 20%–25% of patients with end-stage kidney disease have been reported with an undetermined kidney disease (UKD) [1–3]. Monogenic disease–causing variants are underdiagnosed in patients with CKD, with a prevalence estimation of about 10% [4]. Since 2019, we have proposed exome sequencing (ES) by next-generation sequencing to all patients with UKD. Our strategy is based on an in silico panel analysis of known genes related to kidney disease. We conducted a literature survey to add new published genes to our bioinformatics panel. We reanalyzed all ES-unsolved cases in the light of the newly discovered gene. Recently, Devane et al. [5] reported TUPL3 as a new ciliopathy-associated disease gene. Here, we report two cases that were initially described as ES-unsolved cases last year but that were in fact carriers of TULP3 pathogenic variations. One case carried the same TULP3 pathogenic homozygote variation described by Devane and colleagues and the other carried two new pathogenic biallelic variations which to our knowledge have never been described before in TULP3.
In Patient A, CKD was discovered fortuitously in a 50-year-old Caucasian woman. Renal biopsy showed chronic tubulointerstitial nephropathy of undetermined origin with less than 25% of tubulointerstitial fibrosis, 4/14 sclerosed glomeruli and no deposit. On computed tomography imaging, multi-cystic kidneys were found (Fig. 1). No genetic testing was performed. Other medical events were repeated cystitis, hepatitis B, tonsillectomy, appendectomy, inguinal hernia and carpal tunnel. Her blood pressure was normal. She reached end-stage renal disease when she was 56 years old and was transplanted at the age of 61 years with immediate graft function. She developed abnormal liver tests after kidney transplantation which resolved after cotrimoxazole discontinuation. Cytomegalovirus reactivation with liver involvement was also identified and treated with valganciclovir and decreased immunosuppression. Hepatitis cytolysis completely resolved but gamma-glutamyl transferase remained at 1.5 N. Liver ultrasound was normal but fibrosis-4 (FIB-4) Index for Liver Fibrosis was 2.78, approximating a fibrosis stage between 2 and 3, whilst liver elastography revealed liver stiffness at 16 kPa confirming presence of advanced liver fibrosis. Transthoracic echocardiography performed 1 month after kidney transplant revealed no abnormalities and a normal ejection fraction.
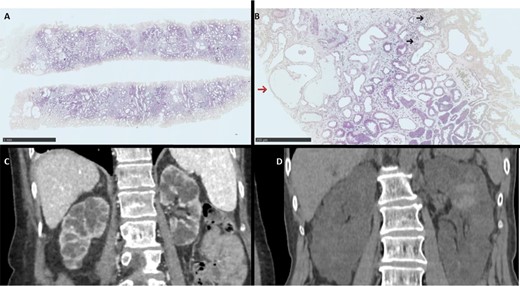
Histological and radiological features from patient. (A, B) Kidney histology H&E staining showing at low magnification (HES ×2.5) low to moderate fibrosis with pseudocystic dilatation of the tubules and at higher magnification (HES ×10) interstitial lymphocytic infiltrate (dark arrow) and pseudocystic tubular dilatation (red arrow). (C, D) Computed tomography (CT) showing renal cysts in Patients A and B, respectively.
In Patient B, the patient was a 66-year-old in whom we discovered end-stage CKD without any symptom other than a general weakness. The workup showed multicystic kidneys with a slightly increased volume without any other cystic localization on abdominal tomography (Fig. 1).
First exome analysis was negative, but we reanalyzed (see our method in the discussion section) the exome following the description of the TULP3 gene by Devane et al. We identified in Patient A biallelic variant in the TULP3 gene (Table 1) classified IV according to the American College of Medical Genetics and Genomics (ACMG) classification [6]. This variation was described in three patients from two Mediterranean families with early disease onset before the age of 20 years characterized by a severe liver phenotype disease without cardiac involvement Table 2 [5]. Only one patient had renal involvement with cortical and medullary microcysts and a slight renal dysfunction. In Patient B, trio exome with the patient's son and daughter reanalysis identified two compound heterozygous variations (in trans) in the TULP3 gene (Table 1 and Fig. 1) classified IV according to the ACMG classification. A reverse phenotype showed left ventricular hypertrophy in relation to long-term high blood pressure but revealed no hepatic disease Table 2. The family tree did not show any renal disease in the siblings (three brothers, one of whom died of unknown reason at age 9 months, and two sisters).
Details of the TULP3 pathogenic variants.
| Patient | Exon | Genomic position | Position | Variant | rs number | Zigoty | gnomAD frequency | Number of HMZ in gnomAD | ACMG classification | DANN |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 6/11 | chr12:g.3040253del | c.544del | p.(Leu182TrpfsTer4) | 924744512 | HMZ | 0.00003185 | 0 | 4 | |
| B | 6/11 | chr12:g.3040236C>T | c.526C>T | p.(Gln176Ter) | rs763840266 | HTZ | 0.00001193 | 0 | 4 | D |
| B | 10/11 | chr12:g.3047348del | c.1093del | p.(Ala365ProfsTer24) | NA | HTZ | NA | NA | 4 |
| Patient | Exon | Genomic position | Position | Variant | rs number | Zigoty | gnomAD frequency | Number of HMZ in gnomAD | ACMG classification | DANN |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 6/11 | chr12:g.3040253del | c.544del | p.(Leu182TrpfsTer4) | 924744512 | HMZ | 0.00003185 | 0 | 4 | |
| B | 6/11 | chr12:g.3040236C>T | c.526C>T | p.(Gln176Ter) | rs763840266 | HTZ | 0.00001193 | 0 | 4 | D |
| B | 10/11 | chr12:g.3047348del | c.1093del | p.(Ala365ProfsTer24) | NA | HTZ | NA | NA | 4 |
NA: not available; D: damaging; HMZ: homozygous; HTZ: heterozygous. DANN: deleterious annotation of genetic variants using neural networks; gnomAD: Genome Aggregation Database.
Details of the TULP3 pathogenic variants.
| Patient | Exon | Genomic position | Position | Variant | rs number | Zigoty | gnomAD frequency | Number of HMZ in gnomAD | ACMG classification | DANN |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 6/11 | chr12:g.3040253del | c.544del | p.(Leu182TrpfsTer4) | 924744512 | HMZ | 0.00003185 | 0 | 4 | |
| B | 6/11 | chr12:g.3040236C>T | c.526C>T | p.(Gln176Ter) | rs763840266 | HTZ | 0.00001193 | 0 | 4 | D |
| B | 10/11 | chr12:g.3047348del | c.1093del | p.(Ala365ProfsTer24) | NA | HTZ | NA | NA | 4 |
| Patient | Exon | Genomic position | Position | Variant | rs number | Zigoty | gnomAD frequency | Number of HMZ in gnomAD | ACMG classification | DANN |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 6/11 | chr12:g.3040253del | c.544del | p.(Leu182TrpfsTer4) | 924744512 | HMZ | 0.00003185 | 0 | 4 | |
| B | 6/11 | chr12:g.3040236C>T | c.526C>T | p.(Gln176Ter) | rs763840266 | HTZ | 0.00001193 | 0 | 4 | D |
| B | 10/11 | chr12:g.3047348del | c.1093del | p.(Ala365ProfsTer24) | NA | HTZ | NA | NA | 4 |
NA: not available; D: damaging; HMZ: homozygous; HTZ: heterozygous. DANN: deleterious annotation of genetic variants using neural networks; gnomAD: Genome Aggregation Database.
TULP3 phenotype observation in affected individuals compared with the Devane et al. report.
| Report | Patients | TULP3 nucleotide/amino acid change | Origin, sex, age | Kidney phenotype | Liver phenotype | Cardiac phenotype |
|---|---|---|---|---|---|---|
| Devane et al. | I.1 | c.544delC (p.Leu182TrpfsTer4) | European, F, 18 years | No | Cirrhosis and portal HTN at 14 years | TTE and ECG normal |
| I.2 | European, M, 16 years | No | Cirrhosis and portal HTN at 12 years | TTE and ECG normal | ||
| II | European, M, 16 years | Non-enlarged kidneys with cortical and medullary microcysts with increased cortical echogenicity, eGFR: 86 | Cholestasis and jaundice at 4 years, cirrhosis and portal HTN | TTE normal at 16 years | ||
| Robert et al. | A | European, F, 50 years | Non-enlarged kidneys with cortical and medullary microcysts, renal transplantation at 60 years | Liver fibrosis at 60 years | Left ventricular hypertrophy and normal ECG at 61 years | |
| B | c.526C>T (p.Gln176Ter)/c.1093del (p.Ala365ProfsTer24) | European, F, 66 years | Slightly enlarged kidneys with cortical and medullary microcysts, end-stage kidney disease 66 years | No | TTE normal and ECG at 66 years |
| Report | Patients | TULP3 nucleotide/amino acid change | Origin, sex, age | Kidney phenotype | Liver phenotype | Cardiac phenotype |
|---|---|---|---|---|---|---|
| Devane et al. | I.1 | c.544delC (p.Leu182TrpfsTer4) | European, F, 18 years | No | Cirrhosis and portal HTN at 14 years | TTE and ECG normal |
| I.2 | European, M, 16 years | No | Cirrhosis and portal HTN at 12 years | TTE and ECG normal | ||
| II | European, M, 16 years | Non-enlarged kidneys with cortical and medullary microcysts with increased cortical echogenicity, eGFR: 86 | Cholestasis and jaundice at 4 years, cirrhosis and portal HTN | TTE normal at 16 years | ||
| Robert et al. | A | European, F, 50 years | Non-enlarged kidneys with cortical and medullary microcysts, renal transplantation at 60 years | Liver fibrosis at 60 years | Left ventricular hypertrophy and normal ECG at 61 years | |
| B | c.526C>T (p.Gln176Ter)/c.1093del (p.Ala365ProfsTer24) | European, F, 66 years | Slightly enlarged kidneys with cortical and medullary microcysts, end-stage kidney disease 66 years | No | TTE normal and ECG at 66 years |
ECG: electrocardiogram; F: female; HTN: hypertension; M: male; TTE: transthoracic echocardiography.
TULP3 phenotype observation in affected individuals compared with the Devane et al. report.
| Report | Patients | TULP3 nucleotide/amino acid change | Origin, sex, age | Kidney phenotype | Liver phenotype | Cardiac phenotype |
|---|---|---|---|---|---|---|
| Devane et al. | I.1 | c.544delC (p.Leu182TrpfsTer4) | European, F, 18 years | No | Cirrhosis and portal HTN at 14 years | TTE and ECG normal |
| I.2 | European, M, 16 years | No | Cirrhosis and portal HTN at 12 years | TTE and ECG normal | ||
| II | European, M, 16 years | Non-enlarged kidneys with cortical and medullary microcysts with increased cortical echogenicity, eGFR: 86 | Cholestasis and jaundice at 4 years, cirrhosis and portal HTN | TTE normal at 16 years | ||
| Robert et al. | A | European, F, 50 years | Non-enlarged kidneys with cortical and medullary microcysts, renal transplantation at 60 years | Liver fibrosis at 60 years | Left ventricular hypertrophy and normal ECG at 61 years | |
| B | c.526C>T (p.Gln176Ter)/c.1093del (p.Ala365ProfsTer24) | European, F, 66 years | Slightly enlarged kidneys with cortical and medullary microcysts, end-stage kidney disease 66 years | No | TTE normal and ECG at 66 years |
| Report | Patients | TULP3 nucleotide/amino acid change | Origin, sex, age | Kidney phenotype | Liver phenotype | Cardiac phenotype |
|---|---|---|---|---|---|---|
| Devane et al. | I.1 | c.544delC (p.Leu182TrpfsTer4) | European, F, 18 years | No | Cirrhosis and portal HTN at 14 years | TTE and ECG normal |
| I.2 | European, M, 16 years | No | Cirrhosis and portal HTN at 12 years | TTE and ECG normal | ||
| II | European, M, 16 years | Non-enlarged kidneys with cortical and medullary microcysts with increased cortical echogenicity, eGFR: 86 | Cholestasis and jaundice at 4 years, cirrhosis and portal HTN | TTE normal at 16 years | ||
| Robert et al. | A | European, F, 50 years | Non-enlarged kidneys with cortical and medullary microcysts, renal transplantation at 60 years | Liver fibrosis at 60 years | Left ventricular hypertrophy and normal ECG at 61 years | |
| B | c.526C>T (p.Gln176Ter)/c.1093del (p.Ala365ProfsTer24) | European, F, 66 years | Slightly enlarged kidneys with cortical and medullary microcysts, end-stage kidney disease 66 years | No | TTE normal and ECG at 66 years |
ECG: electrocardiogram; F: female; HTN: hypertension; M: male; TTE: transthoracic echocardiography.
Interestingly, the phenotype and age of disease onset of Patient A are radically different from the patients described by Devane et al. with the same mutation in the TULP3 gene Table 2. Dysfunction of primary cilium is characterized by multisystem organ fibrosis via alterations in different molecular pathways. Beside the implication of the TULP3 gene in ciliary trafficking, the major mechanism is direct antifibrotic signalling which leads directly to the loss of antifibrotic modulation of WNT and TGF-β signalling pathways. As expected, presence of nonsense mutations may result in a severe phenotype that involves early onset liver disease and late onset of kidney fibrosis with hypertrophic cardiomyopathy. A milder phenotype is observed in our Patient A. Patient B presented only renal involvement which could be potentially explained by the genetic background effects that can be both intrinsic, namely due to interactions between the causal variant and other genetic modifiers, and/or extrinsic, due to an environmental impact inducing epigenetic modification [7]. Very interestingly, Jafari Khamirani et al. reported a homozygous missense mutation in the TULP3 gene in consanguineous patients with a severe phenotype, which highlighted the potential importance of genetic background effects [8]. Furthermore, we described association of one new nonsense mutation and frameshift mutation in the TUPL3 gene with limited renal involvement. In ciliopathies, lack of genotype/phenotype correlation is frequent. The CEP290 gene is a good illustration of this phenomenon with phenotypes ranging from the lethal Meckel-Grüber syndrome to isolated blindness, with various clinical symptoms, highly variable severity and differing ages of onset [9]. Our cases illustrate the clinical and phenotypical severity heterogeneity related to fibrosis, primarily liver fibrosis, for the TULP3-truncating variation. We could hypothesize that loss of function variation in the TULP3 gene is highly variable with a background-dependent fitness change. Modifier gene exploration and environmental exposure may be the next step towards increasing our understanding of mendelian disorders. Our report confirms the difficulty of guiding molecular diagnosis using only phenotyping data and supports the benefits of systematic genomic use when seeking the aetiology of CKD.
A negative genetic test does not exclude a genetic cause of disease, since some variations can be undetected by the assay (for example, lack of coverage, triplets expansion, variants in highly homologous regions) and since the data are interpreted in the light of current knowledge. In the former, ACMG recommends periodic reanalysis of sequencing data [10]. Reanalysis of previous negative ES based on expansion of the kidney panel with newly discovered gene-associated kidney disease or by applying improved bioinformatic pipelines has not been evaluated in kidney disease. In our centre, reanalysis of ES data is performed for each newly discovered gene-associated kidney disease. We perform a bibliography survey using a table of contents subscription from the main kidney and genetic journals, a Twitter social network subscription to the accounts of the most authoritative journals and recognized experts on nephrogenomics, and findings from major American and European congresses. From 2019 to the present, we have added 45 genes to our customized kidney panel from the initial panel published by Groopman et al. (625 genes) [4]. This reanalysis is performed using the signed informed patient consent collected during the initial nephrogenomic consultation. For each new gene-associated kidney disease described in the literature, we collect from a GATK GenomicsDB variant datastore (https://gatk.broadinstitute.org/hc/en-us/articles/360035891051-GenomicsDB) all samples showing rare variants (frequency below 0.1% [10]) in the gene of interest. Then we compare the patient phenotype with the molecular data.
Our strategy of ES and regular reanalysis of genomic data provide a robust approach resolving UKD.
ACKNOWLEDGEMENTS
We thank Mme Ponce Fanny from the bioinformatic team of Eurofins Biomnis.
AUTHORS’ CONTRIBUTIONS
T.R. wrote the report, collected and analyzed the data. B.S., T.L., J.T., L.R., S.B. and M.D. analyzed the data. All authors helped to write the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare they have no conflict of interest regarding this study.


{kind=link}
Comments