





The primary hyperoxalurias (PHs) represent a group of rare autosomal recessive inherited disorders of the glyoxylate metabolism leading to an endogenous overproduction of oxalate by the liver [1, 2]. This insoluble metabolic end-product has to be excreted via the kidneys and hence primarily results in nephrolithiasis and/or nephrocalcinosis, but later in chronic kidney disease and eventually in end-stage renal disease (ESRD) [1, 2]. However, calcium oxalate crystal formations are not only found in kidneys, but also elsewhere in the body, e.g. in the bone, myocardium and the eye.
Three forms of PH with considerable clinical overlap can be differentiated, which are associated with mutations in the AGXT (PH1), GRHPR (PH2) and HOGA1 (PH3) genes [2, 3]. PH1 represents the most common and most severe form of PH. Its clinical presentation is highly heterogeneous and includes infantile oxalosis with ESRD in the first weeks of life, resulting in a significantly increased morbidity and mortality, and the more common non-infantile form with ESRD mostly in early adulthood [2]. In PH2, ESRD usually occurs later in life [4]. Patients with the least severe form, PH3, often have preserved kidney function, although impaired renal function may occur.
Sequential or combined liver–kidney transplantation provides the only causative treatment for PH1 and sometimes also for PH2 [2]. The decline in renal function can be delayed but not prevented by conservative management, and oxalate continues to accumulate even if intensified dialysis is performed [2, 5]. However, therapeutic options are on the horizon, such as RNA interference (RNAi) targeting upstream glycolate oxidase or further downstream liver-specific lactate dehydrogenase A to deplete the substrate for oxalate synthesis, or Oxalobacter formigenes interacting with colonic epithelium and inducing colonic oxalate secretion, as well as gene therapy [5].
OPHTHALMOLOGIC EXAMINATION IN PH
Extra-renal manifestations may cause a severe burden of disease in PH [2]. This underscores the need for ophthalmologic screening, which might also help to identify patients who may require more intensive therapeutic strategies. Best-corrected visual acuity (BCVA) testing, slit-lamp examination with a detailed evaluation of the anterior segment of the eye and dilated indirect ophthalmoscopy are the cornerstones of each eye examination.
High-resolution retinal imaging is essential for documentation, disease monitoring and for visualizing subtle alterations that may otherwise remain unnoticed [6–8]. Two non-invasive imaging modalities are widely and routinely used for characterizing retinal diseases: (i) fundus colour photography is principally used to document ophthalmoscopy findings and (ii) optical coherence tomography (OCT) is informative with regard to the cross-sectional retinal anatomy and is frequently described as the optical analogue of an ultrasound examination. Its high resolution allows identification and longitudinal monitoring of even minor alterations in different layers of the retina [9].
PRONOUNCED OXALATE RETINOPATHY IN AN INFANTILE PH1 PATIENT
A 9-year-old boy presented for a retinal evaluation of PH1. Medical history included progression to ESRD at the age of 4 months followed by peritoneal dialysis and haemodialysis, and sequential liver–kidney transplantation at the age of 10 and 17 months, respectively. Despite an early initiation of therapy and a normal function of transplanted organs, BCVA was 20/100 in the right and 20/40 in the left eye. Fundoscopy revealed extended crystalline oxalate deposits and subretinal fibrosis. On OCT imaging, the retinal structure was severely impaired with subretinal hyper-reflective material, intraretinal cystic spaces, abundant retinal crystals and pronounced thinning of the outer nuclear layer (ONL; Figure 1). The thickness of the ONL, containing cell bodies of photoreceptors, is an important biomarker for retinal degeneration.
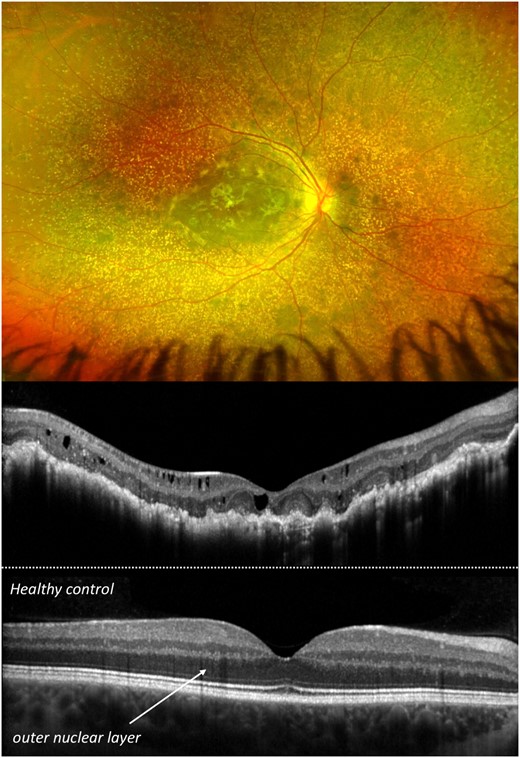
Pseudo-colour fundus image (top) and optical coherence tomography (OCT) (middle) of an infantile primary hyperoxaluria type 1 patient with severe retinal alterations and oxalate deposits. For comparison, OCT of a healthy control is shown (bottom).
RETINAL ALTERATIONS IN PH
The retinal phenotype of patients with PH1 has been documented in various studies; however, systematic investigations of patients with PH2 and PH3 are currently lacking [4, 6–8]. Extra-renal oxalosis has been described in PH2, indicating the need for studies on the retinal phenotype in these patients [4].
Patients with infantile PH1 usually present with severe oxalate deposition resulting in a variable degree of vision loss already at young age [6–8]. Potential explanations for these pronounced oxalate depositions include high oxalate levels before transplantation and a particularly low threshold for subretinal oxalate deposits at a young age and/or a more severe tissue response to such deposits. A further finding in infantile PH1 includes macular subretinal fibrosis. Retinal alterations potentially occur in succession with (i) crystalline deposits, (ii) focal hyperpigmentation and (iii) diffuse macular hyperpigmentation, which may finally lead to (iv) subretinal fibrosis [6].
In sharp contrast, the majority of patients with non-infantile PH1 present with mild or no retinal alterations, which do not significantly affect visual function. This includes small, drusen-like retinal changes that appear as hyper-reflective subretinal lesions on OCT and as hyper-autofluorescent alterations on autofluorescence imaging [6, 7].
REGRESSION OF RETINAL ALTERATIONS?
There is as yet no conclusive evidence regarding if and to what extent retinal oxalate deposition may regress over time. While some studies demonstrated reversal of retinal oxalate deposition after increased frequency of dialysis or (liver–)kidney transplantation, other studies observed no regression of retinal crystals [7, 8, 10]. Cases without crystals surrounding subretinal fibrosis and the lack of crystals within areas of diffuse pigmentation may indicate mechanisms for clearing oxalate deposits from the subretinal space [6]. To determine if and at what rate retinal oxalate deposition may reverse, as it has been described in other parts of the body (e.g. in bones), long-term studies using high-resolution retinal imaging will be crucial. Such studies may also inform whether or not monitoring of retinal PH1-related changes may be of value for assessing therapeutic efficacy. Reversal of subretinal fibrosis remains unlikely, and visual function is lost permanently in cases where OCT imaging reveals photoreceptor degeneration [8].
FUNDING
This work was supported by the Dr. Werner Jackstädt Foundation, Wuppertal, Germany (Grant S0134-10.22 to J.B.), the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC), and the German Research Foundation (DFG; Grant within the SFB/TRR 57 to B.H.). The views expressed are those of the authors and not necessarily those of the National Health Service (NHS), the NIHR or the Department of Health. The sponsor and funding organization had no role in the design or conduct of this research.
CONFLICT OF INTEREST STATEMENT
Johannes Birtel is a consultant of Alnylam Pharmaceuticals, Cambridge, MA, USA. Bernd Hoppe is now also an employee of Dicerna Pharmaceuticals, Cambridge, MA, USA. The other authors declare no conflicts of interest.


{kind=link}
Comments