





Abstract
The nexus of chronic kidney disease (CKD) and cardiovascular disease (CVD) amplifies the morbidity and mortality of CKD, emphasizing the need for defining and establishing therapeutic initiatives to modify and abrogate the progression of CKD and concomitant CV risks. In addition to the traditional CV risk factors, disturbances of mineral metabolism are specific risk factors that contribute to the excessive CV mortality in patients with CKD. These risk factors include dysregulations of circulating factors that modulate phosphate metabolism, including fibroblast growth factor 23 (FGF23) and soluble Klotho. Reduced circulating levels and suppressed renal Klotho expression may be associated with adverse outcomes in CKD patients. While elevated circulating concentrations or locally produced FGF23 in the strained heart exert prohypertrophic mechanisms on the myocardium, Klotho attenuates tissue fibrosis, progression of CKD, cardiomyopathy, endothelial dysfunction, vascular stiffness and vascular calcification. Mineralocorticoid receptor (MR) activation in nonclassical targets, mediated by aldosterone and other ligands, amplifies CVD in CKD. In concert, we detail how the interplay of elevated FGF23, activation of the MR and concomitant reductions of circulating Klotho in CKD may potentiate each other’s deleterious effects on the kidney and heart, thereby contributing to the initiation and progression of kidney and cardiac functional deterioration, acting through multipronged, albeit complementary, mechanistic pathways.
INTRODUCTION
Chronic kidney disease (CKD) is extremely common and has emerged as one of the leading noncommunicable causes of death worldwide. The Global Burden of Disease studies show that CKD has emerged as a leading cause of mortality worldwide [1, 2]. Furthermore, CKD is projected to increase in importance among the various global causes of death. Consequently there is urgency to implement measures that will facilitate the identification, monitoring and treatment of CKD. Implicit in this formulation is the urgent need to fully elucidate the pathogenetic mediators promoting CKD and to leverage these findings in order to develop preventive and therapeutic measures for the rational management of the global burden of CKD.
The nexus of CKD and cardiovascular disease (CVD) amplifies the morbidity and mortality of CKD, imposing increasing demands on health services. CVD is the leading cause of death and morbidity in people with CKD [3]. In 25- to 34-year-old patients with end-stage kidney disease (ESKD), annual mortality is increased 500- to 1000-fold and corresponds to that of the ∼85-year-old general population. Furthermore, CVD is the most common cause of mortality and morbidity in patients with CKD [4]. As an example, a recent study of adults with nondiabetic proteinuric primary nephrotic syndrome reported significantly higher hazard ratios (HRs) of adverse CV outcomes, including death {adjusted HR 1.34 [95% confidence interval (CI) 1.09–1.64]} at a mean age of 49 years compared with matched healthy controls without diabetes, diagnosed nephrotic syndrome or with proteinuria [5].
This relationship extends to pediatric patients as well. Even in children and adolescents (ages 0–19 years), the percentage of CVD-related deaths are exponentially higher in patients with CKD (34%) compared with the general age-matched population (3.6%) based on the more current US Renal Data System data, thus reconfirming that CVD remains the major cause of death in children with CKD [6]. Of interest, this relationship also extends to children and adolescents following successful kidney transplantation [7].
Because of the increasing prevalence of CKD and dialysis-dependent ESKD [1–4], the number of people at high risk of CV events will increase, with increasing demands on health services globally. In concert, these observations emphasize the need for defining and establishing therapeutic initiatives to modify and abrogate progression of CKD and concomitant CV risks. Clearly, addressing this unmet need constitutes a medical and societal priority.
In addition to the traditional CV risk factors [8], disturbances of mineral metabolism are specific risk factors that contribute to the excessive CV mortality in patients with CKD. These risk factors include dysregulation of circulating factors that modulate phosphate metabolism, including fibroblast growth factor 23 (FGF23) and soluble Klotho (sKL) [9].
In recent years, mineralocorticoid receptor (MR) activation, excess FGF23 and Klotho insufficiency have all emerged as important mediators of renal and CV injury and consequently constitute major catalysts for accelerating CKD progression. Most patients with CKD develop cardiac hypertrophy and fibrosis, which contribute to myocardial dysfunction, arrhythmias, heart failure and sudden cardiac death [4]. In this article we will review the mechanisms whereby MR activation and FGF23 promote cardiorenal injury. We will also consider the heretofore unappreciated intersection and interplay of MR activation with FGF23 and Klotho. Implicit in this intersection is the formulation that treatment with MR antagonists (MRAs) may dampen and offset both cascades and consequently serves to enhance the cardiorenoprotective efficacy of the current treatment paradigm.
FOUNDATIONAL BACKGROUND AND EXPERIMENTAL OBSERVATIONS OF FGF23 AND KLOTHO
FGF23
The plasma level of FGF23 is elevated in cardiac and renal failure and is associated with poor clinical prognosis for these disorders [9–13]. FGF23 is a bone-derived hormone that regulates phosphorus balance by enhancing urinary phosphorus excretion and inhibiting the synthesis of 1,25-dihydroxyvitamin D [1,25(OH)2D] [10]. The actions of FGF23 are mediated through FGF receptors (FGFRs) with the participation of its coreceptor α-Klotho, a membrane-bound protein mainly expressed in the kidney. Elevated plasma levels of FGF23 and reduced circulating levels of cleaved sKL are associated with both left ventricular hypertrophy (LVH) and the progression of CKD [9]. These adverse outcomes are attributed in part to FGF23-mediated effects to stimulate LVH by both direct [11] and indirect pathways, but the mechanisms whereby FGF23 promotes LVH remain controversial and may involve both renal and extrarenal effects [12–14].
Elevated FGF23 concentrations are strongly associated with higher left ventricular mass independent of blood pressure (BP) in both CKD patients and in subjects without CKD [11, 15–17]. Furthermore, FGF23 induces hypertrophy of cardiomyocytes in vitro and in vivo through FGFR-mediated activation in a Klotho-independent manner acting through the calcineurin–nuclear factor of activated T cell (NFAT) axis [11] and through disturbances of calcium (Ca) handling in cardiac cells [18].
Compelling evidence mitigating against direct effects of FGF23 on BP derives from studies in a model of X-linked hypophosphatemic rickets in mice with a missense mutation in the phosphate-regulating gene with homologies to endopeptidase on the X chromosome. This model is characterized by increased production and secretion of bone FGF23 and elevated FGF23 levels, but importantly occurring in the absence of hypertension (HTN) [19].
One indirect pathway potentially contributing to LVH entails the effects of FGF23 on renal sodium (Na) handling by its ability to directly upregulate the Na chloride cotransporter (NCC) in the distal tubule thereby promoting Na reabsorption with resultant volume expansion and cardiac hypertrophy [20]. However, this formulation requires confirmation by conducting studies in models with CKD, because the tested animals in the study by Andrukhova et al. [20] that were administered exogenous recombinant FGF23 had preserved renal function, the aldosterone levels were not elevated and, counterintuitively, reduced salt intake aggravated the HTN.
In experimental models with uremia and elevated FGF23 levels, the expression of the NCC was either reduced or did not differ from normal controls [21, 22], suggesting that factors independent of the prevailing concentrations of FGF23 may determine NCC activity in the distal tubule. Of interest, activation in FGF23 signaling was initially demonstrated by modifying the expression of the ‘with no lysine K/kinase’ pathway [23] and by genetically ablating the NCC gene [24]. Elevation of both FGF23 and aldosterone levels in NCC null mice (a model of mild volume depletion) suggested an aldosterone-dependent effect attributable to volume depletion, because this hormone was increased, and the increased FGF23 levels declined in response to treatment with the MRA eplerenone [24]. Mice exposed to the mineralocorticoid deoxycorticosterone acetate displayed elevated FGF23 levels, and in vitro osteoblastic cells exposed to aldosterone revealed increased fgf23 transcript levels via enhancement of store-operated Ca2+ entry. These effects were reversed by the MRAs spironolactone and eplerenone [25].
Clinical studies by Xu et al. [26] and our studies in nondialysis CKD patients [27] demonstrated positive correlations between the circulating FGF23 levels and the fractional excretion of Na, reduced plasma renin activity and increased aldosterone levels across FGF23 tertiles. There was no correlation of the FGF23 levels with BP. In concert, these observations argue against Na retention induced by FGF23 [26, 27].
FGF23 is regulated in an endocrine loop fashion, namely, it reduces serum phosphate, 1,25(OH)2D and parathyroid hormone (PTH) levels, and these factors in turn stimulate bone FGF23 expression (Figure 1). Activated vitamin D appears to be the most potent regulator of FGF23 by direct fgf23 gene transcription in the gene promoter region [28]. Nevertheless, FGF23 can also be regulated by PTH or Ca through pathways independent of 1,25(OH)2D and the vitamin D receptor (VDR), as demonstrated in VDR-ablated animals [29], as well as in patients with vitamin D–dependent rickets type 1 who are unable to synthesize 1,25(OH)2D [30]. The sympathetic nervous system can also stimulate FGF23 production in bone by β-adrenergic signaling pathways, thereby contributing to elevated circulating FGF23 levels [31]. Furthermore, changes in serum or dietary phosphate may also modulate FGF23 production by as yet undetermined mechanisms [32]. More recently, anemia and erythropoietin have been identified as stimulators of bone and bone marrow production of FGF23 [33] that may potentially have important clinical implications in the management of patients with CKD and their CV complications.
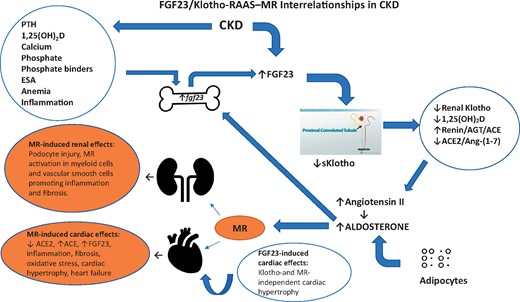
FGF23/Klotho RAAS–MR interrelationships in CKD. In CKD, bone FGF23 secretion is stimulated by various factors (left top oval shape) and medications resulting in higher circulating levels. Both Ang II and aldosterone also directly stimulate FGF23 secretion (thick blue arrow). Reduced renal Klotho and increased blood-borne FGF23 result in reduced 1,25(OH)2D, increased Ang II and reduced ACE2/Ang-(1–7) (right oval shape). The displayed interrelationships, acting in concert with MR activation, play complimentary roles in promoting the initiation and progression of renal and cardiac damage (left bottom orange ovals). ESA, erythropoietin-stimulating agent.
Klotho
α-Klotho (hereinafter simply referred to as Klotho) is expressed mostly in the distal and proximal tubules of the kidney but also in other tissues, including the choroid plexus and parathyroid glands [34]. These cells also express FGFR1c, FGFR3c and/or FGFR4, which can form binary complexes with Klotho and thus are considered targets of FGF23 [35]. The full-length, membrane form of Klotho consists of two similar domains (termed KL1 and KL2) that act as coreceptors with FGFRs to bind FGF23 and mediate phosphaturia [9, 32, 34–36]. Klotho is cleaved by the A-disintegrin and metalloproteinase domain-containing proteins 10 and 17 (ADAM10 and ADAM17, respectively) [37] and released from the kidney into the circulation as a full-length protein or as KL1 and KL2 fragments; these circulating isoforms are collectively called sKl [32, 34].
sKL, simply referred to as a cleaved full fragment of Klotho protein, can not only weakly activate FGFRs to transduce the FGF23 signaling pathway, but also functions as an enzyme and hormonal substance and is the main functional form in the circulation, cerebrospinal fluid and urine of mammals, and exerts multiple biological actions on distant organs [32, 34, 38]. sKL may exert phosphaturic properties as an FGF23-independent hormone, as evidenced following its injection in the FGF23−/− mouse and in cultured cells devoid of FGF23 [39].
Klotho expression is stimulated by VDR activators that bind to vitamin D–responsive elements in the Klotho gene promoter [40]. Erythropoietin, rapamycin, statins, fosinopril and losartan are all reported to increase Klotho messenger RNA (mRNA) expression, but the underlying mechanisms have not yet been elucidated [40, 41]. Factors that suppress Klotho gene transcription include angiotensin II (Ang II), FGF23, inflammation, oxidative stress, uremic toxins, epigenetic methylation and hypoxia, frequently involving upregulation of the Wnt/β-catenin pathway and transforming growth factor-β (TGF-β) [41, 42]. Klotho attenuates HTN and exerts renoprotective properties demonstrated in several preclinical models [42, 43].
Although a hormonal function of Klotho has been demonstrated [39], a specific receptor for sKL has not been identified. [43]. Recent studies have shed further light on this gap in knowledge and have identified monosialogangliosides GM1 and GM3 present in lipid rafts as receptors for sKL [44]. sKL may have the ability to alter the function and abundance of membrane glycoproteins by cleaving terminal sugars from sugar chains through a putative glycosidase activity [45]. Regardless, we must conclude that some of the hormonal and enzymatic functions of Klotho are still a matter of controversy [46].
Because the kidney is the main source for circulating Klotho, it is not surprising that CKD and ESKD patients manifest low renal Klotho expression and low levels of circulating Klotho. Patients on maintenance dialysis, however, may still display detectable circulating Klotho [47], thereby suggesting that renal Klotho expression is not completely suppressed. Alternatively, Klotho may derive from extrarenal sources, although to date its origin has not been elucidated. The very early decline of serum Klotho levels in patients with CKD [48] suggests that this change precedes the increase in serum FGF23 and PTH and reduction in 1,25(OH)2D. Because of a comparable change in levels of renal Klotho expression to those of serum and urinary Klotho protein, serum and urinary Klotho can serve as surrogate markers for renal Klotho [49].
Detection of Klotho in the urine has been demonstrated by some investigators [50] but not by others [46], and reduced circulating levels and suppressed renal Klotho expression may [51] or may not [52] be associated with adverse outcomes in CKD patients. Discrepancies in the results may be attributable in part to technical issues that complicate the handling of the specimens and the high variability in results obtained using commercial enzyme-linked immunosorbent assays (ELISAs) [53]. Importantly, immunoprecipitation–immunoblot methodology compared with ELISA exhibited a stronger direct correlation with estimated glomerular filtration rate (eGFR), better recovery (capture) of added exogenous Klotho, less susceptibility to variability from sample additives (protease inhibitors) and much better differentiation across different kidney disease groups, including acute kidney injury (AKI) and CKD, as compared with healthy volunteers. Consequently, data of Klotho levels or tissue expression from clinical and experimental studies need to be interpreted with caution [37].
Klotho attenuates tissue fibrosis, progression of CKD, cardiomyopathy and vascular calcification [37]. In contrast, MR expressed in vascular smooth muscle cells (VSMCs), when stimulated by aldosterone, triggers osteo-/chondrogenic signaling, leading to expression of osteogenic transcription factors and enzymes and subsequent mineralization [54]. In Klotho hypomorphic (kl/kl) mice that develop hyperphosphatemia, renal Na loss with hyperaldosteronism and vascular calcification, the MR blockade with spironolactone reduced vascular calcification, an effect also observed following adrenalectomy [55]. The inhibitory effect of the MRA in the adrenalectomized kl/kl mice highlights the potential significance of vascular auto-/paracrine MR activation leading to aortic osteo-/chondrogenic transformation independent of circulating aldosterone levels. These effects were reproduced by MR silencing in vitro, confirming a crucial role for ligand-independent MR activation in vascular calcification [55, 56] that also has been reported in uremic rats [57].
Vascular calcification in CKD results mainly from impaired renal phosphate elimination with subsequent hyperphosphatemia and precipitation of Ca–phosphate [58]. Elevated phosphate concentrations were associated with upregulation of vascular CYP11B2 (aldosterone synthase) required for the triggering of vascular osteo-/chondrogenic reprograming in the kl/kl mice, in mice with experimental CKD and in coronary arteries of patients with CKD [55], emphasizing the contributing role of phosphate dysregulation to vascular calcification in CKD [58]. The above-mentioned observations in CKD and non-CKD models suggest an auto-/paracrine crosstalk between the vascular aldosterone synthase–dependent phosphate-induced procalcification genes and the activated vascular MR act to promote vascular calcification independent of circulating aldosterone or Klotho levels.
The Klotho-deficient model also manifests enhanced cardiac and renal fibrosis involving upregulation of the transient receptor potential channel 6–mediated abnormal Ca2+ signaling that can be attenuated by sKL administration [59].
Klotho deficiency also promotes defective endothelial function and impaired vasculogenesis and a reduced circulating sKL level was a significant determinant of vascular stiffness [60]. Klotho protein protects vascular endothelium by inhibition of endothelial inflammation and reduction of oxidative stress. Klotho deficiency–associated uremic cardiomyopathy and oxidative stress–induced renal injury and cardiopulmonary damage can be prevented or attenuated by supplementation of exogenous sKL [61, 62].
In concert, the heretofore described interplay of FGF23 and Klotho may promote CV and renal injury. Specifically, supraphysiological levels of FGF23 and concomitant reductions of circulating Klotho in CKD may potentiate each other’s deleterious effects on the kidney and heart, thereby contributing to the initiation and progression of kidney and cardiac functional deterioration via multipronged, albeit complementary, mechanistic pathways.
FUTURE CONSIDERATIONS: KLOTHO REACTIVATION AND VASCULAR DYSFUNCTION
A consideration of Klotho and the vasculature discloses many associations of interest. Klotho-deficient mice display endothelial dysfunction [63], arterial stiffening [64] and HTN [65].
In the context of the expansive preclinical dataset subtending the effects of Klotho, it is appropriate to inquire if these findings may be relevant in the clinical setting. The following examples support this formulation:
In adults with mild CKD, serum Klotho levels were associated with indicators of vascular dysfunction such as ankle–brachial pulse wave velocity, a marker of arterial stiffness and a major determinant of CV prognosis [60]. However, it remains unclear whether circulating Klotho levels are significantly associated with adverse CV outcomes in pre-dialysis patients with CKD [66]. Consequently it will be of interest to conduct future investigations interrogating whether therapeutic interventions to either maintain or elevate serum Klotho levels could improve arterial stiffness and improve CV outcomes in CKD patients.
In children, subclinical evidence of CVD as well as endothelial dysfunction may be detected as early as the first decade of life in patients undergoing maintenance dialysis and in predialysis CKD Stages 2–4 as well [67]. Circulating levels of Klotho were significantly higher and were associated with higher vitamin D levels and the administration of angiotensin-converting enzyme (ACE) inhibitors in children with CKD [68]. Furthermore, in children with ESKD undergoing peritoneal dialysis, Klotho levels, although reduced compared with normal controls, were readily detected and remained virtually unchanged throughout 1 year of treatment with the VDR activator calcitriol [47].
Klotho insufficiency and hyperaldosteronism are associated with vascular calcification and endothelial dysfunction [56] that can be attenuated by MRAs.
Mechanistically, MRAs suppress aldosterone-mediated osteo-/chondrogenic signaling, heralding the progression of vascular mineralization in endothelial cells [57]; Klotho protein suppresses tumor necrosis factor α (TNF-α)-induced expression of adhesion molecules and nuclear factor–κB (NF-κB) activation that promotes endothelial inflammation [69]; and the Klotho gene is stimulated by VDR activators [40] and VDR activator administration robustly increased circulating Klotho levels and decreased aortic calcification [70].
Thus the combined administration of an MRA and a VDR activator may provide CV protection, including attenuation of endothelial dysfunction and vascular stiffness by reactivation of endogenous Klotho.
Collectively the above-cited studies disclose, and indeed commend, many opportunities for implementing long-term clinical studies in order to evaluate the potential beneficial effects of maintaining appropriate sKL levels in the prevention or possibly attenuation of endothelial dysfunction and vascular stiffness in patients with CKD.
FGF23 AND KLOTHO INTERACTIONS WITH VITAMIN D AND THE RENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM (RAAS)
Chronic activation of the RAAS promotes and maintains diverse clinical disorders including congestive heart failure, HTN and CKD. Elevated circulating and tissue Ang II and aldosterone levels promote a profibrotic, proinflammatory and prohypertrophic milieu that accelerates myocardial remodeling and dysfunction in both cardiac and renal tissues.
Experimental observations have delineated simultaneous activation of the FGF23–Klotho endocrine axis and the RAAS by diverse mechanisms [12, 13, 34, 71]. FGF23 suppresses 1-α hydroxylase and reduces 1,25(OH)2D (which normally represses the renin gene) [72], resulting in increased renin expression and activation of ACE/Ang II and/or reduction of the vasoprotective ACE2/Ang-(1–7) pathway [73, 74]; suppression of ACE2 may contribute to cardiac remodeling and hypertrophy [75]. The potential influence of the ACE2/Ang-(1–7)/Mas-R axis in the control of aldosterone remains incompletely elucidated. Whereas Ang-(1–7) has no direct agonistic activity on aldosterone secretion, the heptapeptide has been reported to act as a negative modulator of aldosterone secretion in vitro and in vivo [76]. Of interest, administration of recombinant FGF23 reverses the induction of renal ACE2 and prevents angiotensin receptor blocker (ARB)-induced Klotho upregulation in murine CKD [77].
Aldosterone can suppress ACE2 transcription and increase ACE expression in the heart and that action can be blocked with the MRA eplerenone [78]. Aldosterone and Ang II also induce upregulation of fgf23 mRNA expression in animal cardiac myocytes, suggesting that the local intracardiac RAAS can induce FGF23 activation [71]. Conversely, FGF23 can induce angiotensinogen (AGT) expression and in turn Ang II stimulates FGF23 production [13, 71, 79] and its expression with the coreceptor Klotho in the heart [71], which normally does not express FGF23 or Klotho at physiologically relevant levels [73]. All the components required for Ang II production are normally present in the heart, including aldosterone [80]. Upregulated cardiac RAAS components including renin and AGT have been demonstrated in the kidney and the myocardium of uremic animals with cardiac hypertrophy and fibrosis [13, 71, 81, 82]. Finally, in the human heart, in addition to upregulation of FGF23 and FGFR4, upregulation of AGT and Ang II–driven pathways including TGF-β, COL1A2, COL3A1 and other genes promoting fibrosis were identified in the myocardial autopsy samples derived from young, deceased patients with long-standing CKD and with evidence of LVH and cardiac fibrosis [71]. The elevated AGT expression correlated positively with the degree of cardiac fibrosis and negatively with the cardiac Klotho protein-positive area quantified by immunohistochemistry, whereas Klotho mRNA was not detected [71]. The latter strongly indicates that the source of the cardiac Klotho protein came from the circulation as the soluble form.
The additive effects of locally activated RAAS and FGF23 with reduced Klotho to those of the elevated bone-derived circulating FGF23 levels jointly promote cardiac fibrosis in CKD. Aldosterone upregulates bone expression of FGF23 [25] and increases circulating FGF23 levels, with additive effects on the heart and kidney. Since FGF23 inhibits 1-α hydroxylase activity [83], the resultant reduced 1,25(OH)2D levels and tissue VDR expression favor renin upregulation with activation of the ACE/Ang II arm of the RAAS [81, 82] and reduction of ACE2/Ang-(1–7) [73, 74], thereby contributing to both renal damage and myocardial hypertrophy. Notably, ACE2 gene expression was reduced in FGF23 transgenic mice [84], adding another likely mechanism by which FGF23 contributes to cardiorenal functional deterioration in CKD. In contrast, VDR activation by paricalcitol increases renal ACE2 and Ang-(1–7) expression [85], suppresses the expression of RAAS genes in the kidney and the heart, improves kidney function, decreases proteinuria and attenuates cardiac hypertrophy in experimental CKD [81, 82] with minimal or no effects on BP.
Obesity, a morbid state affecting >1.9 billion adults and 340 million children worldwide [86], is an additional complication linked to CV complications including HTN and CKD. Aldosterone levels are usually elevated in obese individuals, attributable to leptin-mediated direct transcriptional upregulation of aldosterone synthase CYP11B2 in adrenal glomerulosa cells via Ca-dependent mechanisms [87], thereby contributing to HTN [88]. Importantly, MR blockade with spironolactone blunts leptin-mediated endothelial dysfunction and increased cardiac fibrosis in female mice [87] and reduces BP in obese patients with HTN [89]. Leptin also directly stimulates FGF23 synthesis in bone cells in ob/ob mice [82, 90], suggesting that leptin may be an endocrine or paracrine regulator of FGF23 production in humans. Thus aldosterone and leptin concurrently upregulate bone FGF23 production, thereby contributing to elevated circulating levels of FGF23.
Finally, Klotho associations with the RAAS constitute important determinants of kidney and cardiac integrity. Aldosterone inhibits Klotho gene transcription by inhibiting acetylation of the gene promoter region, resulting in reduced kidney Klotho protein expression levels and increased expression of the renal fibrosis gene fibronectin 1 in experimental CKD [91]. Klotho is downregulated by Ang II, an effect that can be attenuated by AT1R blockade [92], whereas the absence of Klotho upregulates CYP11B2, responsible for aldosterone synthesis [93], which in turn induces FGF23 transcription by the osteoblast [25]. Conversely, Klotho downregulates RAAS activity as a result of suppressing the stimulatory effects of the Wnt/β-catenin pathway on the RAAS. Collectively, these effects of Klotho result in amelioration of kidney injury and fibrosis and attenuation of HTN in experimental CKD models [94].
In summary, the intricate intersections and interrelationships between an activated RAAS (including aldosterone), excess FGF23, insufficient Klotho and suboptimal 1,25(OH)2D act in concert to promote the initiation and progression of renal and cardiac injury in CKD (Figure 1).
EFFECTS OF ALDOSTERONE AND MR ACTIVATION: A REMARKABLE PARADIGM SHIFT OF OUR PRESENT CONCEPTS
Aldosterone plays a major role in the control of both BP and extracellular volume homeostasis by stimulating renal Na reabsorption and potassium excretion. More than 30 years ago, our understanding of the interaction between aldosterone and the MR differed markedly from our present concepts. It is now accepted that Ang II does not constitute the major driver of aldosterone secretion, aldosterone is only one of the physiological ligands for MR and although aldosterone’s Na-retaining effects are relevant in defending volume homeostasis in the setting of hypovolemia and the control of BP, they are not the primary determinants of HTN. Rather, aldosterone raises BP primarily by actions on the vasculature and central nervous system through MR-dependent and -independent mechanisms.
In addition to aldosterone’s classic effects subserving renal electrolyte excretion, a series of parallel studies have documented a wider, nonepithelial role for aldosterone in promoting inflammation, collagen formation, fibrosis and necrosis. More than 30 years ago, Brilla et al. [95] demonstrated that chronically elevated aldosterone, when coupled with salt loading, promoted cardiac fibrosis [96]. In contrast, spironolactone, an MRA, conferred a protective effect against the development of cardiac fibrosis when administered to uninephrectomized rats receiving both a high salt diet and aldosterone. In concert, these findings suggested that MR activation could promote cardiac fibrosis.
MR
The MR is located intracellularly in many tissues: endothelial cells, VSMCs, adipocytes, fibroblasts, cardiomyocytes and macrophages [97, 98]. In the kidney, the MR has been found in the distal tubules, collecting duct, podocytes, fibroblasts and mesangial cells [99]. While aldosterone is the principal steroid for MR activation, cortisol and other ligands such as Ras-related botulism toxin substrate 1 (Rac1) (see below) as well as diseases (i.e. heart failure and albuminuria) can activate MR [100]. After aldosterone binds with the MR, the new complex attaches to DNA in the nucleus, causing transcription of genes [101].
In many pathological conditions, including in diabetes and CKD (see Table 1), overactivation of the MR promotes many detrimental renal effects: injury of podocytes and albuminuria (even in the absence of HTN), reduced renal blood flow, AKI and increased expression of proinflammatory cytokines and profibrotic proteins [98–100]. MR overactivation can initiate and cause progression of chronic renal interstitial inflammation and consequently fibrosis (Figure 1).
The interrelationship of RAAS cascade and FGF23
|
|
The interrelationship of RAAS cascade and FGF23
|
|
Rac1
Rac1 is a member of the Rac group of guanosine triphosphate phosphohydrolases, located intracellularly and acting as a second messenger. Rac1 is a regulator of cellular signaling, cytoskeletal integrity and cell growth. Rac1 can activate MR and multiple kinases. It controls gene expression, inflammation and fibrosis [102].
Of interest, Rac1 has been shown to activate MR without aldosterone, increase epithelial Na channel activity and generate reactive oxygen species (ROS) [103]. In addition, salt sensitivity was experimentally transferred through Rac1 [104]. Upregulation of Rac1 leads to podocyte injury (see below) and, importantly, potentiation of MR and aldosterone actions. Rac1 plays a role in podocyte injury, albuminuria and nephrotic syndrome and promotes ROS generation and TGF-β signaling, all posing a risk for renal interstitial inflammation and fibrosis [105, 106]. Rac1 causes podocyte injury and detachment, leading to glomerulosclerosis and proteinuria, and its activation was observed in kidney biopsies of patients with proteinuric nephrotic syndrome [107].
Recently Rac1 has been proposed to constitute a therapeutic target for management of nephrotic syndrome [108]. Urinary Rac1 was elevated in focal segmental glomerulosclerosis and diabetic kidney disease (DKD) and was suggested, in addition to a target, as a new biomarker for evaluating therapy [109].
Rac1 is also involved in the regulation of bone FGF23 transcription by modulating the actin cytoskeleton restructuring of the osteoblast. Treatment of osteoblasts with a Rac1 inhibitor significantly reduced 1,25(OH)2D-mediated FGF23 transcription, supporting the notion that Rac1 signaling is essential for the 1,25(OH)2D3-induced production of FGF23 [110]. Thus Rac1 may constitute another modulator in the intricate cross-talk between the bone and the kidney.
MECHANISMS LINKING ALDOSTERONE WITH MR, RAC1, RAAS AND FGF23 ACTIVATION THEREBY INDUCING INFLAMMATION AND FIBROSIS: A SYNTHESIS
The initiation of chronic inflammation by aldosterone, MR and Rac1 and its progression to glomerular and interstitial fibrosis is a complex process and only key aspects will be discussed.
Fibrosis, defined as an excessive accumulation of extracellular matrix (i.e. collagen and fibronectin), is caused by chronic tissue injury, chronic inflammation or trauma [111]. As mentioned earlier, reduced renal synthesis of 1,25(OH)2D by FGF23 and decreased kidney and myocardial VDR expression in CKD is associated with RAAS upregulation, cardiac hypertrophy, glomerulotubular damage and fibrosis [12, 81, 82, 85]. A functional VDR has been identified in primary human cardiac fibroblasts [112]. The VDR activators calcitriol and paricalcitol downregulate not only the RAAS, but also other gene products involved in promoting cardiorenal inflammation and fibrogenesis such as TGF-β, vascular endothelial growth factor, inflammatory leukocytes (CD45) and ADAM secretases, resulting in attenuation of cardiac and renal inflammation, fibrosis and dysfunction [12, 81, 113]. Additionally, VDR exerts direct inhibition of NFAT target genes [11, 12], providing an additional explanation for the cardioprotective properties provided by VDR activator administration despite cardiac FGFR activation and persistently elevated FGF23 levels [12]. Finally, VDR activators, by upregulating Klotho [40], further contribute to suppression of the RAAS.
Aldosterone and/or MR activation initiates the inflammatory cycle with the generation of ROS by nicotinamide adenine dinucleotide phosphate in the mitochondria, potentiated by a high salt intake via Rac1 [114]. Aldosterone can also directly, via the AT1 receptor, initiate ROS production and stimulate NF-κB and serum glucocorticoid kinase 1 activity, facilitating inflammation and fibrosis [115].
Macrophages from bone marrow and tissue, attracted by tissue injury, apoptotic cells and cytokines, play a central role in inflammation and are the major producer of TGF-β, instrumental in the generation of fibrosis [111, 115].
Major initial inflammatory pathways include Nod-like receptors (NLRs). NLRs are immune complexes sensing intracellular pathogens or toxic signals. They are also referred to as inflammasomes and as a group of proteins activating intracellular caspase-1 with release of cytokines [interleukin-1β (IL-1β), IL-18] [116]. The best described inflammasome is NLR family pyrin domain containing 3, found with increased glomerular expression in renal biopsies in DKD and correlating with the degree of albuminuria [117].
CKD constitutes a state of systemic inflammation and is identified as a well-established risk factor of morbidity and mortality in CKD [117] and is partly associated with disturbances of FGF23 metabolism [119]. While pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β are potent inducers of FGF23 production, excess levels of circulating FGF23 increase the production of various cytokines in diverse tissues. FGF23, acting via FGFR4/calcineurin/NFAT signaling, induces the expression of inflammatory cytokines IL-6 and C-reactive protein in hepatocytes, which lack Klotho, with the consequent elevated serum levels of these cytokines [120]. FGF23 can also target bronchial epithelial cells with subsequent upregulation of IL-1β and IL-8 in patients with chronic lung disorders and FGF23 may target peritoneal macrophages to upregulate TNF-α production [119]. Concomitantly, IL-6 elevates the transcription and synthesis of FGF23 by bone. The latter can be attenuated by IL-6 deletion, thereby demonstrating that IL-6 is a potent stimulator of FGF23 in CKD [121]. Notably, aldosterone administration causes increased expression of perivascular IL-6 and inflammation in the kidney that can be prevented by MRA administration [122]. Furthermore, aldosterone-independent mechanisms involving Rac1-mediated MR activation can be prevented by the MRA eplerenone without changes in systemic aldosterone levels, identifying MR as a therapeutic target in CKD [123]. In summary, it is apparent that MR activation, inflammation, FGF23 secretion and reduced Klotho activation constitute a potent complementary cascade in CKD (Figure 1) and that interdiction of these mechanistic arms is desirable and now feasible. It is now a propitious time to initiate further studies with MRA intervention and Klotho reactivation to determine whether treatment with these novel modalities can attenuate this deleterious cascade (Figure 2).
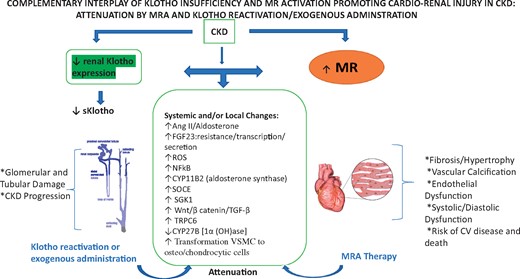
Complementary interplay of Klotho insufficiency and MR activation promoting cardiorenal injury in CKD: attenuation by an MRA and by Klotho reactivation or exogenous administration. In CKD, Klotho insufficiency (left panel) and MR activation (right panel) act on different cell types and through multipronged and complementary systemic and local molecular and signaling mechanisms (midpanel) to promote cardiorenal injury. As detailed in the narrative, Klotho reactivation and/or Klotho supplementation and MRA administration (blue curved arrows) may potentially interdict and attenuate these several cascades that promote cardiorenal injury. SOCE, store-operated Ca entry; SGK1, serum/glucocorticoid regulated kinase 1.
MR OVERACTIVATION AS A DETERMINANT OF CKD PROGRESSION
Increasing evidence over the past decade has implicated pathophysiological overactivation of the MR as a determinant of progression of CKD and its associated morbidity via the above-cited mechanisms of promoting inflammation and fibrosis. This formulation provides a rationale for implementing clinical trials to elucidate the role of blockade of the MR as a novel treatment paradigm to retard the progression of CKD [124–126].
Until recently, there have been no long-term studies on whether MRAs can reduce the rate of progression of CKD [123]. As we have discussed in an in-depth review [127]. a few short-duration clinical trials with MRAs have been conducted that support this hypothesis. The ALdosterone Antagonist Chronic HEModialysis Interventional Survival Trial (spironolactone 25 mg/day; NCT01848639) [127] is ongoing and the Pilot Trial of Hemodialysis Patient Aldosterone antagoniSm With Eplerenone NCT01650012) [128], a pilot, randomized, double-blind, clinical trial studying eplerenone (50 mg/day), has concluded [reviewed in 129].
In contrast to the above-cited studies utilizing steroidal MRAs, the novel nonsteroidal MRA finerenone has recently been shown to slow progression of CKD. Based on the biological plausibility of cardiorenal benefit, two Phase III clinical trials, the FInerenone in reducing kiDnEy faiLure and dIsease prOgression in DKD (FIDELIO-DKD) and FInerenone in reducinG cArdiovascular moRtality and mOrbidity in DKD studies, were initiated in the fall of 2015. The rationale of the trials was to demonstrate and consequently provide new treatment opportunities for improving cardiovascular and renal outcomes in patients with CKD in type 2 diabetes (T2D) [127].
The results of the first of these trials, the FIDELIO-DKD study, were presented at the recent 2020 American Society of Nephrology Annual Meeting and simultaneously published in the New England Journal of Medicine [130]. In summary, patients with CKD and T2D who were treated with finerenone manifested a lower risk of a primary outcome event (kidney failure, a sustained decrease of ≥40% in the eGFR from baseline or death from renal causes) than patients in the comparator placebo arm. In addition to the primary endpoints, patients in the finerenone group also manifested a lower risk of a key secondary outcome event (defined as death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke or hospitalization for heart failure) [130].
CONCLUSIONS
In addition to the traditional CV risk factors and disturbances of mineral metabolism that contribute to the excessive CV mortality in patients with CKD, additional enablers of CV risk include dysregulation of circulating factors that modulate phosphate metabolism, including FGF23 and sKL. In recent years a wide body of experimental evidence has emerged indicating MR activation, excess FGF23 and Klotho insufficiency are important mediators of renal and CV injury and consequently constitute major catalysts for accelerating CKD progression. In this article we have reviewed the mechanisms whereby both MR activation and excess FGF23 promote cardiorenal injury. We have leveraged these findings to highlight and underscore the heretofore unappreciated intersection and interplay of MR activation and alterations of the FGF23/Klotho axis.
To cite a few examples: the administration of recombinant FGF23 reverses the induction of renal ACE2 and prevents ARB-induced Klotho upregulation in murine CKD. Conversely, FGF23 can induce AGT expression and, in turn, Ang II stimulates FGF23 production and its expression with the coreceptor Klotho in the heart, which normally does not express FGF23 or Klotho in physiologically relevant levels.
We propose that when acting in tandem, these parallel endocrine cascades constitute a ‘force multiplier’ to accelerate and exacerbate the trajectory to renal and cardiovascular injury. Implicit in this intersection is the formulation that treatment with MRAs may oppose and dampen these interconnected mediators and consequently serve to enhance the cardiorenoprotective efficacy of current and newly available treatment paradigms.
ACKNOWLEDGEMENTS
M.E. reports personal fees from Alnylam Pharmaceuticals, Bayer and Vifor Pharma outside the submitted article. M.E. received no personal funding for this submitted article.
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
Comments