





ABSTRACT
Anaemia is common in chronic kidney disease (CKD) and assessment of the risks and benefits of new therapies is important.
The Anaemia Study in CKD: Erythropoiesis via a Novel prolyl hydroxylase inhibitor Daprodustat-Non-Dialysis (ASCEND-ND) trial includes adult patients with CKD Stages 3–5, not using erythropoiesis-stimulating agents (ESAs) with screening haemoglobin (Hb) 8–10 g/dL or receiving ESAs with screening Hb of 8–12 g/dL. Participants were randomized to daprodustat or darbepoetin alfa (1:1) in an open-label trial (steering committee- and sponsor-blinded), with blinded endpoint assessment. The co-primary endpoints are mean change in Hb between baseline and evaluation period (average over Weeks 28–52) and time to first adjudicated major adverse cardiovascular (CV) event. Baseline characteristics were compared with those of participants in similar anaemia trials.
Overall, 3872 patients were randomized from 39 countries (median age 67 years, 56% female, 56% White, 27% Asian and 10% Black). The median baseline Hb was 9.9 g/dL, blood pressure was 135/74 mmHg and estimated glomerular filtration rate was 18 mL/min/1.73 m2. Among randomized patients, 53% were ESA non-users, 57% had diabetes and 37% had a history of CV disease. At baseline, 61% of participants were using renin–angiotensin system blockers, 55% were taking statins and 49% were taking oral iron. Baseline demographics were similar to those in other large non-dialysis anaemia trials.
ASCEND-ND will define the efficacy and safety of daprodustat compared with darbepoetin alfa in the treatment of patients with anaemia associated with CKD not on dialysis.
What is already known about this subject?
Anaemia is a common complication of chronic kidney disease (CKD).
Treatment of anaemia with erythropoiesis-stimulating agents (ESAs) has become the standard of care for patients with CKD complicated by anaemia; however, ESAs may be associated with adverse effects on some cardiovascular (CV) outcomes.
Daprodustat is a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI) and it is important to compare the haematological efficacy, and the CV safety and efficacy of daprodustat with darbepoetin alfa in CKD patients not requiring dialysis (CKD-ND).
What this study adds?
The Anaemia Study in CKD: Erythropoiesis via a Novel PHI Daprodustat-Non-Dialysis (ASCEND-ND) trial is a global, randomized, open-label (steering committee- and sponsor-blinded), parallel-group, active-controlled, event-driven Phase 3 trial designed to demonstrate whether daprodustat is non-inferior to the comparator ESA darbepoetin alfa for two co-primary endpoints: haemoglobin efficacy and CV safety in CKD-ND patients.
ASCEND-ND is one of the largest anaemia studies in ND patients (N = 3872) being performed in 39 countries across Europe, North America, Latin America and Asia Pacific region. Baseline characteristics were similar to those of patients enrolled in other large CV outcome trials, thus supporting the generalizability of this study population.
What impact this may have on practice or policy?
This study will determine the efficacy and safety of daprodustat in CKD-ND patients and the large and diverse study population will help ensure the clinical applicability of the results.
If daprodustat is non-inferior to ESAs, it may provide an alternative oral dosing option to existing treatments.
INTRODUCTION
Anaemia frequently accompanies advanced chronic kidney disease (CKD), mainly affecting patients with kidney failure requiring dialysis [1]. However, it is however, also common in patients with Stage 4 or 5 CKD [i.e. estimated glomerular filtration rate (eGFR) <30 mL/min/1.73 m2] and interventions are often required to increase or maintain haemoglobin (Hb) levels in this population. Furthermore, most people with kidney failure will develop anaemia before they start dialysis and anaemia is associated with poor quality of life and high rates of mortality and morbidity [2, 3]. Data regarding the risks and benefits of treatments for anaemia in people with CKD not requiring dialysis (CKD-ND) are therefore important.
Although treatment with recombinant human erythropoietin (rhEPO) and its analogues was initially focussed on patients with kidney failure requiring dialysis, it was rapidly extended to patients with earlier stages of CKD and has become the standard of care in this population [4–6]. The effect of normalizing Hb on cardiovascular (CV) and other outcomes in patients with CKD has been assessed in large, randomized trials, but these did not demonstrate evidence of benefit. Indeed, some of these trials showed an increase in specific CV events [7–10] with higher Hb targets, possibly related to high doses of exogenous rhEPO and its analogues [11–13].
Hypoxia-inducible factor prolyl hydroxylase inhibitors (HIF-PHIs) have been developed to stimulate erythropoiesis through the inhibition of HIF-prolyl hydroxylase (PHD) enzymes [14]. It has been suggested that these oral agents provide a more physiological approach to treat anaemia. HIF-PHIs may have particular advantages for patients with CKD-ND along with patients receiving peritoneal dialysis or kidney transplant recipients for its ease of use (oral therapy), as well as with the removal of injection burden and cold storage requirements with current therapies.
Daprodustat (previously GSK1278863) is a HIF-PHI that is being developed to treat anaemia in CKD. Initial Phase 3 clinical trials in Japan demonstrated that daprodustat is effective at correcting and maintaining Hb and it appears to be well-tolerated [15].
Here we describe the ASCEND-ND (Anaemia Studies in CKD: Erythropoiesis via a Novel PHI Daprodustat-Non-Dialysis) Phase 3 trial, designed to assess the efficacy and safety of daprodustat compared with darbepoetin on Hb and CV outcomes and examine the baseline characteristics of randomized participants.
MATERIALS AND METHODS
Study design
ASCEND-ND is a global, randomized, open-label (steering committee- and sponsor-blinded), parallel-group, active-controlled, event-driven Phase 3 trial comparing the efficacy and safety of daprodustat with darbepoetin alfa in patients with CKD-ND (ClinicalTrials.gov: NCT02876835; EudraCT: 2016-000542-65). The study was approved by the ethics committees or institutional review boards at each participating institution and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki.
The ASCEND-ND study timeline was divided into four periods: a screening period, a placebo run-in period, a treatment period and a follow-up period (Figure 1). The 4-week screening period permitted assessment of eligibility based on clinical and laboratory assessments, while the 4-week run-in period was used to establish the tolerance of and adherence to placebo tablets and study procedures. Participants receiving prior erythropoiesis-stimulating agents (ESAs) continued these during the screening and run-in periods.
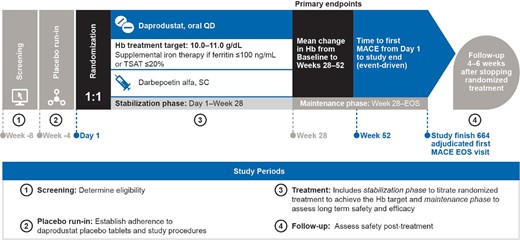
ASCEND-ND study design. Serum and plasma samples are collected at baseline, Week 28 and Week 52 for future analysis of biomarkers of CV risk and iron metabolism. EOS, end of study; Hb, haemoglobin; MACE, major adverse cardiovascular event; QD, once daily; SC, subcutaneous; TSAT, transferrin saturation.
Participants were randomized to oral daprodustat or subcutaneous darbepoetin alfa (1:1) if they successfully completed the run-in period. Thereafter, the treatment period was divided into a stabilization phase, from Day 1 to Week 28, with dose titration to achieve the prespecified Hb target range (10–11 g/dL) and a maintenance phase to maintain Hb, from Week 28 to the end of the study; randomized treatment was discontinued at the end-of-study visit.
Participants attended routine follow-up visits at least every 4 weeks during Year 1 of the study and at least every 12 weeks thereafter until the end-of-study visit was completed. Participants were asked to complete an off-treatment follow-up visit after discontinuing randomized treatment. Serum, plasma and urine samples were collected at baseline, Week 28 and Week 52 for future analysis of biomarkers.
Eligibility criteria
Eligibility was determined at Week −8, with a subset of entry criteria reconfirmed at Day 1 (randomization). Eligible patients had CKD Stage 3–5, were not currently receiving dialysis or scheduled to start dialysis within 90 days after study start, had either a screening Hb 8–10 g/dL if they were not receiving ESAs or a screening Hb of 8–12 g/dL if they were receiving ESAs, demonstrated adherence to daprodustat placebo tablets during the run-in period, were iron-replete (ferritin >100 ng/mL and transferrin saturation >20%) and were able to provide informed consent. The key inclusion and exclusion criteria are provided in Table 1 and complete entry criteria are outlined in Supplementary data, Table S1.
Key inclusion and exclusion criteria
| Inclusion criteria | Exclusion criteria |
|---|---|
Age: 18–≤99 years of ageCKD stage (at screening): KDOQI CKD Stages 3, 4 or 5 defined by eGFR using the CKD Epidemiology Collaboration (CKD-EPI) formula ESAs: ● Group 1 (not using ESAs): No ESA use within the 6 weeks prior to screening and no ESA use between screening and randomization (Day 1) ● Group 2 (ESA users): Use of any approved ESA for the 6 weeks prior to screening and continuing between screening and randomization Hb concentrationa:On Week –8:● Group 1 (not using ESAs): 8–10 g/dL● Group 2 (ESA users): 8–12 g/dL On randomization (Day 1):● Group 1 (not using ESAs): 8–10 g/dL | Dialysis: On dialysis or clinical evidence of impending need to initiate dialysis within 90 days after study start (Day 1) Kidney transplant: Planned living kidney transplant within 52 weeks after study start (Day 1) Iron: Ferritin ≤100 ng/mL (≤100 µg/L), TSAT ≤20%, at screening Evidence of non-renal anaemia: Aplasias, untreated pernicious anaemia, thalassemia major, sickle cell disease or myelodysplastic syndrome, GI bleeding Cardiovascular comorbidities: MI or acute coronary syndrome or stroke or TIA ≤4 weeks of screening, NYHA Class IV heart failure, uncontrolled hypertension (contraindicating rhEPO use) Liver disease (any one of the following): ● Alanine transaminase: >2× ULN at screening ● Bilirubin: >1.5× ULN at screening ● Current unstable liver or biliary disease per investigator assessment |
● Group 2 (ESA users): Hb 8–11 g/dL and receiving at least the minimum rhEPO dose [epoetins (including biosimilars): 1500 U/week IV or 1000 U/week SC; darbepoetin alfa: 20 µg/4 weeks SC/IV; methoxy PEG- epoetin: 30 µg/month SC/IV] Compliance with placebo [randomization (Day 1) only]: ≥80% and ≤120% compliance with placebo during run-in period | Malignancy: History of malignancy within 2 years before screening through to randomization (Day 1) or currently receiving treatment for cancer or complex kidney cyst Females only: Pregnancy (as confirmed by a positive serum human chorionic gonadotrophin test), breastfeeding, or subject is of reproductive potential and does not agree to follow one of the contraceptive options listed in the List of Highly Effective Methods for Avoiding Pregnancy Other conditions: Any other condition, clinical or laboratory abnormality, or examination finding that the investigator considers would put the subject at unacceptable risk, which may affect study compliance (e.g. intolerance to darbepoetin alfa) or prevent understanding of the aims or investigational procedures or possible consequences of the study |
| Inclusion criteria | Exclusion criteria |
|---|---|
Age: 18–≤99 years of ageCKD stage (at screening): KDOQI CKD Stages 3, 4 or 5 defined by eGFR using the CKD Epidemiology Collaboration (CKD-EPI) formula ESAs: ● Group 1 (not using ESAs): No ESA use within the 6 weeks prior to screening and no ESA use between screening and randomization (Day 1) ● Group 2 (ESA users): Use of any approved ESA for the 6 weeks prior to screening and continuing between screening and randomization Hb concentrationa:On Week –8:● Group 1 (not using ESAs): 8–10 g/dL● Group 2 (ESA users): 8–12 g/dL On randomization (Day 1):● Group 1 (not using ESAs): 8–10 g/dL | Dialysis: On dialysis or clinical evidence of impending need to initiate dialysis within 90 days after study start (Day 1) Kidney transplant: Planned living kidney transplant within 52 weeks after study start (Day 1) Iron: Ferritin ≤100 ng/mL (≤100 µg/L), TSAT ≤20%, at screening Evidence of non-renal anaemia: Aplasias, untreated pernicious anaemia, thalassemia major, sickle cell disease or myelodysplastic syndrome, GI bleeding Cardiovascular comorbidities: MI or acute coronary syndrome or stroke or TIA ≤4 weeks of screening, NYHA Class IV heart failure, uncontrolled hypertension (contraindicating rhEPO use) Liver disease (any one of the following): ● Alanine transaminase: >2× ULN at screening ● Bilirubin: >1.5× ULN at screening ● Current unstable liver or biliary disease per investigator assessment |
● Group 2 (ESA users): Hb 8–11 g/dL and receiving at least the minimum rhEPO dose [epoetins (including biosimilars): 1500 U/week IV or 1000 U/week SC; darbepoetin alfa: 20 µg/4 weeks SC/IV; methoxy PEG- epoetin: 30 µg/month SC/IV] Compliance with placebo [randomization (Day 1) only]: ≥80% and ≤120% compliance with placebo during run-in period | Malignancy: History of malignancy within 2 years before screening through to randomization (Day 1) or currently receiving treatment for cancer or complex kidney cyst Females only: Pregnancy (as confirmed by a positive serum human chorionic gonadotrophin test), breastfeeding, or subject is of reproductive potential and does not agree to follow one of the contraceptive options listed in the List of Highly Effective Methods for Avoiding Pregnancy Other conditions: Any other condition, clinical or laboratory abnormality, or examination finding that the investigator considers would put the subject at unacceptable risk, which may affect study compliance (e.g. intolerance to darbepoetin alfa) or prevent understanding of the aims or investigational procedures or possible consequences of the study |
CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ESA, erythropoiesis-stimulating agent; GI, gastrointestinal; IV, intravenous; Hb, haemoglobin; HD, haemodialysis; KDOQI, Kidney Disease Outcomes Quality Initiative; MI, myocardial infarction; NYHA, New York Heart Association; PD, peritoneal dialysis; PEG, polyethylene glycol; rhEPO, recombinant human erythropoietin; SC, subcutaneous; TIA, transient ischaemic attack; TSAT, transferrin saturation; ULN, upper limit of normal.
Determined using HemoCue, a point-of-care test.
Key inclusion and exclusion criteria
| Inclusion criteria | Exclusion criteria |
|---|---|
Age: 18–≤99 years of ageCKD stage (at screening): KDOQI CKD Stages 3, 4 or 5 defined by eGFR using the CKD Epidemiology Collaboration (CKD-EPI) formula ESAs: ● Group 1 (not using ESAs): No ESA use within the 6 weeks prior to screening and no ESA use between screening and randomization (Day 1) ● Group 2 (ESA users): Use of any approved ESA for the 6 weeks prior to screening and continuing between screening and randomization Hb concentrationa:On Week –8:● Group 1 (not using ESAs): 8–10 g/dL● Group 2 (ESA users): 8–12 g/dL On randomization (Day 1):● Group 1 (not using ESAs): 8–10 g/dL | Dialysis: On dialysis or clinical evidence of impending need to initiate dialysis within 90 days after study start (Day 1) Kidney transplant: Planned living kidney transplant within 52 weeks after study start (Day 1) Iron: Ferritin ≤100 ng/mL (≤100 µg/L), TSAT ≤20%, at screening Evidence of non-renal anaemia: Aplasias, untreated pernicious anaemia, thalassemia major, sickle cell disease or myelodysplastic syndrome, GI bleeding Cardiovascular comorbidities: MI or acute coronary syndrome or stroke or TIA ≤4 weeks of screening, NYHA Class IV heart failure, uncontrolled hypertension (contraindicating rhEPO use) Liver disease (any one of the following): ● Alanine transaminase: >2× ULN at screening ● Bilirubin: >1.5× ULN at screening ● Current unstable liver or biliary disease per investigator assessment |
● Group 2 (ESA users): Hb 8–11 g/dL and receiving at least the minimum rhEPO dose [epoetins (including biosimilars): 1500 U/week IV or 1000 U/week SC; darbepoetin alfa: 20 µg/4 weeks SC/IV; methoxy PEG- epoetin: 30 µg/month SC/IV] Compliance with placebo [randomization (Day 1) only]: ≥80% and ≤120% compliance with placebo during run-in period | Malignancy: History of malignancy within 2 years before screening through to randomization (Day 1) or currently receiving treatment for cancer or complex kidney cyst Females only: Pregnancy (as confirmed by a positive serum human chorionic gonadotrophin test), breastfeeding, or subject is of reproductive potential and does not agree to follow one of the contraceptive options listed in the List of Highly Effective Methods for Avoiding Pregnancy Other conditions: Any other condition, clinical or laboratory abnormality, or examination finding that the investigator considers would put the subject at unacceptable risk, which may affect study compliance (e.g. intolerance to darbepoetin alfa) or prevent understanding of the aims or investigational procedures or possible consequences of the study |
| Inclusion criteria | Exclusion criteria |
|---|---|
Age: 18–≤99 years of ageCKD stage (at screening): KDOQI CKD Stages 3, 4 or 5 defined by eGFR using the CKD Epidemiology Collaboration (CKD-EPI) formula ESAs: ● Group 1 (not using ESAs): No ESA use within the 6 weeks prior to screening and no ESA use between screening and randomization (Day 1) ● Group 2 (ESA users): Use of any approved ESA for the 6 weeks prior to screening and continuing between screening and randomization Hb concentrationa:On Week –8:● Group 1 (not using ESAs): 8–10 g/dL● Group 2 (ESA users): 8–12 g/dL On randomization (Day 1):● Group 1 (not using ESAs): 8–10 g/dL | Dialysis: On dialysis or clinical evidence of impending need to initiate dialysis within 90 days after study start (Day 1) Kidney transplant: Planned living kidney transplant within 52 weeks after study start (Day 1) Iron: Ferritin ≤100 ng/mL (≤100 µg/L), TSAT ≤20%, at screening Evidence of non-renal anaemia: Aplasias, untreated pernicious anaemia, thalassemia major, sickle cell disease or myelodysplastic syndrome, GI bleeding Cardiovascular comorbidities: MI or acute coronary syndrome or stroke or TIA ≤4 weeks of screening, NYHA Class IV heart failure, uncontrolled hypertension (contraindicating rhEPO use) Liver disease (any one of the following): ● Alanine transaminase: >2× ULN at screening ● Bilirubin: >1.5× ULN at screening ● Current unstable liver or biliary disease per investigator assessment |
● Group 2 (ESA users): Hb 8–11 g/dL and receiving at least the minimum rhEPO dose [epoetins (including biosimilars): 1500 U/week IV or 1000 U/week SC; darbepoetin alfa: 20 µg/4 weeks SC/IV; methoxy PEG- epoetin: 30 µg/month SC/IV] Compliance with placebo [randomization (Day 1) only]: ≥80% and ≤120% compliance with placebo during run-in period | Malignancy: History of malignancy within 2 years before screening through to randomization (Day 1) or currently receiving treatment for cancer or complex kidney cyst Females only: Pregnancy (as confirmed by a positive serum human chorionic gonadotrophin test), breastfeeding, or subject is of reproductive potential and does not agree to follow one of the contraceptive options listed in the List of Highly Effective Methods for Avoiding Pregnancy Other conditions: Any other condition, clinical or laboratory abnormality, or examination finding that the investigator considers would put the subject at unacceptable risk, which may affect study compliance (e.g. intolerance to darbepoetin alfa) or prevent understanding of the aims or investigational procedures or possible consequences of the study |
CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ESA, erythropoiesis-stimulating agent; GI, gastrointestinal; IV, intravenous; Hb, haemoglobin; HD, haemodialysis; KDOQI, Kidney Disease Outcomes Quality Initiative; MI, myocardial infarction; NYHA, New York Heart Association; PD, peritoneal dialysis; PEG, polyethylene glycol; rhEPO, recombinant human erythropoietin; SC, subcutaneous; TIA, transient ischaemic attack; TSAT, transferrin saturation; ULN, upper limit of normal.
Determined using HemoCue, a point-of-care test.
Study treatments and management strategies
Daprodustat and darbepoetin alfa dosing strategies, along with those for iron therapy, are detailed in Table 2. A protocol-mandated rescue algorithm was in place to minimize the risk of an inadequate Hb response for an extended period and to enable consistency in the application of rescue therapy across the study (Table 3).
Study treatments and management strategies
| Study treatments | Initiation | Protocol-specified dose adjustment algorithma |
|---|---|---|
| Daprodustat | ● Starting dose 4–12 mg based on prior ESA dose at randomization ● Nine dose steps available (1 mg, 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg, 16 mg and 24 mg) | ● Dose adjustments (i.e. increase, decrease, maintain, or withhold if Hb ≥ 12 g/dL) are implemented by the IRT system to maintain Hb concentrations within the range of 10–11 g/dLb - Hb value measured at least every 4 weeks (Day 1 through Week 52) or at least every 12 weeks (post-Week 52 until the end of treatment) - From Week 52 onward, additional 4-weekly study visits to check Hb and dispense randomized treatment are required if 1. Hb is outside the target range 2. Dose has changed 3. A moderate CYP2C8 inhibitor has been started/stopped/changed 4. Participant has transitioned to dialysis 5. Participant has changed from HD to PD 6. Per investigator discretion allows for an early dose adjustment |
| Darbepoetin alfa | ● Starting dose based on patients’ prior ESA dose (converted to darbepoetin alfa) and Hb at the time of randomization ● Pre-defined dose-stepsc: stepwise increases or decreases in weekly dose from 20 to 33% for most steps (20–400 µg as a total 4-weekly dose; doses ≤150 µg are administered every 4 weeks; 200 µg and 300 µg are divided and administered every 2 weeks; 400 µg is divided and administered once a week) | |
| Iron | ● Started if TSAT is ≤20% and/or ferritin is ≤100 ng/mL - Type of iron, dose and route is determined by the investigator based on local clinical practice and the patient's iron status | ● Iron must be stopped if values of ferritin > 800 ng/mL and TSAT > 20% or if TSAT > 40% are present - Investigators are to be guided by local/regional guidelines and may stop administration of iron at a lower ferritin or TSAT level if clinically indicated - The framework for starting and stopping iron is based on a review of global and regional iron guidelines, as well as input from the ASCEND Steering Committees |
| The Hb and Iron Subcommittee of the Steering Committee is monitoring blinded patient Hb and iron data during the trial. Assessment of the quality of clinical care provided to patients was monitored by the Standard of Care Subcommittee of the Steering Committee. | ||
| Study treatments | Initiation | Protocol-specified dose adjustment algorithma |
|---|---|---|
| Daprodustat | ● Starting dose 4–12 mg based on prior ESA dose at randomization ● Nine dose steps available (1 mg, 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg, 16 mg and 24 mg) | ● Dose adjustments (i.e. increase, decrease, maintain, or withhold if Hb ≥ 12 g/dL) are implemented by the IRT system to maintain Hb concentrations within the range of 10–11 g/dLb - Hb value measured at least every 4 weeks (Day 1 through Week 52) or at least every 12 weeks (post-Week 52 until the end of treatment) - From Week 52 onward, additional 4-weekly study visits to check Hb and dispense randomized treatment are required if 1. Hb is outside the target range 2. Dose has changed 3. A moderate CYP2C8 inhibitor has been started/stopped/changed 4. Participant has transitioned to dialysis 5. Participant has changed from HD to PD 6. Per investigator discretion allows for an early dose adjustment |
| Darbepoetin alfa | ● Starting dose based on patients’ prior ESA dose (converted to darbepoetin alfa) and Hb at the time of randomization ● Pre-defined dose-stepsc: stepwise increases or decreases in weekly dose from 20 to 33% for most steps (20–400 µg as a total 4-weekly dose; doses ≤150 µg are administered every 4 weeks; 200 µg and 300 µg are divided and administered every 2 weeks; 400 µg is divided and administered once a week) | |
| Iron | ● Started if TSAT is ≤20% and/or ferritin is ≤100 ng/mL - Type of iron, dose and route is determined by the investigator based on local clinical practice and the patient's iron status | ● Iron must be stopped if values of ferritin > 800 ng/mL and TSAT > 20% or if TSAT > 40% are present - Investigators are to be guided by local/regional guidelines and may stop administration of iron at a lower ferritin or TSAT level if clinically indicated - The framework for starting and stopping iron is based on a review of global and regional iron guidelines, as well as input from the ASCEND Steering Committees |
| The Hb and Iron Subcommittee of the Steering Committee is monitoring blinded patient Hb and iron data during the trial. Assessment of the quality of clinical care provided to patients was monitored by the Standard of Care Subcommittee of the Steering Committee. | ||
ESA, erythropoiesis-stimulating agent; Hb, haemoglobin; HD, haemodialysis; IRT, interactive response technology; IV, intravenous; PD, peritoneal dialysis; TSAT, transferrin saturation.
During the trial, overrides of the dose adjustment algorithm for exceptional circumstances associated with a safety concern are permitted if approved by the sponsor.
Based on the HemoCue Hb value.
Complete details of darbepoetin alfa dose steps (dose and frequency) are outlined in Supplementary data, Table S5.
Study treatments and management strategies
| Study treatments | Initiation | Protocol-specified dose adjustment algorithma |
|---|---|---|
| Daprodustat | ● Starting dose 4–12 mg based on prior ESA dose at randomization ● Nine dose steps available (1 mg, 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg, 16 mg and 24 mg) | ● Dose adjustments (i.e. increase, decrease, maintain, or withhold if Hb ≥ 12 g/dL) are implemented by the IRT system to maintain Hb concentrations within the range of 10–11 g/dLb - Hb value measured at least every 4 weeks (Day 1 through Week 52) or at least every 12 weeks (post-Week 52 until the end of treatment) - From Week 52 onward, additional 4-weekly study visits to check Hb and dispense randomized treatment are required if 1. Hb is outside the target range 2. Dose has changed 3. A moderate CYP2C8 inhibitor has been started/stopped/changed 4. Participant has transitioned to dialysis 5. Participant has changed from HD to PD 6. Per investigator discretion allows for an early dose adjustment |
| Darbepoetin alfa | ● Starting dose based on patients’ prior ESA dose (converted to darbepoetin alfa) and Hb at the time of randomization ● Pre-defined dose-stepsc: stepwise increases or decreases in weekly dose from 20 to 33% for most steps (20–400 µg as a total 4-weekly dose; doses ≤150 µg are administered every 4 weeks; 200 µg and 300 µg are divided and administered every 2 weeks; 400 µg is divided and administered once a week) | |
| Iron | ● Started if TSAT is ≤20% and/or ferritin is ≤100 ng/mL - Type of iron, dose and route is determined by the investigator based on local clinical practice and the patient's iron status | ● Iron must be stopped if values of ferritin > 800 ng/mL and TSAT > 20% or if TSAT > 40% are present - Investigators are to be guided by local/regional guidelines and may stop administration of iron at a lower ferritin or TSAT level if clinically indicated - The framework for starting and stopping iron is based on a review of global and regional iron guidelines, as well as input from the ASCEND Steering Committees |
| The Hb and Iron Subcommittee of the Steering Committee is monitoring blinded patient Hb and iron data during the trial. Assessment of the quality of clinical care provided to patients was monitored by the Standard of Care Subcommittee of the Steering Committee. | ||
| Study treatments | Initiation | Protocol-specified dose adjustment algorithma |
|---|---|---|
| Daprodustat | ● Starting dose 4–12 mg based on prior ESA dose at randomization ● Nine dose steps available (1 mg, 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg, 16 mg and 24 mg) | ● Dose adjustments (i.e. increase, decrease, maintain, or withhold if Hb ≥ 12 g/dL) are implemented by the IRT system to maintain Hb concentrations within the range of 10–11 g/dLb - Hb value measured at least every 4 weeks (Day 1 through Week 52) or at least every 12 weeks (post-Week 52 until the end of treatment) - From Week 52 onward, additional 4-weekly study visits to check Hb and dispense randomized treatment are required if 1. Hb is outside the target range 2. Dose has changed 3. A moderate CYP2C8 inhibitor has been started/stopped/changed 4. Participant has transitioned to dialysis 5. Participant has changed from HD to PD 6. Per investigator discretion allows for an early dose adjustment |
| Darbepoetin alfa | ● Starting dose based on patients’ prior ESA dose (converted to darbepoetin alfa) and Hb at the time of randomization ● Pre-defined dose-stepsc: stepwise increases or decreases in weekly dose from 20 to 33% for most steps (20–400 µg as a total 4-weekly dose; doses ≤150 µg are administered every 4 weeks; 200 µg and 300 µg are divided and administered every 2 weeks; 400 µg is divided and administered once a week) | |
| Iron | ● Started if TSAT is ≤20% and/or ferritin is ≤100 ng/mL - Type of iron, dose and route is determined by the investigator based on local clinical practice and the patient's iron status | ● Iron must be stopped if values of ferritin > 800 ng/mL and TSAT > 20% or if TSAT > 40% are present - Investigators are to be guided by local/regional guidelines and may stop administration of iron at a lower ferritin or TSAT level if clinically indicated - The framework for starting and stopping iron is based on a review of global and regional iron guidelines, as well as input from the ASCEND Steering Committees |
| The Hb and Iron Subcommittee of the Steering Committee is monitoring blinded patient Hb and iron data during the trial. Assessment of the quality of clinical care provided to patients was monitored by the Standard of Care Subcommittee of the Steering Committee. | ||
ESA, erythropoiesis-stimulating agent; Hb, haemoglobin; HD, haemodialysis; IRT, interactive response technology; IV, intravenous; PD, peritoneal dialysis; TSAT, transferrin saturation.
During the trial, overrides of the dose adjustment algorithm for exceptional circumstances associated with a safety concern are permitted if approved by the sponsor.
Based on the HemoCue Hb value.
Complete details of darbepoetin alfa dose steps (dose and frequency) are outlined in Supplementary data, Table S5.
Rescue algorithm for anaemia management
| Evaluate subject for rescue if: HemoCue Hb remains <9 g/dL (at a scheduled study visit, Week 4 onwards) despite threea consecutive dose increases above the starting or post-rescueb dose (where HemoCue Hb is <9 g/dL before each dose increase) or HemoCue Hb is <7.5 g/dL despite a dose increase at the prior study visit. | |
Step 1: Initial intervention | While continuing randomized treatment (increase dose if HemoCue Hb <7.5 g/dL; otherwise maintain current dose), intervene with one or more of the following as dictated by clinical comorbidities • Single course of IV iron up to 1000 mg (in addition to the iron management criteria) • Transfusion of up to two units of PRBC if clinically indicated • Allow additional 4 weeks on randomised treatment (Note: this is a required choice; can be combined with either or both of the above) |
| Step 2: Rescue | Check HemoCue Hb 4 weeks ± 1 week from last study visit; earlier checks of Hb may be obtained to advise further intervention as clinically indicated Randomized treatment should be permanently discontinued and the subject should be rescued according to local clinical practice if either • HemoCue Hb remains <9 g/dL despite initial intervention based on the average of two HemoCue Hb valuesc or • More than two units of PRBC were needed for transfusion (and was not related to acute bleeding) |
| Evaluate subject for rescue if: HemoCue Hb remains <9 g/dL (at a scheduled study visit, Week 4 onwards) despite threea consecutive dose increases above the starting or post-rescueb dose (where HemoCue Hb is <9 g/dL before each dose increase) or HemoCue Hb is <7.5 g/dL despite a dose increase at the prior study visit. | |
Step 1: Initial intervention | While continuing randomized treatment (increase dose if HemoCue Hb <7.5 g/dL; otherwise maintain current dose), intervene with one or more of the following as dictated by clinical comorbidities • Single course of IV iron up to 1000 mg (in addition to the iron management criteria) • Transfusion of up to two units of PRBC if clinically indicated • Allow additional 4 weeks on randomised treatment (Note: this is a required choice; can be combined with either or both of the above) |
| Step 2: Rescue | Check HemoCue Hb 4 weeks ± 1 week from last study visit; earlier checks of Hb may be obtained to advise further intervention as clinically indicated Randomized treatment should be permanently discontinued and the subject should be rescued according to local clinical practice if either • HemoCue Hb remains <9 g/dL despite initial intervention based on the average of two HemoCue Hb valuesc or • More than two units of PRBC were needed for transfusion (and was not related to acute bleeding) |
Hb, haemoglobin; PRBC, packed red blood cells.
Two consecutive dose increases if starting/post-rescue dose is daprodustat 12 mg or darbepoetin alfa 200 µg over 4 weeks, one dose increase if starting/post-rescue dose is daprodustat 16 mg or darbepoetin alfa 300 µg over 4 weeks; and no prior dose increase if starting/post-rescue dose is daprodustat 24 mg or darbepoetin alfa 400 µg over 4 weeks (top dose).
For subjects who previously were evaluated for rescue and who are able to continue in the trial, ‘post-rescue’ dose is the dose of randomized treatment that a subject is receiving at the study visit after initial intervention.
Repeat HemoCue Hb at the same study visit to confirm Hb (using the same sample); take average of two values.
Rescue algorithm for anaemia management
| Evaluate subject for rescue if: HemoCue Hb remains <9 g/dL (at a scheduled study visit, Week 4 onwards) despite threea consecutive dose increases above the starting or post-rescueb dose (where HemoCue Hb is <9 g/dL before each dose increase) or HemoCue Hb is <7.5 g/dL despite a dose increase at the prior study visit. | |
Step 1: Initial intervention | While continuing randomized treatment (increase dose if HemoCue Hb <7.5 g/dL; otherwise maintain current dose), intervene with one or more of the following as dictated by clinical comorbidities • Single course of IV iron up to 1000 mg (in addition to the iron management criteria) • Transfusion of up to two units of PRBC if clinically indicated • Allow additional 4 weeks on randomised treatment (Note: this is a required choice; can be combined with either or both of the above) |
| Step 2: Rescue | Check HemoCue Hb 4 weeks ± 1 week from last study visit; earlier checks of Hb may be obtained to advise further intervention as clinically indicated Randomized treatment should be permanently discontinued and the subject should be rescued according to local clinical practice if either • HemoCue Hb remains <9 g/dL despite initial intervention based on the average of two HemoCue Hb valuesc or • More than two units of PRBC were needed for transfusion (and was not related to acute bleeding) |
| Evaluate subject for rescue if: HemoCue Hb remains <9 g/dL (at a scheduled study visit, Week 4 onwards) despite threea consecutive dose increases above the starting or post-rescueb dose (where HemoCue Hb is <9 g/dL before each dose increase) or HemoCue Hb is <7.5 g/dL despite a dose increase at the prior study visit. | |
Step 1: Initial intervention | While continuing randomized treatment (increase dose if HemoCue Hb <7.5 g/dL; otherwise maintain current dose), intervene with one or more of the following as dictated by clinical comorbidities • Single course of IV iron up to 1000 mg (in addition to the iron management criteria) • Transfusion of up to two units of PRBC if clinically indicated • Allow additional 4 weeks on randomised treatment (Note: this is a required choice; can be combined with either or both of the above) |
| Step 2: Rescue | Check HemoCue Hb 4 weeks ± 1 week from last study visit; earlier checks of Hb may be obtained to advise further intervention as clinically indicated Randomized treatment should be permanently discontinued and the subject should be rescued according to local clinical practice if either • HemoCue Hb remains <9 g/dL despite initial intervention based on the average of two HemoCue Hb valuesc or • More than two units of PRBC were needed for transfusion (and was not related to acute bleeding) |
Hb, haemoglobin; PRBC, packed red blood cells.
Two consecutive dose increases if starting/post-rescue dose is daprodustat 12 mg or darbepoetin alfa 200 µg over 4 weeks, one dose increase if starting/post-rescue dose is daprodustat 16 mg or darbepoetin alfa 300 µg over 4 weeks; and no prior dose increase if starting/post-rescue dose is daprodustat 24 mg or darbepoetin alfa 400 µg over 4 weeks (top dose).
For subjects who previously were evaluated for rescue and who are able to continue in the trial, ‘post-rescue’ dose is the dose of randomized treatment that a subject is receiving at the study visit after initial intervention.
Repeat HemoCue Hb at the same study visit to confirm Hb (using the same sample); take average of two values.
Objectives and endpoints
The trial has two primary objectives: to compare the effects of daprodustat and darbepoetin alfa on Hb response (efficacy outcome) and CV events (safety outcome) using non-inferiority margins agreed to in consultation with regulatory agencies (see Statistical analysis section). Hb response will be evaluated as the mean change in Hb from baseline to the evaluation period (EP; Weeks 28–52). The CV safety outcome is the first adjudicated major adverse CV event [MACE; i.e. the composite of all-cause mortality, non-fatal myocardial infarction (MI) or non-fatal stroke]. All deaths and potential non-fatal CV events are adjudicated by an external, independent and blinded endpoints committee led by the Duke Clinical Research Institute (Durham, NC, USA) in collaboration with George Clinical (Sydney, NSW, Australia). The principal secondary endpoints, which include superiority assessment of effects on MACEs and CKD progression, are listed in Table 4 along with other secondary endpoints (also to be tested for superiority).
Primary and secondary objectives and endpoints
| Objectives | Endpoints |
|---|---|
| Co-primary objectives | Co-primary endpoints (tested in parallel for non-inferiority) |
| • To compare daprodustat with darbepoetin alfa for CV safety (non inferiority) | • Time to first occurrence of adjudicated MACE (composite of all-cause mortality, non-fatal MI and non-fatal stroke) |
| • To compare daprodustat with darbepoetin alfa for Hb efficacy (non inferiority) | • Mean change in Hb between baseline and EP (mean over Weeks 28 to 52) |
| Principal secondary objectives | Principal secondary endpoints(tested for superiority, adjusted for multiplicity) |
| • To compare daprodustat with darbepoetin alfa on CV safety endpoints | • Time to first occurrence of adjudicated - MACE - MACE or a thromboembolic event (vascular access thrombosis, symptomatic deep vein thrombosis or symptomatic pulmonary embolism) - MACE or a hospitalization for heart failure |
| • To compare daprodustat with darbepoetin alfa on the progression of CKD | • Time to progression of CKDa |
| Secondary objectives | Secondary endpoints(tested for superiorityb, no multiplicity adjustment) |
| • To compare daprodustat with darbepoetin alfa on additional CV safety endpoints | • All-cause mortality, CV mortality, fatal or non-fatal MI, fatal or non-fatal strokec • MACE or hospitalization for heart failurec (recurrent events analysis) • CV mortality or non-fatal MIc • All-cause hospitalization • All-cause hospital re-admission within 30 days • MACE or hospitalization for heart failure or thromboembolic eventsc • Hospitalization for heart failurec • Thromboembolic eventsc • Individual components of CKD progressionc |
| • To compare daprodustat with darbepoetin alfa on Hb variability | • Hb change from baseline to Week 52b • N (%) responders, defined as mean Hb within the Hb analysis range 10–11.5 g/dL during EPd |
| • Percent time Hb in analysis range (10–11.5 g/dL) during the evaluation period (EP, Weeks 28–52) and during the maintenance phase (MP; Weeks 28 to end of trial) (non-inferiority analysis that will use a margin of 15% less time in range)b | |
| • To compare daprodustat with darbepoetin alfa on BP | • Change from baseline in SBP, DBP and MAP at Week 52 and at end of treatment • Number of BP exacerbation events per 100 patient-years • N (%) with at least one BP exacerbation event during study |
| • To compare daprodustat with darbepoetin alfa on the time to rescue (defined as permanently stopping randomized treatment due to meeting rescue criteria) | • Time to stopping randomized treatment due to meeting rescue criteria |
| • To compare daprodustat with darbepoetin alfa on HRQoL and utility score | • Mean change in SF-36 HRQoL scores (PCS, MCS and eight health domains) between baseline and Weeks 8, 12, 28 and 52; of particular interest are the changes from baseline in the vitality and physical functioning domains at Week 28 and 52. • Change from baseline in Health Utility (EQ-5D-5L) score at Week 52 • Change from baseline in EQ VAS at Week 52 |
| • To compare daprodustat with darbepoetin alfa on the symptom severity and change | • Change from Baseline at Weeks 8, 12, 28 and 52 by domain and overall symptom score on the CKD-AQ • Change from Baseline at Week 8,12, 28 and 52 in PGI-S |
| Objectives | Endpoints |
|---|---|
| Co-primary objectives | Co-primary endpoints (tested in parallel for non-inferiority) |
| • To compare daprodustat with darbepoetin alfa for CV safety (non inferiority) | • Time to first occurrence of adjudicated MACE (composite of all-cause mortality, non-fatal MI and non-fatal stroke) |
| • To compare daprodustat with darbepoetin alfa for Hb efficacy (non inferiority) | • Mean change in Hb between baseline and EP (mean over Weeks 28 to 52) |
| Principal secondary objectives | Principal secondary endpoints(tested for superiority, adjusted for multiplicity) |
| • To compare daprodustat with darbepoetin alfa on CV safety endpoints | • Time to first occurrence of adjudicated - MACE - MACE or a thromboembolic event (vascular access thrombosis, symptomatic deep vein thrombosis or symptomatic pulmonary embolism) - MACE or a hospitalization for heart failure |
| • To compare daprodustat with darbepoetin alfa on the progression of CKD | • Time to progression of CKDa |
| Secondary objectives | Secondary endpoints(tested for superiorityb, no multiplicity adjustment) |
| • To compare daprodustat with darbepoetin alfa on additional CV safety endpoints | • All-cause mortality, CV mortality, fatal or non-fatal MI, fatal or non-fatal strokec • MACE or hospitalization for heart failurec (recurrent events analysis) • CV mortality or non-fatal MIc • All-cause hospitalization • All-cause hospital re-admission within 30 days • MACE or hospitalization for heart failure or thromboembolic eventsc • Hospitalization for heart failurec • Thromboembolic eventsc • Individual components of CKD progressionc |
| • To compare daprodustat with darbepoetin alfa on Hb variability | • Hb change from baseline to Week 52b • N (%) responders, defined as mean Hb within the Hb analysis range 10–11.5 g/dL during EPd |
| • Percent time Hb in analysis range (10–11.5 g/dL) during the evaluation period (EP, Weeks 28–52) and during the maintenance phase (MP; Weeks 28 to end of trial) (non-inferiority analysis that will use a margin of 15% less time in range)b | |
| • To compare daprodustat with darbepoetin alfa on BP | • Change from baseline in SBP, DBP and MAP at Week 52 and at end of treatment • Number of BP exacerbation events per 100 patient-years • N (%) with at least one BP exacerbation event during study |
| • To compare daprodustat with darbepoetin alfa on the time to rescue (defined as permanently stopping randomized treatment due to meeting rescue criteria) | • Time to stopping randomized treatment due to meeting rescue criteria |
| • To compare daprodustat with darbepoetin alfa on HRQoL and utility score | • Mean change in SF-36 HRQoL scores (PCS, MCS and eight health domains) between baseline and Weeks 8, 12, 28 and 52; of particular interest are the changes from baseline in the vitality and physical functioning domains at Week 28 and 52. • Change from baseline in Health Utility (EQ-5D-5L) score at Week 52 • Change from baseline in EQ VAS at Week 52 |
| • To compare daprodustat with darbepoetin alfa on the symptom severity and change | • Change from Baseline at Weeks 8, 12, 28 and 52 by domain and overall symptom score on the CKD-AQ • Change from Baseline at Week 8,12, 28 and 52 in PGI-S |
BP, blood pressure; CKD-AQ, Chronic Kidney Disease-Anaemia Questionnaire; CV, cardiovascular; DBP, diastolic BP; EP, evaluation phase; EQ-5D-5L, EuroQoL 5-dimension 5-level; EQ VAS, EuroQoL visual analogue scale; Hb, haemoglobin; HRQoL, health-related quality of life; MACE, major adverse cardiac event; MAP, mean arterial pressure; MCS, mental component score; MP, maintenance phase; PCS, physical component score; PGI-S, patient global impression of severity; SBP, systolic BP; SF-36, 36-item Short Form.
Conversion factor from g/dL to g/L is 10 and from g/dL to mmol/L is 0.6206 (e.g. Hb of 10–11 g/dL is equivalent to 100–110 g/L or 6.2–6.8 mmol/L).
Progression of CKD defined as 40% decline in eGFR from baseline (confirmed 4–13 weeks later) or end-stage renal disease as defined by initiating chronic dialysis for ≥90 days or not initiating chronic dialysis when dialysis is indicated or kidney transplantation.
Hb change from baseline to Week 52 is tested for non-inferiority using the −0.75 g/dL margin used in the co-primary analysis. % time in range is tested first for non-inferiority, then for superiority.
Events adjudicated; for CKD progression only, two components to be adjudicated.
To account for within-subject variability, 0.5 g/dL was added to the upper end of the target range to create a defined analysis range of 10.0–11.5 g/dL.
Primary and secondary objectives and endpoints
| Objectives | Endpoints |
|---|---|
| Co-primary objectives | Co-primary endpoints (tested in parallel for non-inferiority) |
| • To compare daprodustat with darbepoetin alfa for CV safety (non inferiority) | • Time to first occurrence of adjudicated MACE (composite of all-cause mortality, non-fatal MI and non-fatal stroke) |
| • To compare daprodustat with darbepoetin alfa for Hb efficacy (non inferiority) | • Mean change in Hb between baseline and EP (mean over Weeks 28 to 52) |
| Principal secondary objectives | Principal secondary endpoints(tested for superiority, adjusted for multiplicity) |
| • To compare daprodustat with darbepoetin alfa on CV safety endpoints | • Time to first occurrence of adjudicated - MACE - MACE or a thromboembolic event (vascular access thrombosis, symptomatic deep vein thrombosis or symptomatic pulmonary embolism) - MACE or a hospitalization for heart failure |
| • To compare daprodustat with darbepoetin alfa on the progression of CKD | • Time to progression of CKDa |
| Secondary objectives | Secondary endpoints(tested for superiorityb, no multiplicity adjustment) |
| • To compare daprodustat with darbepoetin alfa on additional CV safety endpoints | • All-cause mortality, CV mortality, fatal or non-fatal MI, fatal or non-fatal strokec • MACE or hospitalization for heart failurec (recurrent events analysis) • CV mortality or non-fatal MIc • All-cause hospitalization • All-cause hospital re-admission within 30 days • MACE or hospitalization for heart failure or thromboembolic eventsc • Hospitalization for heart failurec • Thromboembolic eventsc • Individual components of CKD progressionc |
| • To compare daprodustat with darbepoetin alfa on Hb variability | • Hb change from baseline to Week 52b • N (%) responders, defined as mean Hb within the Hb analysis range 10–11.5 g/dL during EPd |
| • Percent time Hb in analysis range (10–11.5 g/dL) during the evaluation period (EP, Weeks 28–52) and during the maintenance phase (MP; Weeks 28 to end of trial) (non-inferiority analysis that will use a margin of 15% less time in range)b | |
| • To compare daprodustat with darbepoetin alfa on BP | • Change from baseline in SBP, DBP and MAP at Week 52 and at end of treatment • Number of BP exacerbation events per 100 patient-years • N (%) with at least one BP exacerbation event during study |
| • To compare daprodustat with darbepoetin alfa on the time to rescue (defined as permanently stopping randomized treatment due to meeting rescue criteria) | • Time to stopping randomized treatment due to meeting rescue criteria |
| • To compare daprodustat with darbepoetin alfa on HRQoL and utility score | • Mean change in SF-36 HRQoL scores (PCS, MCS and eight health domains) between baseline and Weeks 8, 12, 28 and 52; of particular interest are the changes from baseline in the vitality and physical functioning domains at Week 28 and 52. • Change from baseline in Health Utility (EQ-5D-5L) score at Week 52 • Change from baseline in EQ VAS at Week 52 |
| • To compare daprodustat with darbepoetin alfa on the symptom severity and change | • Change from Baseline at Weeks 8, 12, 28 and 52 by domain and overall symptom score on the CKD-AQ • Change from Baseline at Week 8,12, 28 and 52 in PGI-S |
| Objectives | Endpoints |
|---|---|
| Co-primary objectives | Co-primary endpoints (tested in parallel for non-inferiority) |
| • To compare daprodustat with darbepoetin alfa for CV safety (non inferiority) | • Time to first occurrence of adjudicated MACE (composite of all-cause mortality, non-fatal MI and non-fatal stroke) |
| • To compare daprodustat with darbepoetin alfa for Hb efficacy (non inferiority) | • Mean change in Hb between baseline and EP (mean over Weeks 28 to 52) |
| Principal secondary objectives | Principal secondary endpoints(tested for superiority, adjusted for multiplicity) |
| • To compare daprodustat with darbepoetin alfa on CV safety endpoints | • Time to first occurrence of adjudicated - MACE - MACE or a thromboembolic event (vascular access thrombosis, symptomatic deep vein thrombosis or symptomatic pulmonary embolism) - MACE or a hospitalization for heart failure |
| • To compare daprodustat with darbepoetin alfa on the progression of CKD | • Time to progression of CKDa |
| Secondary objectives | Secondary endpoints(tested for superiorityb, no multiplicity adjustment) |
| • To compare daprodustat with darbepoetin alfa on additional CV safety endpoints | • All-cause mortality, CV mortality, fatal or non-fatal MI, fatal or non-fatal strokec • MACE or hospitalization for heart failurec (recurrent events analysis) • CV mortality or non-fatal MIc • All-cause hospitalization • All-cause hospital re-admission within 30 days • MACE or hospitalization for heart failure or thromboembolic eventsc • Hospitalization for heart failurec • Thromboembolic eventsc • Individual components of CKD progressionc |
| • To compare daprodustat with darbepoetin alfa on Hb variability | • Hb change from baseline to Week 52b • N (%) responders, defined as mean Hb within the Hb analysis range 10–11.5 g/dL during EPd |
| • Percent time Hb in analysis range (10–11.5 g/dL) during the evaluation period (EP, Weeks 28–52) and during the maintenance phase (MP; Weeks 28 to end of trial) (non-inferiority analysis that will use a margin of 15% less time in range)b | |
| • To compare daprodustat with darbepoetin alfa on BP | • Change from baseline in SBP, DBP and MAP at Week 52 and at end of treatment • Number of BP exacerbation events per 100 patient-years • N (%) with at least one BP exacerbation event during study |
| • To compare daprodustat with darbepoetin alfa on the time to rescue (defined as permanently stopping randomized treatment due to meeting rescue criteria) | • Time to stopping randomized treatment due to meeting rescue criteria |
| • To compare daprodustat with darbepoetin alfa on HRQoL and utility score | • Mean change in SF-36 HRQoL scores (PCS, MCS and eight health domains) between baseline and Weeks 8, 12, 28 and 52; of particular interest are the changes from baseline in the vitality and physical functioning domains at Week 28 and 52. • Change from baseline in Health Utility (EQ-5D-5L) score at Week 52 • Change from baseline in EQ VAS at Week 52 |
| • To compare daprodustat with darbepoetin alfa on the symptom severity and change | • Change from Baseline at Weeks 8, 12, 28 and 52 by domain and overall symptom score on the CKD-AQ • Change from Baseline at Week 8,12, 28 and 52 in PGI-S |
BP, blood pressure; CKD-AQ, Chronic Kidney Disease-Anaemia Questionnaire; CV, cardiovascular; DBP, diastolic BP; EP, evaluation phase; EQ-5D-5L, EuroQoL 5-dimension 5-level; EQ VAS, EuroQoL visual analogue scale; Hb, haemoglobin; HRQoL, health-related quality of life; MACE, major adverse cardiac event; MAP, mean arterial pressure; MCS, mental component score; MP, maintenance phase; PCS, physical component score; PGI-S, patient global impression of severity; SBP, systolic BP; SF-36, 36-item Short Form.
Conversion factor from g/dL to g/L is 10 and from g/dL to mmol/L is 0.6206 (e.g. Hb of 10–11 g/dL is equivalent to 100–110 g/L or 6.2–6.8 mmol/L).
Progression of CKD defined as 40% decline in eGFR from baseline (confirmed 4–13 weeks later) or end-stage renal disease as defined by initiating chronic dialysis for ≥90 days or not initiating chronic dialysis when dialysis is indicated or kidney transplantation.
Hb change from baseline to Week 52 is tested for non-inferiority using the −0.75 g/dL margin used in the co-primary analysis. % time in range is tested first for non-inferiority, then for superiority.
Events adjudicated; for CKD progression only, two components to be adjudicated.
To account for within-subject variability, 0.5 g/dL was added to the upper end of the target range to create a defined analysis range of 10.0–11.5 g/dL.
Randomization and stratification
Participants were stratified by baseline use of an ESA, by region and by participation in the ambulatory blood pressure monitoring substudy. The region groupings used for stratification were developed to enable balance across different parts of the world with similar standards of care (see Supplementary data, Table S2 for details).
Following stratification, participants were randomized 1:1 to receive oral daprodustat or darbepoetin alfa. A central randomization approach was used to help protect against bias due to the open-label design.
Statistical analysis
A sample size of 4500 was originally planned for this event-driven trial, based on a target of 945 adjudicated first MACE events. With the non-inferiority margin defined as a hazard ratio (HR) of 1.2, this provided ∼90% power to establish non-inferiority for time to first adjudicated MACE, assuming a true underlying 3% lower relative risk of a MACE in favour of daprodustat (i.e. a true underlying HR of 0.97). In addition, this also provided 80% power for non-inferiority, assuming the true underlying risk of a MACE is the same in both groups (i.e. a true underlying HR of 1.00). In July 2020, prior to completion of recruitment and study unblinding, the MACE non-inferiority margin was changed to 1.25, resulting in 664 MACEs being required to maintain 90% power. The change was made after discussion with regulatory authorities and with approval from the academic-led steering committee and the Independent Data Monitoring Committee. The rationale for the non-inferiority margin change was to accelerate study closeout in the context of the coronavirus disease 2019 (COVID-19) pandemic and to align with the non-inferiority margin used in other HIF-PHI clinical studies [16]. Additionally, due to the COVID-19 pandemic, a decision was made to end the study screening in September 2020, which resulted in <4500 participants being randomized.
The study size also provided >99% power to perform the intention-to-treat (ITT) non-inferiority test for the treatment difference of mean change in Hb between baseline and EP (daprodustat − darbepoetin alfa) based on a non-inferiority margin of −0.75 g/dL. Multiple imputations using a missing-at-random assumption will be used to impute missing Hb values.
Conditional on the co-primary endpoints achieving non-inferiority at the one-sided 2.5% level, statistical testing will progress to the principal secondary endpoints (Table 4), including superiority for MACEs and CKD progression. CKD progression is defined as a 40% decline in eGFR from baseline (confirmed 4–13 weeks later) or end-stage kidney disease, which is defined as initiating chronic dialysis (≥90 days or adjudicated as intended to be chronic if the duration of dialysis is <90 days); dialysis being indicated but not initiated (as reported by the investigator and confirmed by adjudication); or kidney transplantation. Statistical testing for the principal secondary endpoints will be adjusted for multiplicity using the Holm–Bonferroni procedure [17].
Descriptive statistics in the form of number and percentage of participants or median and 25th (P25) and 75th (P75) percentiles are provided for baseline variables. Baseline values are presented for the ITT population, overall and by baseline ESA use.
Study oversight
ASCEND-ND was developed in collaboration with steering committees, which provided academic and scientific leadership as well as oversight during the study, as previously described for the ASCEND-D trial [18]. An external, independent Clinical Events Classification group blinded to the treatment assignment, led by the Duke Clinical Research Institute in collaboration with George Clinical, is responsible for adjudicating predefined events (all-cause mortality, MI, stroke, hospitalization for heart failure, thromboembolic events and select progression of CKD events). Committee members and their respective affiliations are presented in Supplementary data, Table S3.
Comparison with other large ND anaemia trials
To assess comparability to prior anaemia studies in the ND population, we compared baseline characteristics from ASCEND-ND participants with baseline characteristics from participants enrolled in similar anaemia trials in a CKD-ND population. Included in the comparison were the HIF-PHI trials with roxadustat (OLYMPUS; NCT02174627) [19] and vadadustat (PRO2TECT; NCT02648347) [20], as well as the placebo-controlled TREAT (NCT00093015) [21] study of darbepoetin alfa.
RESULTS
ASCEND-ND is being conducted at 506 research sites in 39 countries. The study completed recruitment in December 2020; country-level/region participant distribution is listed in Figure 2. In total, 30% of participants originated in Europe, the Middle East and Africa (EMEA); 26% in North America (predominantly the USA); 15% in Latin America and 28% in the Asia Pacific region.
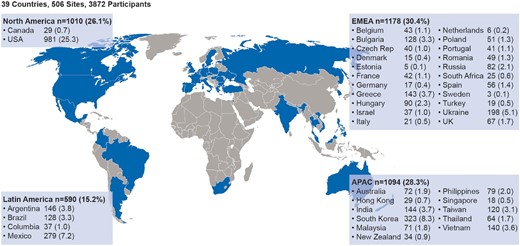
ASCEND-ND country-level participant distribution. APAC; Asia Pacific; EMEA: Europe, Middle East and Africa.
Screening, run-in, and randomization
A total of 10 498 patients were screened, including patients who were rescreened, of whom 6626 (63%) did not meet entry criteria and were not randomized. The reasons for screening failure are listed in Supplementary data, Table S4.
A total of 3872 participants were randomized. One additional participant was randomized but had not provided valid informed consent, so was removed from the total of randomized participants.
Participant characteristics
Baseline characteristics are summarized in Table 5. The randomized participants had a median age of 67 years and 56% were female. The majority of participants were white (56%); 27% self-identified as Asian, 10% as Black, 23% as Hispanic or Latino and 5% as American Indian or Alaskan native background. Of note, within the USA, 33% of participants were black (post hoc). At randomization, 81% of participants had Stage 4 or 5 CKD, 57% had diabetes and 37% had a history of CV disease.
Baseline characteristics of the overall ITT population and by ESA user status
| ESA user status | |||
|---|---|---|---|
| Characteristics | ITT population (N = 3872) | Yes (n = 1810) | No (n = 2062) |
| Age (years), median (25th–75th percentile) | 67 (57–75) | 68 (57–75) | 66 (57–74) |
| Women (%) | 56 | 58 | 55 |
| Race (%) | |||
| White | 56 | 58 | 54 |
| Asian | 27 | 27 | 28 |
| Black | 10 | 5 | 13 |
| American Indian or Alaska Native | 5 | 7 | 3 |
| Native Hawaiian or Other Pacific Islander | <1 | <1 | <1 |
| Multiple | 2 | 3 | 2 |
| Ethnicity, Hispanic or Latino (%) | 23 | 27 | 20 |
| Baseline weight (kg), median (25th–75th percentile) | 71.2 (60.0–84.5) | 70.9 (59.1–83.5) | 71.6 (60.4–85.9) |
| Baseline body mass index (kg/m2), median (25th–75th percentile) | 26.7 (23.3–31.1) | 26.7 (23.3–30.8) | 26.7 (23.3–31.3) |
CKD stage (%) Stage 1 and 2 Stage 3 Stage 4 Stage 5 Missing | <1 18 46 36 <1 | <1 18 47 35 <1 | <1 18 45 36 0 |
| Renal comorbidities (%) | |||
| Hypertension | 94 | 94 | 95 |
| Diabetes | 57 | 54 | 59 |
| Glomerulonephritis | 10 | 11 | 9 |
| Autosomal dominant polycystic kidney disease | 3 | 2 | 4 |
| Cardiovascular disease history (%)a | 37 | 37 | 37 |
| Coronary artery disease | 20 | 20 | 19 |
| Heart failure | 13 | 12 | 14 |
| Valvular heart disease | 8 | 9 | 6 |
| Angina pectoris | 8 | 8 | 7 |
| Atrial fibrillation | 5 | 6 | 5 |
| Myocardial infarction | 7 | 8 | 6 |
| Stroke | 7 | 6 | 7 |
| Transient ischaemic attack | 4 | 3 | 4 |
| Cardiac arrest | 1 | <1 | 1 |
| Thromboembolic events (%)b | 4 | 4 | 4 |
| Cancer (%) | 5 | 6 | 4 |
| Smoking status | |||
| Current smoker (%) | 6 | 5 | 7 |
| Former smoker (%) | 22 | 22 | 22 |
| Baseline blood pressure (mmHg), median (25th–75th percentile) | |||
| Systolic | 135.0 (125.0–146.7) | 135.0 (124.3–146.7) | 135.3 (125.3–147.0) |
| Diastolic | 74.0 (66.0–81.3) | 74.0 (66.7–81.3) | 73.7 (65.3–81.0) |
| Mean arterial pressure | 94.7 (87.2–101.6) | 94.7 (87.4–101.6) | 94.6 (86.9–101.6) |
| Baseline laboratory values, median (25th–75th percentile) | |||
| Haemoglobin (g/dL) | 9.9 (9.2–10.5) | 10.4 (9.7–10.9) | 9.5 (9.0–10.0) |
| <9 g/dL (%) | 17 | 8 | 24 |
| 9–10 g/dL (%) | 38 | 25 | 49 |
| 10–11 g/dL (%) | 36 | 48 | 25 |
| >11 g/dL (%) | 10 | 18 | 2 |
| eGFR (mL/min/1.73m2) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) |
| Serum creatinine (mg/dL) | 3.0 (2.2–4.3) | 3.0 (2.1–4.2) | 3.1 (2.2–4.3) |
| hsCRP (mg/L) | 2.0 (0.8–5.4) | 2.1 (0.8–5.5) | 2.0 (0.8–5.3) |
| Albumin (g/dL) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) |
| Haemoglobin A1c (%) (in patients with diabetes) | 6.5 (5.8–7.6) | 6.5 (5.8–7.5) | 6.6 (5.8–7.6) |
| White blood cells (×109/L) | 6.5 (5.3–7.8) | 6.4 (5.2–7.8) | 6.5 (5.3–7.9) |
| Platelets (×109/L) | 215.0 (175.0–262.0) | 211.0 (171.0–260.0) | 220.0 (178.0–264.0) |
| Transferrin saturation (%) | 29.0 (23.0–37.0) | 31.0 (24.0–39.0) | 28.0 (23.0–35.0) |
| Ferritin (µg/L) | 271.0 (168.0–453.0) | 280.0 (173.0–472.0) | 264.0 (162.0–438.0) |
| Hepcidin (µg/L) | 105.3 (61.4–168.7) | 110.6 (62.8–175.8) | 101.4 (59.8–160.0) |
| iPTH (ng/L) | 125.4 (67.4–242.2) | 121.6 (65.5–241.2) | 128.2 (67.4–243.1) |
| Total cholesterol (mg/dL) | 158.3 (131.3–193.1) | 156.4 (129.3–189.2) | 160.2 (131.3–195.0) |
| Low-density lipoprotein cholesterol | 84.2 (64.1–110.8) | 81.9 (63.0–108.9) | 85.0 (66.0–112.0) |
| High-density lipoprotein cholesterol | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) |
| Medications (%) | |||
| Diabetes medications | 49 | 48 | 51 |
| Insulin | 31 | 30 | 31 |
| Antihypertensives | 95 | 95 | 95 |
| ACE inhibitor or ARB | 61 | 60 | 61 |
| Beta blocker | 48 | 49 | 48 |
| SGLT2 inhibitor | <1 | <1 | <1 |
| Statin | 55 | 54 | 56 |
| Anticoagulant | 8 | 8 | 7 |
| Antiplatelet | 37 | 37 | 38 |
| Aspirin | 30 | 29 | 31 |
| Vitamin K antagonist | 3 | 4 | 3 |
Phosphate bindersc Iron-based Calcium-based Non-calcium and non-iron based | 21 1 17 3 | 23 1 18 5 | 19 1 16 2 |
| Vitamin D | 33 | 35 | 32 |
| Calcimimetics | <1 | <1 | <1 |
| Oral irond | 49 | 48 | 51 |
| IV iron | 12 | 13 | 10 |
| Prior ESA use (%) | 47 | 100 | 0 |
Prior ESA type at randomization (%) Darbepoetin alfa only Epoetin only Methoxy PEG-epoetin beta only Multiple | 31 58 10 1 | 31 58 10 1 | 0 0 0 0 |
| Prior ESA dose standardized to IV epoetin (U/week)e | 3934 (2493–6310) | 3934 (2493–6310) | N/A |
| ESA user status | |||
|---|---|---|---|
| Characteristics | ITT population (N = 3872) | Yes (n = 1810) | No (n = 2062) |
| Age (years), median (25th–75th percentile) | 67 (57–75) | 68 (57–75) | 66 (57–74) |
| Women (%) | 56 | 58 | 55 |
| Race (%) | |||
| White | 56 | 58 | 54 |
| Asian | 27 | 27 | 28 |
| Black | 10 | 5 | 13 |
| American Indian or Alaska Native | 5 | 7 | 3 |
| Native Hawaiian or Other Pacific Islander | <1 | <1 | <1 |
| Multiple | 2 | 3 | 2 |
| Ethnicity, Hispanic or Latino (%) | 23 | 27 | 20 |
| Baseline weight (kg), median (25th–75th percentile) | 71.2 (60.0–84.5) | 70.9 (59.1–83.5) | 71.6 (60.4–85.9) |
| Baseline body mass index (kg/m2), median (25th–75th percentile) | 26.7 (23.3–31.1) | 26.7 (23.3–30.8) | 26.7 (23.3–31.3) |
CKD stage (%) Stage 1 and 2 Stage 3 Stage 4 Stage 5 Missing | <1 18 46 36 <1 | <1 18 47 35 <1 | <1 18 45 36 0 |
| Renal comorbidities (%) | |||
| Hypertension | 94 | 94 | 95 |
| Diabetes | 57 | 54 | 59 |
| Glomerulonephritis | 10 | 11 | 9 |
| Autosomal dominant polycystic kidney disease | 3 | 2 | 4 |
| Cardiovascular disease history (%)a | 37 | 37 | 37 |
| Coronary artery disease | 20 | 20 | 19 |
| Heart failure | 13 | 12 | 14 |
| Valvular heart disease | 8 | 9 | 6 |
| Angina pectoris | 8 | 8 | 7 |
| Atrial fibrillation | 5 | 6 | 5 |
| Myocardial infarction | 7 | 8 | 6 |
| Stroke | 7 | 6 | 7 |
| Transient ischaemic attack | 4 | 3 | 4 |
| Cardiac arrest | 1 | <1 | 1 |
| Thromboembolic events (%)b | 4 | 4 | 4 |
| Cancer (%) | 5 | 6 | 4 |
| Smoking status | |||
| Current smoker (%) | 6 | 5 | 7 |
| Former smoker (%) | 22 | 22 | 22 |
| Baseline blood pressure (mmHg), median (25th–75th percentile) | |||
| Systolic | 135.0 (125.0–146.7) | 135.0 (124.3–146.7) | 135.3 (125.3–147.0) |
| Diastolic | 74.0 (66.0–81.3) | 74.0 (66.7–81.3) | 73.7 (65.3–81.0) |
| Mean arterial pressure | 94.7 (87.2–101.6) | 94.7 (87.4–101.6) | 94.6 (86.9–101.6) |
| Baseline laboratory values, median (25th–75th percentile) | |||
| Haemoglobin (g/dL) | 9.9 (9.2–10.5) | 10.4 (9.7–10.9) | 9.5 (9.0–10.0) |
| <9 g/dL (%) | 17 | 8 | 24 |
| 9–10 g/dL (%) | 38 | 25 | 49 |
| 10–11 g/dL (%) | 36 | 48 | 25 |
| >11 g/dL (%) | 10 | 18 | 2 |
| eGFR (mL/min/1.73m2) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) |
| Serum creatinine (mg/dL) | 3.0 (2.2–4.3) | 3.0 (2.1–4.2) | 3.1 (2.2–4.3) |
| hsCRP (mg/L) | 2.0 (0.8–5.4) | 2.1 (0.8–5.5) | 2.0 (0.8–5.3) |
| Albumin (g/dL) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) |
| Haemoglobin A1c (%) (in patients with diabetes) | 6.5 (5.8–7.6) | 6.5 (5.8–7.5) | 6.6 (5.8–7.6) |
| White blood cells (×109/L) | 6.5 (5.3–7.8) | 6.4 (5.2–7.8) | 6.5 (5.3–7.9) |
| Platelets (×109/L) | 215.0 (175.0–262.0) | 211.0 (171.0–260.0) | 220.0 (178.0–264.0) |
| Transferrin saturation (%) | 29.0 (23.0–37.0) | 31.0 (24.0–39.0) | 28.0 (23.0–35.0) |
| Ferritin (µg/L) | 271.0 (168.0–453.0) | 280.0 (173.0–472.0) | 264.0 (162.0–438.0) |
| Hepcidin (µg/L) | 105.3 (61.4–168.7) | 110.6 (62.8–175.8) | 101.4 (59.8–160.0) |
| iPTH (ng/L) | 125.4 (67.4–242.2) | 121.6 (65.5–241.2) | 128.2 (67.4–243.1) |
| Total cholesterol (mg/dL) | 158.3 (131.3–193.1) | 156.4 (129.3–189.2) | 160.2 (131.3–195.0) |
| Low-density lipoprotein cholesterol | 84.2 (64.1–110.8) | 81.9 (63.0–108.9) | 85.0 (66.0–112.0) |
| High-density lipoprotein cholesterol | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) |
| Medications (%) | |||
| Diabetes medications | 49 | 48 | 51 |
| Insulin | 31 | 30 | 31 |
| Antihypertensives | 95 | 95 | 95 |
| ACE inhibitor or ARB | 61 | 60 | 61 |
| Beta blocker | 48 | 49 | 48 |
| SGLT2 inhibitor | <1 | <1 | <1 |
| Statin | 55 | 54 | 56 |
| Anticoagulant | 8 | 8 | 7 |
| Antiplatelet | 37 | 37 | 38 |
| Aspirin | 30 | 29 | 31 |
| Vitamin K antagonist | 3 | 4 | 3 |
Phosphate bindersc Iron-based Calcium-based Non-calcium and non-iron based | 21 1 17 3 | 23 1 18 5 | 19 1 16 2 |
| Vitamin D | 33 | 35 | 32 |
| Calcimimetics | <1 | <1 | <1 |
| Oral irond | 49 | 48 | 51 |
| IV iron | 12 | 13 | 10 |
| Prior ESA use (%) | 47 | 100 | 0 |
Prior ESA type at randomization (%) Darbepoetin alfa only Epoetin only Methoxy PEG-epoetin beta only Multiple | 31 58 10 1 | 31 58 10 1 | 0 0 0 0 |
| Prior ESA dose standardized to IV epoetin (U/week)e | 3934 (2493–6310) | 3934 (2493–6310) | N/A |
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ERI, erythropoietin resistance index; ESA, erythropoiesis-stimulating agent; hsCRP, high-sensitivity C-reactive protein; iPTH, intact parathyroid hormone; ITT, intent-to-treat; IV, intravenous; N/A, not applicable; PEG, polyethylene glycol; SGLT2, sodium-glucose cotransporter 2.
Results are based on the in-stream database as of 23 April 2021. Until the time of database lock, data entered into the electronic case report form may be updated by investigator site staff. Therefore, final data may change with continued data updates.
Continuous variables are expressed as median (25th and 75th percentiles). All baseline laboratory tests were performed by a central laboratory except for haemoglobin, which uses central laboratory values if available, or a point-of-care HemoCue value if the central laboratory value is missing.
Haemoglobin A1C was only collected for patients with diabetes.
CVD in ASCEND-ND was defined as angina pectoris, MI, stroke, coronary artery disease, transient ischaemic attack, heart failure, atrial fibrillation, cardiac arrest and valvular heart disease.
Thromboembolic events include pulmonary embolism, deep vein thrombosis, retinal vein occlusion, arteriovenous graft thrombosis, arteriovenous fistula thrombosis and central venous catheter thrombosis.
Subjects may be counted in multiple rows.
Includes ferric citrate.
See Supplementary data, Table S6 for ESA dose conversion details.
Baseline characteristics of the overall ITT population and by ESA user status
| ESA user status | |||
|---|---|---|---|
| Characteristics | ITT population (N = 3872) | Yes (n = 1810) | No (n = 2062) |
| Age (years), median (25th–75th percentile) | 67 (57–75) | 68 (57–75) | 66 (57–74) |
| Women (%) | 56 | 58 | 55 |
| Race (%) | |||
| White | 56 | 58 | 54 |
| Asian | 27 | 27 | 28 |
| Black | 10 | 5 | 13 |
| American Indian or Alaska Native | 5 | 7 | 3 |
| Native Hawaiian or Other Pacific Islander | <1 | <1 | <1 |
| Multiple | 2 | 3 | 2 |
| Ethnicity, Hispanic or Latino (%) | 23 | 27 | 20 |
| Baseline weight (kg), median (25th–75th percentile) | 71.2 (60.0–84.5) | 70.9 (59.1–83.5) | 71.6 (60.4–85.9) |
| Baseline body mass index (kg/m2), median (25th–75th percentile) | 26.7 (23.3–31.1) | 26.7 (23.3–30.8) | 26.7 (23.3–31.3) |
CKD stage (%) Stage 1 and 2 Stage 3 Stage 4 Stage 5 Missing | <1 18 46 36 <1 | <1 18 47 35 <1 | <1 18 45 36 0 |
| Renal comorbidities (%) | |||
| Hypertension | 94 | 94 | 95 |
| Diabetes | 57 | 54 | 59 |
| Glomerulonephritis | 10 | 11 | 9 |
| Autosomal dominant polycystic kidney disease | 3 | 2 | 4 |
| Cardiovascular disease history (%)a | 37 | 37 | 37 |
| Coronary artery disease | 20 | 20 | 19 |
| Heart failure | 13 | 12 | 14 |
| Valvular heart disease | 8 | 9 | 6 |
| Angina pectoris | 8 | 8 | 7 |
| Atrial fibrillation | 5 | 6 | 5 |
| Myocardial infarction | 7 | 8 | 6 |
| Stroke | 7 | 6 | 7 |
| Transient ischaemic attack | 4 | 3 | 4 |
| Cardiac arrest | 1 | <1 | 1 |
| Thromboembolic events (%)b | 4 | 4 | 4 |
| Cancer (%) | 5 | 6 | 4 |
| Smoking status | |||
| Current smoker (%) | 6 | 5 | 7 |
| Former smoker (%) | 22 | 22 | 22 |
| Baseline blood pressure (mmHg), median (25th–75th percentile) | |||
| Systolic | 135.0 (125.0–146.7) | 135.0 (124.3–146.7) | 135.3 (125.3–147.0) |
| Diastolic | 74.0 (66.0–81.3) | 74.0 (66.7–81.3) | 73.7 (65.3–81.0) |
| Mean arterial pressure | 94.7 (87.2–101.6) | 94.7 (87.4–101.6) | 94.6 (86.9–101.6) |
| Baseline laboratory values, median (25th–75th percentile) | |||
| Haemoglobin (g/dL) | 9.9 (9.2–10.5) | 10.4 (9.7–10.9) | 9.5 (9.0–10.0) |
| <9 g/dL (%) | 17 | 8 | 24 |
| 9–10 g/dL (%) | 38 | 25 | 49 |
| 10–11 g/dL (%) | 36 | 48 | 25 |
| >11 g/dL (%) | 10 | 18 | 2 |
| eGFR (mL/min/1.73m2) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) |
| Serum creatinine (mg/dL) | 3.0 (2.2–4.3) | 3.0 (2.1–4.2) | 3.1 (2.2–4.3) |
| hsCRP (mg/L) | 2.0 (0.8–5.4) | 2.1 (0.8–5.5) | 2.0 (0.8–5.3) |
| Albumin (g/dL) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) |
| Haemoglobin A1c (%) (in patients with diabetes) | 6.5 (5.8–7.6) | 6.5 (5.8–7.5) | 6.6 (5.8–7.6) |
| White blood cells (×109/L) | 6.5 (5.3–7.8) | 6.4 (5.2–7.8) | 6.5 (5.3–7.9) |
| Platelets (×109/L) | 215.0 (175.0–262.0) | 211.0 (171.0–260.0) | 220.0 (178.0–264.0) |
| Transferrin saturation (%) | 29.0 (23.0–37.0) | 31.0 (24.0–39.0) | 28.0 (23.0–35.0) |
| Ferritin (µg/L) | 271.0 (168.0–453.0) | 280.0 (173.0–472.0) | 264.0 (162.0–438.0) |
| Hepcidin (µg/L) | 105.3 (61.4–168.7) | 110.6 (62.8–175.8) | 101.4 (59.8–160.0) |
| iPTH (ng/L) | 125.4 (67.4–242.2) | 121.6 (65.5–241.2) | 128.2 (67.4–243.1) |
| Total cholesterol (mg/dL) | 158.3 (131.3–193.1) | 156.4 (129.3–189.2) | 160.2 (131.3–195.0) |
| Low-density lipoprotein cholesterol | 84.2 (64.1–110.8) | 81.9 (63.0–108.9) | 85.0 (66.0–112.0) |
| High-density lipoprotein cholesterol | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) |
| Medications (%) | |||
| Diabetes medications | 49 | 48 | 51 |
| Insulin | 31 | 30 | 31 |
| Antihypertensives | 95 | 95 | 95 |
| ACE inhibitor or ARB | 61 | 60 | 61 |
| Beta blocker | 48 | 49 | 48 |
| SGLT2 inhibitor | <1 | <1 | <1 |
| Statin | 55 | 54 | 56 |
| Anticoagulant | 8 | 8 | 7 |
| Antiplatelet | 37 | 37 | 38 |
| Aspirin | 30 | 29 | 31 |
| Vitamin K antagonist | 3 | 4 | 3 |
Phosphate bindersc Iron-based Calcium-based Non-calcium and non-iron based | 21 1 17 3 | 23 1 18 5 | 19 1 16 2 |
| Vitamin D | 33 | 35 | 32 |
| Calcimimetics | <1 | <1 | <1 |
| Oral irond | 49 | 48 | 51 |
| IV iron | 12 | 13 | 10 |
| Prior ESA use (%) | 47 | 100 | 0 |
Prior ESA type at randomization (%) Darbepoetin alfa only Epoetin only Methoxy PEG-epoetin beta only Multiple | 31 58 10 1 | 31 58 10 1 | 0 0 0 0 |
| Prior ESA dose standardized to IV epoetin (U/week)e | 3934 (2493–6310) | 3934 (2493–6310) | N/A |
| ESA user status | |||
|---|---|---|---|
| Characteristics | ITT population (N = 3872) | Yes (n = 1810) | No (n = 2062) |
| Age (years), median (25th–75th percentile) | 67 (57–75) | 68 (57–75) | 66 (57–74) |
| Women (%) | 56 | 58 | 55 |
| Race (%) | |||
| White | 56 | 58 | 54 |
| Asian | 27 | 27 | 28 |
| Black | 10 | 5 | 13 |
| American Indian or Alaska Native | 5 | 7 | 3 |
| Native Hawaiian or Other Pacific Islander | <1 | <1 | <1 |
| Multiple | 2 | 3 | 2 |
| Ethnicity, Hispanic or Latino (%) | 23 | 27 | 20 |
| Baseline weight (kg), median (25th–75th percentile) | 71.2 (60.0–84.5) | 70.9 (59.1–83.5) | 71.6 (60.4–85.9) |
| Baseline body mass index (kg/m2), median (25th–75th percentile) | 26.7 (23.3–31.1) | 26.7 (23.3–30.8) | 26.7 (23.3–31.3) |
CKD stage (%) Stage 1 and 2 Stage 3 Stage 4 Stage 5 Missing | <1 18 46 36 <1 | <1 18 47 35 <1 | <1 18 45 36 0 |
| Renal comorbidities (%) | |||
| Hypertension | 94 | 94 | 95 |
| Diabetes | 57 | 54 | 59 |
| Glomerulonephritis | 10 | 11 | 9 |
| Autosomal dominant polycystic kidney disease | 3 | 2 | 4 |
| Cardiovascular disease history (%)a | 37 | 37 | 37 |
| Coronary artery disease | 20 | 20 | 19 |
| Heart failure | 13 | 12 | 14 |
| Valvular heart disease | 8 | 9 | 6 |
| Angina pectoris | 8 | 8 | 7 |
| Atrial fibrillation | 5 | 6 | 5 |
| Myocardial infarction | 7 | 8 | 6 |
| Stroke | 7 | 6 | 7 |
| Transient ischaemic attack | 4 | 3 | 4 |
| Cardiac arrest | 1 | <1 | 1 |
| Thromboembolic events (%)b | 4 | 4 | 4 |
| Cancer (%) | 5 | 6 | 4 |
| Smoking status | |||
| Current smoker (%) | 6 | 5 | 7 |
| Former smoker (%) | 22 | 22 | 22 |
| Baseline blood pressure (mmHg), median (25th–75th percentile) | |||
| Systolic | 135.0 (125.0–146.7) | 135.0 (124.3–146.7) | 135.3 (125.3–147.0) |
| Diastolic | 74.0 (66.0–81.3) | 74.0 (66.7–81.3) | 73.7 (65.3–81.0) |
| Mean arterial pressure | 94.7 (87.2–101.6) | 94.7 (87.4–101.6) | 94.6 (86.9–101.6) |
| Baseline laboratory values, median (25th–75th percentile) | |||
| Haemoglobin (g/dL) | 9.9 (9.2–10.5) | 10.4 (9.7–10.9) | 9.5 (9.0–10.0) |
| <9 g/dL (%) | 17 | 8 | 24 |
| 9–10 g/dL (%) | 38 | 25 | 49 |
| 10–11 g/dL (%) | 36 | 48 | 25 |
| >11 g/dL (%) | 10 | 18 | 2 |
| eGFR (mL/min/1.73m2) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) | 18.0 (12.0–26.0) |
| Serum creatinine (mg/dL) | 3.0 (2.2–4.3) | 3.0 (2.1–4.2) | 3.1 (2.2–4.3) |
| hsCRP (mg/L) | 2.0 (0.8–5.4) | 2.1 (0.8–5.5) | 2.0 (0.8–5.3) |
| Albumin (g/dL) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) | 4.0 (3.7–4.2) |
| Haemoglobin A1c (%) (in patients with diabetes) | 6.5 (5.8–7.6) | 6.5 (5.8–7.5) | 6.6 (5.8–7.6) |
| White blood cells (×109/L) | 6.5 (5.3–7.8) | 6.4 (5.2–7.8) | 6.5 (5.3–7.9) |
| Platelets (×109/L) | 215.0 (175.0–262.0) | 211.0 (171.0–260.0) | 220.0 (178.0–264.0) |
| Transferrin saturation (%) | 29.0 (23.0–37.0) | 31.0 (24.0–39.0) | 28.0 (23.0–35.0) |
| Ferritin (µg/L) | 271.0 (168.0–453.0) | 280.0 (173.0–472.0) | 264.0 (162.0–438.0) |
| Hepcidin (µg/L) | 105.3 (61.4–168.7) | 110.6 (62.8–175.8) | 101.4 (59.8–160.0) |
| iPTH (ng/L) | 125.4 (67.4–242.2) | 121.6 (65.5–241.2) | 128.2 (67.4–243.1) |
| Total cholesterol (mg/dL) | 158.3 (131.3–193.1) | 156.4 (129.3–189.2) | 160.2 (131.3–195.0) |
| Low-density lipoprotein cholesterol | 84.2 (64.1–110.8) | 81.9 (63.0–108.9) | 85.0 (66.0–112.0) |
| High-density lipoprotein cholesterol | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) | 46.3 (36.7–56.0) |
| Medications (%) | |||
| Diabetes medications | 49 | 48 | 51 |
| Insulin | 31 | 30 | 31 |
| Antihypertensives | 95 | 95 | 95 |
| ACE inhibitor or ARB | 61 | 60 | 61 |
| Beta blocker | 48 | 49 | 48 |
| SGLT2 inhibitor | <1 | <1 | <1 |
| Statin | 55 | 54 | 56 |
| Anticoagulant | 8 | 8 | 7 |
| Antiplatelet | 37 | 37 | 38 |
| Aspirin | 30 | 29 | 31 |
| Vitamin K antagonist | 3 | 4 | 3 |
Phosphate bindersc Iron-based Calcium-based Non-calcium and non-iron based | 21 1 17 3 | 23 1 18 5 | 19 1 16 2 |
| Vitamin D | 33 | 35 | 32 |
| Calcimimetics | <1 | <1 | <1 |
| Oral irond | 49 | 48 | 51 |
| IV iron | 12 | 13 | 10 |
| Prior ESA use (%) | 47 | 100 | 0 |
Prior ESA type at randomization (%) Darbepoetin alfa only Epoetin only Methoxy PEG-epoetin beta only Multiple | 31 58 10 1 | 31 58 10 1 | 0 0 0 0 |
| Prior ESA dose standardized to IV epoetin (U/week)e | 3934 (2493–6310) | 3934 (2493–6310) | N/A |
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ERI, erythropoietin resistance index; ESA, erythropoiesis-stimulating agent; hsCRP, high-sensitivity C-reactive protein; iPTH, intact parathyroid hormone; ITT, intent-to-treat; IV, intravenous; N/A, not applicable; PEG, polyethylene glycol; SGLT2, sodium-glucose cotransporter 2.
Results are based on the in-stream database as of 23 April 2021. Until the time of database lock, data entered into the electronic case report form may be updated by investigator site staff. Therefore, final data may change with continued data updates.
Continuous variables are expressed as median (25th and 75th percentiles). All baseline laboratory tests were performed by a central laboratory except for haemoglobin, which uses central laboratory values if available, or a point-of-care HemoCue value if the central laboratory value is missing.
Haemoglobin A1C was only collected for patients with diabetes.
CVD in ASCEND-ND was defined as angina pectoris, MI, stroke, coronary artery disease, transient ischaemic attack, heart failure, atrial fibrillation, cardiac arrest and valvular heart disease.
Thromboembolic events include pulmonary embolism, deep vein thrombosis, retinal vein occlusion, arteriovenous graft thrombosis, arteriovenous fistula thrombosis and central venous catheter thrombosis.
Subjects may be counted in multiple rows.
Includes ferric citrate.
See Supplementary data, Table S6 for ESA dose conversion details.
The median Hb at randomization was 9.9 g/dL, eGFR was 18 mL/min/1.73 m2 and participants were iron-replete (transferrin saturation 29% and median ferritin 271 µg/L). Fifty-three percent of participants were not receiving ESAs prior to the study. Participants receiving ESAs were predominantly receiving epoetin (58%) or darbepoetin (31%). Oral iron was prescribed for 49% of participants at baseline; 12% received intravenous iron. Sixty-one percent of participants were using renin–angiotensin system blockers.
Although ESA usage at entry was well balanced across most of the baseline characteristics, there was a notable difference of less ESA use in the Black population at entry (5% of ESA users were Black compared with 13% of ESA non users). Participants receiving ESA therapy at entry had higher median Hb levels (10.4 versus 9.5 g/dL); other clinical and laboratory parameters as well as other concomitant medications by ESA status at entry are listed in Table 5.
ASCEND-ND compared with other large ND anaemia trials
Participants enrolled in ASCEND-ND generally had similar demographic characteristics as the participants in other HIF-PHI ND trials [19, 20], as well as TREAT [21]. ASCEND-ND and the PRO2TECT ND [20] trials had an active comparator, whereas the OLYMPUS [19] and TREAT [21] trials compared the respective agents with placebo (Table 6).
Comparison of ASCEND-ND baseline characteristics with characteristics of patients enrolled in large cardiovascular outcome trials in a non-dialysis population
| Characteristics | ASCEND-ND (N = 3872) | Roxadustat ND (N = 2781) Roxa versus placebo[19] | Vadadustat ND ESA-untreated (N = 1751)[20] | Vadadustat ND ESA-treated (N = 1725)[20] | TREAT (N = 4044)[21] | |
|---|---|---|---|---|---|---|
| Design | ||||||
| Population | Anaemia of CKD not on dialysis | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD and Type 2 diabetes mellitus | |
| Blinding | Open-label (sponsor- blind) | Double-blinded (safety personnel not blinded) | Open-label (sponsor and executive steering committee blinded) | Open-label (sponsor and executive steering committee blinded) | Double-blinded | |
| Intervention | Daprodustat | Roxadustat | Vadadustat | Vadadustat | Darbepoetin alfa | |
| Control | Active-controlled (darbepoetin alfa) | Placebo-controlled | Active-controlled (darbepoetin alfa) | Active-controlled (darbepoetin alfa) | Placebo-controlled | |
| Location | 30% EMEA, 26% NA (predominantly USA), 15% LA, 28% APAC | 30% EMEA, 35% NA, 16% LA, 19% APAC | 61% USA, 39% other | 38% USA, 62% other | 26% EMEA, 62% NA, 11% LA, APAC 1% | |
| Demographics | ||||||
| Age (years) | 67.0 | 60.9 | 62.4 | 65.0 | 66.9 | 68 |
| Women, % | 56.0 | 59.2 | 56.2 | 56.0 | 55.4 | 57.3 |
| BMI (kg/m2) | 26.7 | 26.7 | 26.9 | 29.7 | 29.4 | 30.3 |
| Race, % | ||||||
| White | 56 | 45 | 44 | 64 | 72 | 64 |
| Asian | 27 | 39 | 39 | 5 | 7 | NR |
| Black | 10 | 8 | 8 | 21 | 13 | 20 |
| Other | 7 | 8 | 8 | 10 | 7 | 16 |
CKD stage, % Stage 1 and 2 Stage 3 Stage 4 Stage 5 | <1 18 46 36 | NR NR NR NR | NR NR NR NR | NR 21 44 34 | NR 24 48 28 | NR NR NR NR |
| Missing | <1 | NR | NR | 1 | <1 | NR |
| Comorbidities, % | ||||||
| Cardiovascular disease | 37 | NR | NR | 47 | 45 | 65 |
| Diabetes mellitus | 57 | 53 | 56 | 58 | 51 | 100 |
| Heart failure | 13 | 11 | 11 | EX | EX | 33 |
| Hypertension | 94 | 92 | 93 | 57 | 54 | NR |
| Myocardial infarction | 7 | EX | EX | EX | EX | 18 |
| Stroke | 7 | EX | EX | EX | EX | 11 |
Blood pressure, mmHg Systolic Diastolic | 135.0 74.0 | 134.4 74.5 | 135.5 74.1 | 139.2 73.6 | 136.7 73.8 | 136 71 |
Laboratory values Haemoglobin, g/dL eGFR (mL/min/1.73 m2) Serum creatinine hsCRP (mg/dL) Ferritin (µg/L) Transferrin saturation (%) Hepcidin (µg/L) | 9.9 18.0 3.0 2.0 271.0 29.0 105.3 | 9.1 19.7 NR 0.7 NR NR 163.2 | 9.1 20.0 NR 0.7 NR NR 155.5 | 9.1 21.5 3.4 0.8 364.2 30.9 105.1 | 10.4 22.7 3.1 0.8 375.8 32.9 103.3 | 10.4 34 1.9 NR 133 23 NR |
Concomitant medications, % ACEi/ARB Aspirin Oral iron Statin | 61 30 49 55 | 66 NR 53 51 | 61 NR 53 51.1 | 60 35 29 NR | 64 33 24 NR | 80 42 42 58 |
| ESA use, % | 47 | EXa | EXa | N/A | 100b | 9.5 |
| Prior ESA dose standardized to IV epoetin (U/week) | 3934 | N/A | N/A | N/A | 104.7c | NR |
| Characteristics | ASCEND-ND (N = 3872) | Roxadustat ND (N = 2781) Roxa versus placebo[19] | Vadadustat ND ESA-untreated (N = 1751)[20] | Vadadustat ND ESA-treated (N = 1725)[20] | TREAT (N = 4044)[21] | |
|---|---|---|---|---|---|---|
| Design | ||||||
| Population | Anaemia of CKD not on dialysis | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD and Type 2 diabetes mellitus | |
| Blinding | Open-label (sponsor- blind) | Double-blinded (safety personnel not blinded) | Open-label (sponsor and executive steering committee blinded) | Open-label (sponsor and executive steering committee blinded) | Double-blinded | |
| Intervention | Daprodustat | Roxadustat | Vadadustat | Vadadustat | Darbepoetin alfa | |
| Control | Active-controlled (darbepoetin alfa) | Placebo-controlled | Active-controlled (darbepoetin alfa) | Active-controlled (darbepoetin alfa) | Placebo-controlled | |
| Location | 30% EMEA, 26% NA (predominantly USA), 15% LA, 28% APAC | 30% EMEA, 35% NA, 16% LA, 19% APAC | 61% USA, 39% other | 38% USA, 62% other | 26% EMEA, 62% NA, 11% LA, APAC 1% | |
| Demographics | ||||||
| Age (years) | 67.0 | 60.9 | 62.4 | 65.0 | 66.9 | 68 |
| Women, % | 56.0 | 59.2 | 56.2 | 56.0 | 55.4 | 57.3 |
| BMI (kg/m2) | 26.7 | 26.7 | 26.9 | 29.7 | 29.4 | 30.3 |
| Race, % | ||||||
| White | 56 | 45 | 44 | 64 | 72 | 64 |
| Asian | 27 | 39 | 39 | 5 | 7 | NR |
| Black | 10 | 8 | 8 | 21 | 13 | 20 |
| Other | 7 | 8 | 8 | 10 | 7 | 16 |
CKD stage, % Stage 1 and 2 Stage 3 Stage 4 Stage 5 | <1 18 46 36 | NR NR NR NR | NR NR NR NR | NR 21 44 34 | NR 24 48 28 | NR NR NR NR |
| Missing | <1 | NR | NR | 1 | <1 | NR |
| Comorbidities, % | ||||||
| Cardiovascular disease | 37 | NR | NR | 47 | 45 | 65 |
| Diabetes mellitus | 57 | 53 | 56 | 58 | 51 | 100 |
| Heart failure | 13 | 11 | 11 | EX | EX | 33 |
| Hypertension | 94 | 92 | 93 | 57 | 54 | NR |
| Myocardial infarction | 7 | EX | EX | EX | EX | 18 |
| Stroke | 7 | EX | EX | EX | EX | 11 |
Blood pressure, mmHg Systolic Diastolic | 135.0 74.0 | 134.4 74.5 | 135.5 74.1 | 139.2 73.6 | 136.7 73.8 | 136 71 |
Laboratory values Haemoglobin, g/dL eGFR (mL/min/1.73 m2) Serum creatinine hsCRP (mg/dL) Ferritin (µg/L) Transferrin saturation (%) Hepcidin (µg/L) | 9.9 18.0 3.0 2.0 271.0 29.0 105.3 | 9.1 19.7 NR 0.7 NR NR 163.2 | 9.1 20.0 NR 0.7 NR NR 155.5 | 9.1 21.5 3.4 0.8 364.2 30.9 105.1 | 10.4 22.7 3.1 0.8 375.8 32.9 103.3 | 10.4 34 1.9 NR 133 23 NR |
Concomitant medications, % ACEi/ARB Aspirin Oral iron Statin | 61 30 49 55 | 66 NR 53 51 | 61 NR 53 51.1 | 60 35 29 NR | 64 33 24 NR | 80 42 42 58 |
| ESA use, % | 47 | EXa | EXa | N/A | 100b | 9.5 |
| Prior ESA dose standardized to IV epoetin (U/week) | 3934 | N/A | N/A | N/A | 104.7c | NR |
ACE, angiotensin-converting enzyme; APAC, Asia Pacific; ARB, angiotensin receptor blocker; BMI, body mass index; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; EMEA, Europe, Middle East and Africa; ESA, erythropoiesis-stimulating agent; EX, excluded; hsCRP, high-sensitivity C-reactive protein; IV, intravenous; LA, Latin America; N/A, not applicable; NA, North America; NDD, non-dialysis-dependent; NR, not reported.
Continuous variables are expressed as medians (ASCEND-ND and TREAT) and means (roxadustat and vadadustat). CVD definition varies by study (ASCEND-ND: angina pectoris, MI, stroke, coronary artery disease, transient ischaemic attack, heart failure, atrial fibrillation, cardiac arrest and valvular heart disease; TREAT: coronary artery disease, MI, peripheral arterial disease, stroke, transient ischaemic attack, heart failure, prior arterial revascularization, valvular heart disease, ventricular tachycardia and/or fibrillation and an automatic implantable cardioverter defibrillator or pacemaker).
Exclusion criteria, no ESA use within last 6 weeks.
Assumption based on eligibility criteria.
ESA dose standardized to IV epoetin units per kg/week calculated using baseline weight.
Comparison of ASCEND-ND baseline characteristics with characteristics of patients enrolled in large cardiovascular outcome trials in a non-dialysis population
| Characteristics | ASCEND-ND (N = 3872) | Roxadustat ND (N = 2781) Roxa versus placebo[19] | Vadadustat ND ESA-untreated (N = 1751)[20] | Vadadustat ND ESA-treated (N = 1725)[20] | TREAT (N = 4044)[21] | |
|---|---|---|---|---|---|---|
| Design | ||||||
| Population | Anaemia of CKD not on dialysis | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD and Type 2 diabetes mellitus | |
| Blinding | Open-label (sponsor- blind) | Double-blinded (safety personnel not blinded) | Open-label (sponsor and executive steering committee blinded) | Open-label (sponsor and executive steering committee blinded) | Double-blinded | |
| Intervention | Daprodustat | Roxadustat | Vadadustat | Vadadustat | Darbepoetin alfa | |
| Control | Active-controlled (darbepoetin alfa) | Placebo-controlled | Active-controlled (darbepoetin alfa) | Active-controlled (darbepoetin alfa) | Placebo-controlled | |
| Location | 30% EMEA, 26% NA (predominantly USA), 15% LA, 28% APAC | 30% EMEA, 35% NA, 16% LA, 19% APAC | 61% USA, 39% other | 38% USA, 62% other | 26% EMEA, 62% NA, 11% LA, APAC 1% | |
| Demographics | ||||||
| Age (years) | 67.0 | 60.9 | 62.4 | 65.0 | 66.9 | 68 |
| Women, % | 56.0 | 59.2 | 56.2 | 56.0 | 55.4 | 57.3 |
| BMI (kg/m2) | 26.7 | 26.7 | 26.9 | 29.7 | 29.4 | 30.3 |
| Race, % | ||||||
| White | 56 | 45 | 44 | 64 | 72 | 64 |
| Asian | 27 | 39 | 39 | 5 | 7 | NR |
| Black | 10 | 8 | 8 | 21 | 13 | 20 |
| Other | 7 | 8 | 8 | 10 | 7 | 16 |
CKD stage, % Stage 1 and 2 Stage 3 Stage 4 Stage 5 | <1 18 46 36 | NR NR NR NR | NR NR NR NR | NR 21 44 34 | NR 24 48 28 | NR NR NR NR |
| Missing | <1 | NR | NR | 1 | <1 | NR |
| Comorbidities, % | ||||||
| Cardiovascular disease | 37 | NR | NR | 47 | 45 | 65 |
| Diabetes mellitus | 57 | 53 | 56 | 58 | 51 | 100 |
| Heart failure | 13 | 11 | 11 | EX | EX | 33 |
| Hypertension | 94 | 92 | 93 | 57 | 54 | NR |
| Myocardial infarction | 7 | EX | EX | EX | EX | 18 |
| Stroke | 7 | EX | EX | EX | EX | 11 |
Blood pressure, mmHg Systolic Diastolic | 135.0 74.0 | 134.4 74.5 | 135.5 74.1 | 139.2 73.6 | 136.7 73.8 | 136 71 |
Laboratory values Haemoglobin, g/dL eGFR (mL/min/1.73 m2) Serum creatinine hsCRP (mg/dL) Ferritin (µg/L) Transferrin saturation (%) Hepcidin (µg/L) | 9.9 18.0 3.0 2.0 271.0 29.0 105.3 | 9.1 19.7 NR 0.7 NR NR 163.2 | 9.1 20.0 NR 0.7 NR NR 155.5 | 9.1 21.5 3.4 0.8 364.2 30.9 105.1 | 10.4 22.7 3.1 0.8 375.8 32.9 103.3 | 10.4 34 1.9 NR 133 23 NR |
Concomitant medications, % ACEi/ARB Aspirin Oral iron Statin | 61 30 49 55 | 66 NR 53 51 | 61 NR 53 51.1 | 60 35 29 NR | 64 33 24 NR | 80 42 42 58 |
| ESA use, % | 47 | EXa | EXa | N/A | 100b | 9.5 |
| Prior ESA dose standardized to IV epoetin (U/week) | 3934 | N/A | N/A | N/A | 104.7c | NR |
| Characteristics | ASCEND-ND (N = 3872) | Roxadustat ND (N = 2781) Roxa versus placebo[19] | Vadadustat ND ESA-untreated (N = 1751)[20] | Vadadustat ND ESA-treated (N = 1725)[20] | TREAT (N = 4044)[21] | |
|---|---|---|---|---|---|---|
| Design | ||||||
| Population | Anaemia of CKD not on dialysis | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD | Anaemia of NDD- CKD and Type 2 diabetes mellitus | |
| Blinding | Open-label (sponsor- blind) | Double-blinded (safety personnel not blinded) | Open-label (sponsor and executive steering committee blinded) | Open-label (sponsor and executive steering committee blinded) | Double-blinded | |
| Intervention | Daprodustat | Roxadustat | Vadadustat | Vadadustat | Darbepoetin alfa | |
| Control | Active-controlled (darbepoetin alfa) | Placebo-controlled | Active-controlled (darbepoetin alfa) | Active-controlled (darbepoetin alfa) | Placebo-controlled | |
| Location | 30% EMEA, 26% NA (predominantly USA), 15% LA, 28% APAC | 30% EMEA, 35% NA, 16% LA, 19% APAC | 61% USA, 39% other | 38% USA, 62% other | 26% EMEA, 62% NA, 11% LA, APAC 1% | |
| Demographics | ||||||
| Age (years) | 67.0 | 60.9 | 62.4 | 65.0 | 66.9 | 68 |
| Women, % | 56.0 | 59.2 | 56.2 | 56.0 | 55.4 | 57.3 |
| BMI (kg/m2) | 26.7 | 26.7 | 26.9 | 29.7 | 29.4 | 30.3 |
| Race, % | ||||||
| White | 56 | 45 | 44 | 64 | 72 | 64 |
| Asian | 27 | 39 | 39 | 5 | 7 | NR |
| Black | 10 | 8 | 8 | 21 | 13 | 20 |
| Other | 7 | 8 | 8 | 10 | 7 | 16 |
CKD stage, % Stage 1 and 2 Stage 3 Stage 4 Stage 5 | <1 18 46 36 | NR NR NR NR | NR NR NR NR | NR 21 44 34 | NR 24 48 28 | NR NR NR NR |
| Missing | <1 | NR | NR | 1 | <1 | NR |
| Comorbidities, % | ||||||
| Cardiovascular disease | 37 | NR | NR | 47 | 45 | 65 |
| Diabetes mellitus | 57 | 53 | 56 | 58 | 51 | 100 |
| Heart failure | 13 | 11 | 11 | EX | EX | 33 |
| Hypertension | 94 | 92 | 93 | 57 | 54 | NR |
| Myocardial infarction | 7 | EX | EX | EX | EX | 18 |
| Stroke | 7 | EX | EX | EX | EX | 11 |
Blood pressure, mmHg Systolic Diastolic | 135.0 74.0 | 134.4 74.5 | 135.5 74.1 | 139.2 73.6 | 136.7 73.8 | 136 71 |
Laboratory values Haemoglobin, g/dL eGFR (mL/min/1.73 m2) Serum creatinine hsCRP (mg/dL) Ferritin (µg/L) Transferrin saturation (%) Hepcidin (µg/L) | 9.9 18.0 3.0 2.0 271.0 29.0 105.3 | 9.1 19.7 NR 0.7 NR NR 163.2 | 9.1 20.0 NR 0.7 NR NR 155.5 | 9.1 21.5 3.4 0.8 364.2 30.9 105.1 | 10.4 22.7 3.1 0.8 375.8 32.9 103.3 | 10.4 34 1.9 NR 133 23 NR |
Concomitant medications, % ACEi/ARB Aspirin Oral iron Statin | 61 30 49 55 | 66 NR 53 51 | 61 NR 53 51.1 | 60 35 29 NR | 64 33 24 NR | 80 42 42 58 |
| ESA use, % | 47 | EXa | EXa | N/A | 100b | 9.5 |
| Prior ESA dose standardized to IV epoetin (U/week) | 3934 | N/A | N/A | N/A | 104.7c | NR |
ACE, angiotensin-converting enzyme; APAC, Asia Pacific; ARB, angiotensin receptor blocker; BMI, body mass index; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; EMEA, Europe, Middle East and Africa; ESA, erythropoiesis-stimulating agent; EX, excluded; hsCRP, high-sensitivity C-reactive protein; IV, intravenous; LA, Latin America; N/A, not applicable; NA, North America; NDD, non-dialysis-dependent; NR, not reported.
Continuous variables are expressed as medians (ASCEND-ND and TREAT) and means (roxadustat and vadadustat). CVD definition varies by study (ASCEND-ND: angina pectoris, MI, stroke, coronary artery disease, transient ischaemic attack, heart failure, atrial fibrillation, cardiac arrest and valvular heart disease; TREAT: coronary artery disease, MI, peripheral arterial disease, stroke, transient ischaemic attack, heart failure, prior arterial revascularization, valvular heart disease, ventricular tachycardia and/or fibrillation and an automatic implantable cardioverter defibrillator or pacemaker).
Exclusion criteria, no ESA use within last 6 weeks.
Assumption based on eligibility criteria.
ESA dose standardized to IV epoetin units per kg/week calculated using baseline weight.
Participants in the OLYMPUS trial [19] of roxadustat were somewhat younger than those in the other trials, but the distribution of kidney function appears similar in the trials that have reported these data. TREAT [21] was conducted entirely among people with diabetes, whereas slightly more than half of participants in the HIF-PHI trials had diabetes. Ferritin and transferrin levels were lower in TREAT [21] than in the HIF-PHI trials [19, 20], but eGFR and Hb levels were higher. Other concomitant medications, clinical characteristics and laboratory parameters were similar across the various trials.
DISCUSSION
Anaemia is common in patients with advanced CKD and its treatment remains an area of considerable uncertainty. ESA therapy is widely used to maintain Hb levels in patients with CKD not requiring dialysis; however, parenteral treatment can be inconvenient and painful. HIF-PHIs such as daprodustat potentially offer a more convenient oral alternative and have demonstrated promising safety and efficacy profiles in studies reported to date [15, 22]. The ASCEND-ND trial, as part of the broader group of ASCEND trials including prevalent (ASCEND-D) and incident (ASCEND-ID) dialysis studies, will specifically define the benefit–risk profile of daprodustat when used in patients with anaemia of CKD.
The ASCEND-ND trial compares oral daprodustat with darbepoetin alfa because ESA therapy is currently the standard of care in this population and is recommended in relevant guidelines [4–6]. A difficulty in the design of a trial like ASCEND-ND is blinding participants to the randomized treatment given that the active comparator is administered parenterally. In view of the practical complexities of a double-blind, double-dummy design for a trial of this magnitude, ASCEND-ND has an open-label, sponsor-blinded, design. Throughout the study, the steering and adjudication committees also remained blinded to treatment assignment [23]. Although we acknowledge that the selected ESA comparator (darbepoetin alfa) and the specific dose steps and frequency of administration may differ from local ESA protocols, a protocol-mandated dose adjustment algorithm was used for both treatment groups to prevent differential treatment of each group as had occurred in earlier daprodustat clinical trials [24].
Implementation of a global Hb target range of 10–11 g/dL also enabled uniform treatment of anaemia for participants in all regions. This range accommodates most anaemia guidelines and ESA labelling worldwide, except for the USA, where labelling is more restricted [25]. Other standardizations included a single comparator with defined dose steps and frequency, as well as common iron management protocols, and an anaemia rescue algorithm that was used for both treatment groups. For anaemia rescue, early intervention with intravenous iron and/or transfusions was allowed to help increase Hb before declaring a patient had met the rescue endpoint, which in turn led to permanent discontinuation of randomized treatment. These standardizations will help ensure a more unbiased comparison between the randomized treatment groups.
The trial uses co-primary outcomes that reflect the major questions regarding the potential role of daprodustat in clinical practice. The first is to demonstrate that it is an effective treatment for the anaemia of CKD and is not inferior to ESA therapy as the current standard of care. The second co-primary outcome assesses the CV safety of daprodustat, defined as non-inferiority to ESA for the outcome of a MACE. If non-inferiority is confirmed, the potential for daprodustat to achieve superiority in reducing the risk of MACE and CKD progression will also be assessed. The more physiological circulating levels of erythropoietin likely to be achieved with daprodustat have the potential to lead to better CV outcomes. However, this was not found in pooled ND studies of another HIF-PHI, vadadustat, in which a HR of 1.17 [95% confidence interval (CI) 1.01–1.36] for CV safety did not meet the prespecified non-inferiority criterion of 1.25 for this agent compared with darbepoetin alfa [26]. Another programme assessing the HIF-PHI roxadustat included a pooled CV safety analysis across three placebo-controlled trials with a MACE HR of 1.10 (95% CI 0.96–1.27) being observed [27]; a smaller active-controlled study (Dolomites; NCT02021318) was not powered for CV safety but reported a HR of 0.81 (95% CI 0.52–1.25) [28]. At this time, it is not known what will be seen with other HIF-PHIs.
The optimal Hb target in the CKD population treated with ESAs has been discussed extensively in the scientific kidney community and the lower Hb target—as assessed in both arms of the daprodustat ND trial—should provide a useful comparison of the two therapies. Additionally, the progression of CKD in both treatment arms will be assessed.
ASCEND-ND was designed to allow generalization of the results to the broadest possible CKD population by ensuring that participants are representative of the overall CKD population. Sites were selected from around the world in order to achieve a broad geographical representation in recruitment. A placebo run-in period was established to confirm adherence to an oral medication and to minimize the withdrawal of consent post-randomization that occurred in a prior daprodustat study [29]. While this helped ensure that participants were adherent with randomized treatment, it may limit the general applicability of the results, particularly considering that the oral route of administration is a major advantage of the potential treatment. The baseline characteristics of randomized participants are very similar to those of previously reported trials in participants with anaemia related to CKD, allowing some degree of comparison across trials and comparators (active or placebo).
ASCEND-ND has a number of strengths. It is one of the largest prospective randomized cardiovascular outcomes trials in this field. ASCEND-ND recruited a diverse population with respect to race. Participants include members of minority groups such as those who self-identified as Black, Native American and Latino. Furthermore, one-third of participants within the USA were Black. This diversity of the study population will be important in generalizing the safety and efficacy of daprodustat.
In conclusion, ASCEND-ND has enrolled 3872 participants who are generally representative of patients with anaemia of CKD not on dialysis. The study will test the hypothesis that daprodustat is non-inferior to the comparator darbepoetin alfa for two co-primary endpoints—Hb efficacy and CV safety—as well as a range of secondary efficacy outcomes. The results from this trial, expected in late 2021, will define the benefits and risks of a potential new treatment option for anaemia in patients with CKD.
ACKNOWLEDGEMENTS
Editorial support with writing assistance, assembling figures, grammer editing and referencing was provided by Jonathan Plumb, of Fishawack Indicia, part of Fishawack Health, and was funded by GlaxoSmithKline.
FUNDING
ASCEND-ND is funded by GlaxoSmithKline.
DATA AVAILABILITY STATEMENT
Anonymized individual patient data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
AUTHORS’ CONTRIBUTIONS
V.P. wrote the first draft of the manuscript and contributed to the design, interpretation of data, supervision, management of the research and writing and critical review of the manuscript. All authors contributed to the design, interpretation of data, management of research and writing and critical review of the manuscript. All authors affirm that authorship is merited based on the International Committee of Medical Journal Editors authorship criteria. V.P. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
CONFLICT OF INTEREST STATEMENT
V.P. reports consultancy agreements with AbbVie, Bayer, Boehringer Ingelheim, Chinook, GlaxoSmithKline, Janssen, Pfizer, Astellas, AstraZeneca, Bayer, Baxter, Bristol-Myers Squibb, Durect, Eli Lilly, Gilead, Janssen, Merck, Mitsubishi Tanabe, Mundipharma, Novartis, Novo Nordisk, Pharmalink, Relypsa, Retrophin, Roche, Sanofi, Servier and Vitae; research funding from Pfizer (supplied drug and seed funding for TESTING trial) and GlaxoSmithKline; honoraria from AbbVie, Bayer, Boehringer Ingelheim, GlaxoSmithKline, Janssen, Pfizer, Astellas, AstraZeneca, Baxter, Bristol-Myers Squibb, Chinook, Durect, Eli Lilly, Gilead, Merck, Mitsubishi, Tanabe, Mundipharma, Novartis, Novo Nordisk, Pharmalink, Relypsa, Retrophin, Roche, Sanofi, Servier and Vitae; serves as a scientific advisor on steering committees for trials funded by AbbVie, AstraZeneca, Bayer, Boehringer Ingelheim, Chinook, Eli Lilly, Gilead, GlaxoSmithKline, Janssen, Novartis, Novo Nordisk and Retrophin; and is a board director for George Clinical, George Institute, Garvan Institute, Mindgardens Network, Children's Cancer Institute and Victor Chang Cardiac Research Institute. K.C. reports consultancy fees from GlaxoSmithKline. V.J. reports consultancy fees from GlaxoSmithKline. K.L.J. reports consultancy fees from GlaxoSmithKline and is an advisory board member for Akebia. R.D.L. reports grants and personal fees from Bristol-Myers Squibb and Pfizer; personal fees from Boehringer Ingelheim and Bayer AG and research grants from Amgen, GlaxoSmithKline, Medtronic PLC and Sanofi Aventis. I.C.M. reports research grants, consultancy fees and honoraria from GlaxoSmithKline and Vifor Pharma. J.J.V.M. reports personal fees from Abbott, Hikma, Sun Pharmaceuticals and Servier and his employer has received fees from Alnylam Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Cardurion, Cytokinetics, Dal-Cor, GlaxoSmithKline, Ionis, Novartis, Pfizer and Theracos. G.T.O. reports personal fees from Roche Mexico, Johnson & Johnson, Vifor and AbbVie. S.S. reports grants and consultancy fees from Alnylam, AstraZeneca, Bayer, Bristol-Myers Squibb, Cytokinetics, Gilead, GlaxoSmithKline, Lilly, MyoKardia, Novartis, Respicardia, Sanofi Pasteur and Theracos; grants from Bellerophon, Celladon, Eidos, Ionis, Lone Star Heart, Mesoblast, National Institutes of Health/National Heart, Lung, and Blood Institute and Neurotronik and consultancy fees from Akros, Amgen, Arena, Cardior, Cardurion, Corvia, Daiichi-Sankyo, Ironwood, Merck Sharp & Dohme, Roche, Takeda, Quantum Genetics, AoBiome, Janssen, Cardiac Dimensions, Tenaya, Dinaqor, Tremeau, CellProThera and Moderna. C.W. reports consultancy fees from Akebia, Astellas, AstraZeneca, Bayer, Chiesi, FMC Idorsia, Mundipharma, GlaxoSmithKline, Merck Sharp & Dohme, Reata, Gilead, Tricida, Vifor, Lilly and Takeda and grants and consultancy fees from Boehringer Ingelheim, Sanofi Genzyme and Shire. S.S.W. reports personal fees from Public Health Advocacy Institute, CVS, Roth Capital Partners, Kantum Pharma, Mallinckrodt, Wolters Kluewer, GE Healthcare, GlaxoSmithKline, Mass Medical International, Barron and Budd (versus Fresenius), Johnson & Johnson, Venbio, Strataca, Takeda, Cerus, Pfizer, Bunch and James, Harvard Clinical Research Institute, Oxidien, Sironax, Metro Biotechnology, Biomarin and Bain and grants and personal fees from Allena Pharmaceuticals. D.C.W. reports honoraria and/or consultancy fees from AstraZeneca, Amgen, Astellas, Bayer, Boehringer Ingelheim, GlaxoSmithKline, Jansen, Merck Sharp & Dohme, Mundipharma, Napp, Pharmacosmos, Reata, Tricida and Vifor Fresenius. A.W. reports personal fees from Roche, Bayer, Fresenius and Medice. A.K.S. reports consultancy fees from GlaxoSmithKline. A.B., B.C., A.R.C., R.D., T.L.D., L.K., A.M.M. and L.T. are employees of and stockholders in GlaxoSmithKline.
Notes
This trial is registered with ClinicalTrials.gov: NCT02876835; EudraCT: 2016-000542-65; sponsor protocol number: 200808


{kind=link}
{kind=link}
{kind=link}
Comments