





Abstract
Chronic kidney disease–mineral bone disorder (CKD-MBD) plays a pivotal role in the excess of cardiovascular morbidity and mortality associated with CKD. There is now a growing awareness that pathways involved in CKD-MBD, like canonical Wnt signalling, are activated from the earliest stages of CKD, playing a role in the development of adynamic bone disease with unknown consequences on vasculature. These changes occur before the classic changes in mineral metabolism: secondary hyperparathyroidism, calcitriol deficiency and hyperphosphataemia. Furthermore, vascular calcification is frequently associated and evolves with decreased bone mineral density and deranged bone turnover, while bone and arterial mineralization share common pathways. Therefore, results of clinical trials focused on mineral bone disorder, aimed at preserving bone and cardiovascular health, are considered unsatisfactory. In order to identify more effective therapeutic strategies, it is necessary to clarify the pathways modulating the cross-talk between bone and vasculature and identify new mediators involved in the pathogenesis of CKD-MBD. Much attention has been paid recently to the role of the transforming growth factor-beta superfamily members in renal disease, and in particular of activin A (ActA). Preclinical studies demonstrate an upgrade of ActA signalling in kidney, skeleton, vasculature and heart during CKD. This supports the idea that an endocrine factor produced in the kidney during renal disease, in addition to promoting the progression of kidney damage, deranges other organs’ homoeostasis and participates in CKD-MBD. In this review, we analyse the contribution of ActA to kidney fibrosis and inflammation as well as its role in the development of CKD-MBD.
INTRODUCTION
Chronic kidney disease–mineral bone disorder (CKD-MBD) is characterized by changes in mineral metabolism, a high risk of bone fractures, cardiovascular calcification and left ventricular hypertrophy (LVH). CKD-MBD plays a pivotal role in the excess of cardiovascular morbidity and mortality associated with CKD either directly or indirectly, when the single component of this syndrome interacts with several traditional and renal-related cardiovascular risk factors, such as diabetes, inflammation, anaemia, renin–angiotensin–aldosterone system and fluid overload [1–4].
In recent years, our increasing knowledge of the pathways involved in the onset and progression of CKD-MBD has highlighted that they are rapidly regulated by soluble inhibitors sclerostin and dickkopf-related protein 1 (DKK-1), which could play a pathogenetic role in the development of adynamic bone disease, with unknown consequences on the vascular wall. The expression of sclerostin and DKK-1 is not restricted to the bone but, to varying degrees between the two molecules, takes place in other organs, in particular in the kidney (DKK-1), calcifying vascular smooth muscle cells and aortic valves (sclerostin) [5].
The number of osteocytes expressing sclerostin and circulating sclerostin levels decrease in parallel with the severity of the developing hyperparathyroidism [5, 6].
These phenomena not only help to supersede the old ‘intact nephron hypothesis’, but they also occur well before the development of the well-known ‘triad’ of CKD-MBD: secondary hyperparathyroidism, calcitriol deficiency and hyperphosphataemia. However, the puzzle is still incomplete: what is lacking is a common pathogenetic pathway capable of binding together the early alterations previously described. Not surprisingly, the results of clinical trials aimed at preserving bone health and reducing the cardiovascular burden related to mineral and bone disorders are considered unsatisfactory, with possible unfavourable consequences for cardiovascular health. It is largely debated that some features make CKD-MBD even more complex: vascular calcification (VC) is frequently associated and evolves in parallel with decreased bone mineral density (BMD) and deranged bone turnover, while bone and vessel mineralization share common pathways.
In light of this, and in order to identify more effective therapeutic strategies, it is necessary to clarify the pathways modulating this cross-talk between bone and vasculature, as well as to identify new mediators acting in the early stages of CKD and involved in the pathogenesis of CKD-MBD.
Much attention has been paid recently to the role of the transforming growth factor-beta (TGF-β) superfamily members in renal disease, and in particular of activin A (ActA) [7]. ActA is an important regulator of the normal development of foetal kidneys. While it is not expressed in healthy adult kidneys, it seems to be involved in the progression of several renal diseases and related complications. Indeed, ActA is an important contributor to fibrosis and inflammation in CKD, also promoting mineral bone disorders, VC and LVH [7, 8] Recent studies also support the hypothesis of activin involvement in cardiac hypertrophy [9], although it is likely that this effect can be mediated by other ligands of the TGF-β superfamily able to cross-link activin receptor Type IIA (ActRIIA) (see below).
Like other members of TGF-β superfamily, activin signalling follows the binding with specific receptors belonging to a serine/threonine kinase family. These receptors were distinguished on the basis of their molecular weights and identified as Type I, of low molecular weight, and Type II, of high molecular weight, designed as ActRIIA and ActRIIB [10].
Preclinical studies demonstrate activation of ActRIIA signalling in the kidney, skeleton, vasculature and heart during CKD [11–13]. This supports the idea that an endocrine factor produced in the kidney during renal disease, in addition to promoting the progression of kidney damage, also deranges other organs’ homoeostasis and participates in the pathogenesis of CKD-MBD. During kidney disease, there is a reactivation of those nephrogenic pathways that we usually encounter in the foetus, during early kidney development. Clearly, the activation of the ActA signalling and Wnt pathway is part of this process. More specifically, TGF-β1 induces the secretion of ActA in renal fibroblasts, mesangial and tubular cells. The following activation of ActRIIA signalling lowers klotho levels and increases both renal and circulating DKK-1, thus proving that Wnt activation happens downstream of the ActRIIA signalling [11]. In preclinical studies, inhibition of ActRIIA signalling is effective in reducing the features of CKD-MBD as well as the Wnt signalling that follows the upregulation of activin in the kidney during the renal repair [11–13].
In this review, we analyse the contribution of ActA to kidney fibrosis and inflammation as well as its role in the development of CKD-MBD. Furthermore, we analyse experimental studies that show how ActA inhibition, using a ligand trap for the receptor RAP-011, is protective against CKD progression as well as the markers of CKD-MBD in mice models of kidney disease.
ACTIVINS
Activins are dimeric polypeptides members of the TGF-β superfamily [10], like inhibins, bone morphogenetic proteins (BMPs) and growth differentiation factors (GDFs).
Activins are widely expressed factors involved in cell differentiation, proliferation and inflammation, and they activate signal transduction by binding receptor heteromultimers. They are dimers of two inhibin β chains (βA, βB, βC, βD or βE) linked by disulphide bonds. Among the six known isoforms (activins A, AB, B, C, D and E), ActA, composed of two βA subunits, is the most abundant, with a well-defined biological activity and recently well studied in the field of CKD [10, 14]. Activin subunits are synthesized as large precursor polypeptides containing an NH2-terminal prodomain with 250–350 residues and a COOH-terminal mature domain (Figure 1a). The precursor is cleaved by furin-like proteases, thereby releasing the mature domain that becomes biologically active [10, 14].

(a) Activins are dimers of two inhibin chains (A, B, C, D or E) linked by disulphide bonds. Among the six known isoforms (activins A, AB, B, C, D and E), ActA, composed of two βA subunits, is the most abundant, with a well-defined biological activity, and recently well studied in CKD. Activin subunits are synthesized as large precursor polypeptides containing an NH2-terminal prodomain with 250–350 residues and a COOH-terminal mature domain. Precursor cleavage by furin-like proteases releases the mature domain that becomes biologically active. (b) ActRIIA and ActRIIB are the most important Type II receptors, capable of binding several members of the TGF-β superfamily (see the text for details). The ctivin pathway is triggered when an activating dimer binds to one of the two cell surface Type II receptors. This high-affinity binding leads to a modified ligand conformation that allows its interaction with the Type I receptor. The resulting hormone/receptor complex enables the activated kinase domain of ActRII to recruit and phosphorylate the coupled Type I receptor leading to transduction signal cascade.
All TGF-β family ligands bind to Type II receptors that associate and activate Type I receptors, starting signal transduction. Besides TGF-β receptor II (TGFβRII) and BMP Type II receptor (BMPRII), ActRIIA and ActRIIB are the most important Type II receptors, capable of binding several members of the TGF-β superfamily. Although ActA, whose circulating levels increase several folds in CKD models, is viewed as the primary ligand of ActRIIA [11], it must be emphasized that the Type II TGF-β family receptors ActRIIA, ActRIIB and BMPRII can interact with several overlapping ligands and that, in particular, ActA directly inhibits signalling by the low-affinity ligands BMP-2, BMP-7 and BMP-9 [15] (Figure 1b).
ActA exerts its biologic functions through binding to a Type II receptor, ActRIIA or ActRIIB, which then recruits phosphorylates and activates the Type I receptor ActRI and termed activin receptor-like kinase (ALK), of which seven variants have been identified. Of these, ALK4 (also named ActRIB) has been consistently described as the predominant Type I receptor for activin signalling. Once activated, ALK4 phosphorylates the transcription factors Smads 2 and 3, which are translocated into the nucleus in a complex with Smad 4, to regulate the transcription of genes involved in proliferation, differentiation and apoptosis [10]. The ActRIIA, after linking ActA, undergoes endocytosis during signal transduction and may be downregulated, depending on the effects of activin receptor-interacting proteins (ARIP1 and ARIP2). Specifically, ARIP1 and two isoforms of ARIP2, named ARIP2b and ARIP2c, interact with Smad proteins and ActRIIA enhancing activin signalling, while ARIP2 induces receptor endocytosis and degradation and so the damping of the activin signalling [11, 14, 16–18]. ARIP2 is highly expressed in the vasculature, while ARIP1 expression is much higher than ARIP2 in the kidney [16, 17]. The different distribution pattern of both ARIP1 and ARIP2 in the tissues is one of the factors that may affect the different expression of ActRIIA [10] as well as the effects of ActA upregulation (see also below).
An increase of ActA in the kidney and in the bloodstream has been observed in several experimental models of renal fibrosis and in human samples [19–21]. ActA, whose expression is induced by TGF-β1 or by ActA itself (in a paracrine manner), promotes cell proliferation, differentiation and Collagen I production by renal interstitial fibroblasts [22], and matrix protein production by mesangial cells [23]. TGF-β1 is a key mediator of renal fibrosis and its expression is known to be upregulated in fibrotic kidneys. However, the downstream mechanism of TGF-β1 in fibroblast activation is not totally clear and may be at least partially mediated by ActA [22].
ActA is also involved in the increase of Wnt signalling, a further nephrogenic pathway reactivated in renal repair that plays an important role in tubular epithelial reconstitution and fibrosis. Therefore, by means of canonical signalling, ActA also induces the expression of a family of Wnt inhibitory proteins, DKK-1 and sclerostin [10, 24].
During nephrogenesis DKK-1 has a pivotal role in regulating epithelial proliferation in the loop of Henle and collecting duct. It also increases in kidney disease during renal repair, as it is involved in the fibrotic process [25]. CKD increases the kidney’s sclerostin expression, although it is unclear whether renal sclerostin contributes to the increase in the circulating levels and whether it performs a secondary role in kidney fibrosis, which seems likely [25].
Fang et al. [26] demonstrated an increased renal production and higher levels of circulating DKK-1 and sclerostin in a model of CKD atherosclerotic ldlr–/– mice. In this study, the neutralization of DKK-1 in CKD Stage 2 mice by administration of a monoclonal antibody after renal injury stimulated bone formation rates, corrected the osteodystrophy and prevented CKD-stimulated VC.
Enhanced ActA expression was found in several models of insulin-deficient diabetic kidney disease (DKD), as well as in humans. Serum ActA increases as early as CKD Stage 2, well before any notable increase in parathormone (PTH) and fibroblast growth factor 23 (FGF23) [20, 27].
The role of ActA in renal osteodystrophy, VC, cardiac hypertrophy and CKD progression was subsequently evaluated by Hruska et al. [25] in very elegant experimental models: (i) the diabetic ldlr–/– mouse model with CKD and stimulated atherosclerotic calcification through a high-fat diet (high-fat-fed ldlr–/– mouse with ablative CKD) [11, 12]; and (ii) a murine homologue of X-linked Alport syndrome, which is a deficiency in the gene for the a5 chain of Type IV collagen, Col4a5 [13]. These studies show that ActRIIA signalling is stimulated in the kidney, in the skeleton and in the vasculature (Figure 2). Indeed, the experimental design of these studies contemplated the use of a ligand trap (RAP-011) for ActRIIA, which proved effective in preventing osteodystrophy, VC and cardiac hypertrophy, and in delaying the progression of CKD. RAP-011 is a soluble extracellular domain of ActRIIA, fused to a murine IgG-Fc fragment; the drug traps circulating members of the TGF-β superfamily, thereby preventing their action on the endogenous, membranous, ActRIIA [11–13].
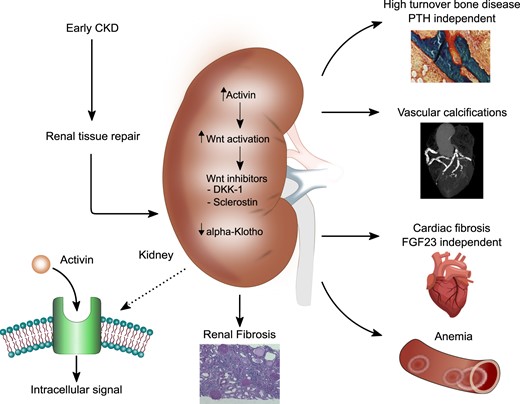
Schematic overview of the effects of ActA systemic activation on the organ involved CKD-MBD.
ACTIVIN, ActRIIA AND RENAL PATHOLOGY
Several experimental studies have demonstrated that ActA expression is one of the developmental processes involved in nephrogenesis that are activated during renal repair. In a rat model of unilateral ureteral obstruction, ActA was highly expressed by interstitial alpha-smooth muscle actin-positive myofibroblasts [28]. In anti-Thy1 glomerulonephritis, glomerular ActA and TGF-β1 were co-expressed and associated with the development of fibrosis [23].
Renal tubular cells release ActA, which acts as a paracrine factor and promotes fibroblast proliferation, contributing to kidney fibrosis and CKD progression [22].
In an experimental model of high-fat-fed ldlr–/– mouse with ablative CKD, Agapova et al. [11] found an increase of ActA (the primary ligand for ActRIIA), ActRIIA signalling (as demonstrated by the contemporary increase in activated Smad2/3, p-Smad2/3), and renal and circulating DKK-1 levels, without any changes in ActRIIA expression. These findings reflect the higher ARIP1, ARIP2a and ARIP2c expression in the kidney (Figure 3a).
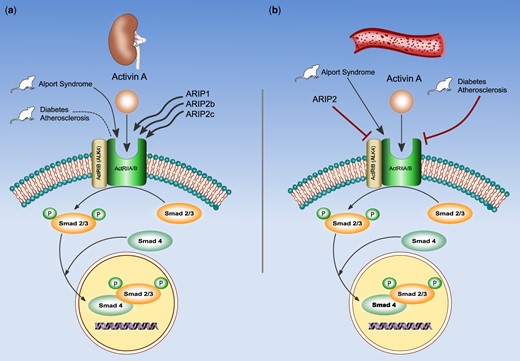
ActA exerts its biologic functions by binding to activin receptors, heterodimeric complexes including Type II (ActRIIA or ActRIIB) and the Type I receptor ActRI, also termed activin receptor-like kinase. Seven variants of ALK have been identified, of these, ALK4 (also named ActRIB) has been consistently described as the predominant Type I receptor for activin signalling. Once activated, ALK4 phosphorylates the transcription factors Smads 2 and 3, which are translocated into the nucleus in a complex with Smad 4, to regulate the transcription of genes involved in proliferation, differentiation and apoptosis. ActRIIA may be downregulated after linking ActA, depending on the effects of activin receptor-interacting proteins (ARIP1 and ARIP2, ARIP2b and ARIP2c). ARIP1 expression is much higher than ARIP2 in the kidney (a), while ARIP2 is highly expressed in the vasculature (b). In high-fat-fed ldlr–/– mice model, ActRIIA signalling is increased, while ActRIIA expression is not affected by CKD, instead, in mice model with Alport syndrome, ActRIIA is overexpressed. These findings mirror the activity of ARIP1, ARIP2a and ARIP2c associated with an effective signalling, but not with ActRIIA downregulation. The different pattern of ActRIIA expression in two mice models could be related to a different pathological context of two mice models.
ActRIIA Inhibition by RAP-011 treatment decreased renal ActA expression without influencing ActRIIAs. These effects were associated with a decrease of renal fibrosis, proteinuria, Type I collagen and fibronectin levels and an increase of renal α-klotho. These results have also been confirmed by Williams et al. [13] in a mice model with Alport syndrome demonstrating a higher expression of ActRIIA, fibronectin and monocyte chemoattractant protein-1 in the kidney and a significant interstitial fibrosis. All these changes were significantly reduced by RAP-011 treatment. The reasons why, in the two murine models, CKD induces a different pattern of ActRIIA expression are unknown, unless we hypothesize the effect of different pathological backgrounds.
However, unlike in the case of high-fat-fed ldlr–/– mouse, ligand trap treatment does not increase klotho expression in the murine homolog of the X-linked Alport syndrome. Therefore, at least in this model, klotho was not responsible for the effects of RAP-011 on the target organs.
The studies evaluating the role of ActA in human kidney disease are still limited [29] and centred on DKD [30].
Interestingly, Bian et al. [21] showed that, in a cohort of diabetic patients and controls, circulating ActA is increased in human DKD and correlates with reduced kidney function; also, albuminuria and tumour necrosis factor receptor-1 are associated with the highest activin levels. Indeed, DKD-injured human renal tubule cells develop a profibrotic and inflammatory phenotype with ActA upregulation.
RENAL OSTEODYSTROPHY
In the experimental studies carried out on two different mice models, (i) ldlr–/– high-fat-fed mice with ablative CKD [11] and (ii) murine X-linked Alport syndrome [13], CKD induced bone resorption following osteoclasts activation and osteoblast dysfunction. These findings are associated with high circulating ActA levels.
In addition to being produced in the kidney during the renal repair phase, ActA is secreted in the bone by osteoblasts and osteoclasts, and is abundant in extracellular bone matrix. It plays an important role in both foetal and adult bone homoeostasis [12] and it seems to be a positive regulator for osteoclastic development and bone resorption, and a negative regulator for osteoblastic bone formation.
The effect of ActA on osteoclastogenesis develops in parallel with that induced by the receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL) [12], although their pathways are independent. ActA induces the phosphorylation of the downstream target Smad2; Smad2 then translocates in the nucleus where it is acetylated by p300/CBP. The role of Smad3 appears to be secondary in osteoclastogenesis because mice lacking Smad3 showed no variation in the number of osteoclasts [31] (Figure 4).
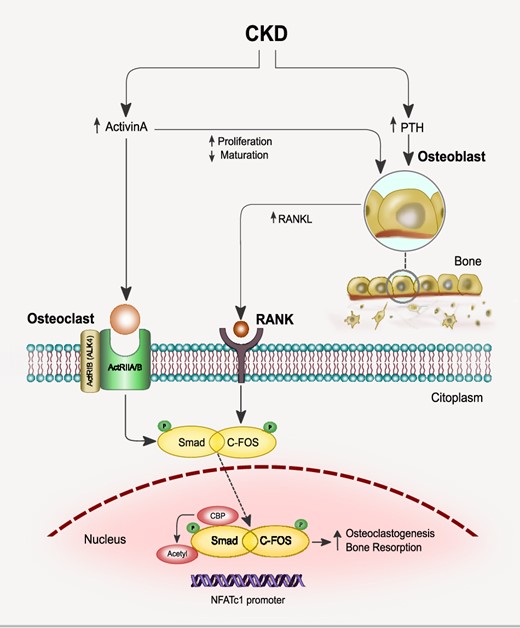
The effect of ActA on osteoclastogenesis develops in parallel with those induced by the receptor activator of NF-κB ligand (RANKL), although their pathways are independent. ActA induces the phosphorylation of the downstream target Smad2, then Smad2 translocates in the nucleus where it is acetylated by p300/CBP. RANKL stimulates the expression and phosphorylation of c-Fos, which upregulates RANK in osteoclasts precursors, then translocates into the nucleus where it plays a pivotal role in the early induction of nuclear factor of NFATc1. The combined effect of RANKL and ActA induces the formation of a complex of activated c-Fos and activated Smad2 and the translocation of this complex into the nucleus, as well as the DNA binding of activated c-Fos to the NFATc1 promoter.
RANKL stimulates the expression and phosphorylation of c-Fos, which upregulates RANK in osteoclasts precursors, then translocates into the nucleus where it plays a pivotal role in the early induction of nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) [12]. Both c-FOS and NFATc1 are involved in osteoclasts differentiation [8]. The combined effect of RANKL and ActA induces the formation of a complex of activated c-Fos and activated Smad2 and the translocation of this complex into the nucleus, as well as the DNA binding of activated c-Fos to the NFATc1 promoter [8] (Figure 4). This could be the pathway through which the osteoclastogenesis and bone resorption are enhanced in later stages of CKD. A synergistic effect of ActA and RANKL in osteoclastogenesis was also demonstrated by Fuller et al. [32] in culture of bone marrow precursors. However, the hypothesis of a complementary effect of both ActA and RANKL on osteoclastogenesis conflicts with the findings of Fowler et al. [33], suggesting instead that ActA provides an additional pathway to limit the extent of osteoclasts differentiation and/or bone resorption. On the contrary, ActA signalling stimulates osteoblast proliferation but reduces their maturation, hence delaying bone mineralization [34]. However, the molecular mechanisms remain still unclear. Like TGF-β, ActA signalling pathways may negatively regulate Runt-related transcription factor 2 (Runx2) functions through a Smad-dependent mechanism. However, the molecular mechanisms remain still unclear. An involvement of Runx2, a master gene and multifunctional regulator of bone formation has been hypothesized. Runx2 induces the differentiation of multipotent mesenchymal cells into immature osteoblasts, addressing the formation of immature bone; however, Runx2 inhibits osteoblast maturation and mature bone formation. Like TGF-β, ActA signalling pathways may negatively regulate Runx2 functions, so impairing osteoblastogenesis, through a Smad-dependent mechanism [8].
In both these models, ActRIIA inhibition prevents osteoclast stimulation and increased osteoblast function and bone formation, notwithstanding changes in PTH and FGF23 [11–13] (Figure 4). The results suggest that features of renal osteodystrophy appear to follow ActRIIA signalling regardless of the effect of PTH on the expression of the RANKL by osteoblasts and osteocytes.
The reversal of bone remodelling by ligand trap may also cause the restoring of Wnt signalling, by decreasing circulating levels of its inhibitor DKK-1. A previous study found that DKK-1 neutralization prevents the development of renal osteodystrophy. In addition to the direct effects of reduced DKK-1 on inhibition of skeletal Wnt activity, another possible mechanism may be that DKK-1 neutralization downregulates the osteocytic production of sclerostin, another Wnt inhibitor, and thus is a powerful inhibitor of bone formation [26].
However, there are some concerns regarding the ldlr–/– mouse model, considering that in a previous study with the same model CKD seemed to induce a further decrease of bone turnover. High ActA levels have moreover been reported, in conjunction with a low bone turnover, in patients with Type 2 diabetes mellitus [35].
CKD-RELATED ActA EFFECTS ON AORTA IN MICE MODELS
As demonstrated by two different experimental models, lldlr/high-fat-fed atherosclerotic diabetic mice and Alport mouse, the effects of ActA on aortic vascular smooth muscle cell (VSMC) are complex and depend on (i) whether or not diabetes and atherosclerosis coexist with CKD and (ii) the different expression of the ARIP. In diabetic atherosclerotic ldlr/high-fat-fed mice, CKD decreases VSMC-specific protein expression as well as ActRIIA signalling; the reversing of the contractile phenotype VSMC (dedifferentiation) contributes to osteoblastic transition, leading to VC. It is important to bear in mind that these are two independent aspects, both inhibited, however, by the administration of an ActRIIA ligand trap [11].
The ActRIIA downregulation in the aorta follows the internalization, degradation and decreased ActRIIA signalling and is mediated by ARIP1 and ARIP2. As we already saw, ARIP2 expression is greater in the vascular wall while ARIP1 expression is much higher in the kidney [16] (Figure 3b).
In contrast, in the aorta of the Alport mouse model with severe CKD both the expression and signalling of ActRIIA increased, as well as ActA levels [13]. These findings are associated with the presence of a non-proliferative contractile phenotype in VSMC and calcium deposits in the tunica media (Figure 3b). Despite the increase in vascular smooth muscle proteins, in the Alport mice CKD induced osteoblastic transition in the aortas and VC. They all decreased after RAP-011 treatment without affecting elevated PTH and FGF23 levels [36].
These apparently contradictory results in the two mice models could be traced back to the involvement, besides VSMCs, of aortic adventitial mesenchymal stem cells. These may differentiate towards VSMCs while expressing an osteogenic phenotype once migrated to the intima as well as the derangement, ActA mediated, of stem cell renewal and differentiation in the bone marrow [37, 38]. ActA, which is produced by renal fibroblasts, entails an increase of both renal and circulating DKK-1 levels, an effect blunted by RAP-011 and that contributes to its efficacy in restoring vascular function during CKD. These results agree with those of Fang et al. [26], demonstrating that neutralization of high DKK-1 levels prevents VC.
The positive effect of DKK-1 neutralization on vascular health is somewhat unexpected since other studies have shown a link between VC and Wnt activity, pointing to the fact that the Wnt inhibitors, DKK-1 and sclerostin, would favour osteoblast transition and VC [24]. A series of previous experimental studies would seem to point towards a protective vascular role played by DKK-1 neutralization against VC. First, it was demonstrated that the canonical Wnt pathway downregulation promotes lipid and macrophage retention in the vessel wall and increases leucocyte-driven systemic inflammation [39]. Secondly, the DKK-1-mediated inhibition of aortic Wnt7b stimulates Smad-mediated aortic endothelial-mesenchymal transition (EndMT) and VC [40].
CARDIAC HYPERTROPHY
Changes in mass and left ventricular structure are known to be a hallmark of CVD in both animal models and patients with CKD. In addition to traditional factors (hypertension, fluid overload and anaemia), a relevant pathophysiological role in the onset of LVH has been assigned to FGF23 and PTH [41].
In the CKD Alport model, mice develop cardiac hypertrophy; ActRIIA inhibition with RAP-011 reverses cardiac hypertrophy despite no changes in FGF23, PTH and phosphate levels. The reduction of cardiac hypertrophy by the ligand trap may be explained by the fact that the latter competes with ActA in binding ActRII, hence causing a reduced signalling. However, the role of ActRIIA in cardiac development, as well as in cardiac hypertrophy, may be mediated by other ligands of the TGF-β superfamily able to bind the ActRII, such as BMPs [42], GDF11 [43] and myostatin.
As previously reported, the most important Type II receptors of the TGF-β superfamily besides TGFβRII are BMPRII, ActRIIA and ActRIIB. Whilst BMPRII is selective for BMPs, ActRIIA and ACTRIIB are activated in response to activins, BMPs, MSNT and GDF11. It is not yet known which TGF-β superfamily ligands are mainly involved in derangement of ActRIIA signalling relatively to cardiac hypertrophy, but it is possible to speculate that the effects on it are secondary to a cross-linking of ACTRII receptors with ActA, GDF11, MSNT and BMPs.
Blocking ACTRII signalling could therefore provide potential therapeutic benefits for the heart [42–44].
It is a known fact that ActA activates fibroblasts, promoting cell proliferation and collagen synthesis through p38 and extracellular signal-regulated kinases mitogen-activated protein kinase pathways. However, in opposition to the detrimental effects of ActRII signalling on cardiac hypertrophy, a rapid upregulation of ActA has been reported in a model of reperfused myocardial infarction. Furthermore, experiments in vivo and in vitro suggested that ActA overexpression may protect ischaemic cardiomyocytes from apoptosis [45].
FUTURE PERSPECTIVES
The studies currently available demonstrate the activation of ActRIIA signalling in the kidney, skeleton, vasculature and heart during CKD. These results support a working hypothesis based on the production of an endocrine factor during kidney disease-related repair capable of inducing CKD progression as well as conditioning the development of CKD-MBD. In this model, CKD-MBD is the consequence of the pathways put in place during kidney repair.
This interpretation does not necessarily represent yet an alternative, but rather is a complementary analysis, to the traditional ‘nephron intact hypothesis’, according to which CKD-MBD is the result of the endocrine trade-offs required by the nephrons attempting to preserve mineral metabolism and, more generally, the internal milieu.
Some questions concerning the changes of ActRIIA signalling in CKD as well as the effects of ActRII signalling inhibition through the treatment with RAP-011 remain open. Patients with Type 2 diabetes mellitus show increased ActA levels together with a low bone turnover, and several clinical studies have found evidence of reduced bone formation and resorption in diabetes. How to reconcile these findings with those from ldlr–/– mice, showing an increase in the histomorphometric parameters of bone turnover? Further studies will have to ascertain whether ActA is necessary to develop osteoclasts together with RANKL under physiological conditions.
It is likely that their action is synergistic in CKD, as the synthesis of PTH in this context unhooked by a feedback mechanism.
The role of ActA in VC is far from being clearly defined. The involvement of TGF-β1 in the induction of medial VC has recently been reported in a CKD mouse model [46].
Since several molecular and cellular mechanisms are involved in the pathogenesis of VC, it may be interesting to carry on exploratory studies in vitro on the role of ActA in VSMCs apoptosis and/or autophagy. This may help clarify whether ActA-dependent Smad signals are implicated in the formation of VC in CKD.
Furthermore, the association between ActA and VC could be traced back to their link with inflammation. Especially in DKD and CKD, inflammation is in fact involved in the progression of VC [47] while ActA plays a regulatory role in the inflammatory process, during which it is produced and released into the bloodstream [48].
Another question concerns the difference in the kidney expression of ActRIIA and in the effects of the ligand trap in two murine models. In the high-fat-fed ldlr–/– mouse model with ablative CKD [11] neither changes in ActRIIA expression nor ligand trap effects were observed. The model with Alport syndrome, on the contrary, demonstrated a high ActRIIA expression, and its reduction following ligand trap treatments could possibly explain the different patterns of expression of the ActRIIA, as well as the different effects of ligand trap treatment, based on the presence or absence of diabetes.
Although ActRII ligand traps have beneficial antifibrotic, vascular and bone effects in preclinical models, their increase in haematocrit could limit their usefulness, especially in earlier stages of CKD. Activins (A and B) and GDF11 inhibit the later stages of erythropoiesis by blocking erythrocyte cell differentiation and maturation. Their neutralization using a ligand trap (ActRIIA-Fc) stimulate erythropoiesis and promotes the survival of erythrocyte precursors in preclinical studies, in healthy humans and also specifically in post-menopausal women [49].
Two ActRII ligand traps, namely sotatercept and luspatercept, are being evaluated in clinical trials for the treatment of various types of anaemias, including CKD, myelodysplastic syndrome and thalassaemia. The pathways that underpin the stimulation of erythropoiesis by sotatercept and luspatercept are poorly understood. Sotatercept, the human orthologue of RAP-011, is a recombinant fusion protein consisting of the extracellular domain of the human ActRIIA linked to the human immunoglobulin G1 Fc domain. It binds a number of TGF-β superfamily ligands including ActA, activin B, GDF-11 and BMP-10. Experimental studies showed that GDF11 and ActA induce inhibition of late-stage erythropoiesis whilst their inhibition may contribute to the erythropoietic stimulatory effects of RAP-011 [50, 51]. Luspatercept (ACE-536) is an Act-RIIB with the modified extracellular domain of ActRIIB fused to the Fc fragment. Luspatercept does not bind either GDF11 or ActA, but may bind other ligands of the TGF-β family or it has even been speculated that it may interfere with the physiological pathways that normally limit the number of mature erythroblasts [50, 52]. Human studies about safety and effects of ligand traps on erythropoiesis are still limited. Recently, in patients with end-stage kidney disease, two Phase II, multicentre, randomized studies, REN-001 and REN-002, examined the safety, tolerability and effects of sotatercept on haemoglobin concentration; REN-001 also explored effects on BMD and abdominal aortic VC [53]. The results of both studies showed an acceptable safety profile and a trend towards a reduction in VC, whilst the erythropoiesis stimulant effect of the sotatercept appears to be limited in end-stage kidney disease. It is presumable that these findings may be related to the small number of patients enrolled in the studies. Although these preliminary data sound encouraging for modulating activin biology in CKD, further preclinical and clinical studies targeting ActRIIA activity in CKD may be warranted. It must be noted that although several trials with the mentioned ligand traps are ongoing, so far none of these compounds has been approved for therapeutic use on humans.
CONFLICT OF INTEREST STATEMENT
None declared. The results presented in this article have not been published previously in whole or part.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments