





Abstract
For many years renal biopsy has been the gold standard for diagnosis in many forms of kidney disease. It provides rapid, accurate and clinically useful information in most individuals with kidney disease. However, in recent years, other diagnostic modalities have become available that may provide more detailed and specific diagnostic information in addition to, or instead of, renal biopsy. Genomics is one of these modalities. Previously prohibitively expensive and time consuming, it is now increasingly available and practical in a clinical setting for the diagnosis of inherited kidney disease. Inherited kidney disease is a significant cause of kidney disease, in both the adult and paediatric populations. While individual inherited kidney diseases are rare, together they represent a significant burden of disease. Because of the heterogenicity of inherited kidney disease, diagnosis and management can be a challenge and often multiple diagnostic modalities are needed to arrive at a diagnosis. We present updates in genomic medicine for renal disease, how genetic testing integrates with our knowledge of renal histopathology and how the two modalities may interact to enhance patient care.

INTRODUCTION
Percutaneous renal biopsy was first described by Iversen and Brun in 1951 [1]. It remains the gold standard for diagnosis in chronic kidney disease (CKD) [2, 3]. It helped to transform nephrology from a specialty where clinical syndromes could only be described to one where the pathology and underlying mechanisms of disease could be understood [4, 5].
Over the last 65 years, the science of interpreting kidney biopsies has developed gradually, aided by the development of three major diagnostic modalities: light microscopy (LM), immunofluorescence (IF) and electron microscopy (EM) [6, 7]. It has become a valuable tool in the diagnosis of native kidney disease and the management of renal transplant. It is useful in showing disease activity, such as crescent formation and inflammatory cell proliferation, and in establishing chronicity and degree of fibrosis [8]. It has a high diagnostic yield and is thought to give useful information in up to 80% of cases [9, 10]. A prospective study by Turner et al. [11] showed that renal biopsy altered the diagnosis in 44% and the therapeutic approach in 31% of cases. Other studies have shown that treatment is modified in up to 54% of patients [12]. It is an invasive procedure, but is generally considered very safe, though not entirely without risk, particularly the risk of bleeding [13, 14]. A recent study by Tøndel et al. [9] showed the risk of major bleeding to be ∼2% and the need for transfusion to be ∼1%. The study reported no deaths in >9000 patients.
While a useful test, renal biopsy has drawbacks. Despite international guidelines and attempts at standardization, interobserver agreement between experienced renal pathologists may be as low as 45% in some cases [15, 16]. Certain anatomic abnormalities, such as a horseshoe kidney, single kidney or atrophic kidneys with thin renal cortices, may also preclude renal biopsy in everything but exceptional circumstances [2]. The accumulation of extracellular matrix in the interstitium or fibrosis is common to all CKD, and while it is a useful indicator of prognosis, it may obliterate useful pathological architecture and make diagnosis difficult [17, 18]. Therefore renal biopsy may be a less useful diagnostic tool in late-presenting CKD. Approximately 15% of all incident patients in the UK who reach end-stage renal disease (ESRD) lack a primary diagnosis [19].
Often in clinical practice, specific patterns of injury seen on biopsy are discussed as if they are diseases rather than descriptions of the histology at a given time. Focal segmental glomerulosclerosis (FSGS), for instance, is often treated as a single disease entity because of a relatively uniform histological pattern. However, the onset, severity and response to treatment in cases of FSGS can vary widely in patients with a very similar histological pattern. Some cases of FSGS respond to corticosteroids, while some do not. It is much more likely that what we see are a number of disease entities unified by similar pathological findings rather than a single disease.
Overall, though it has been and remains a very useful diagnostic tool, renal biopsy can lack precision and may sometimes be inadequate for making a precise renal diagnosis.
Compared with renal biopsy, genetic testing for renal disease is a relatively new innovation. Previously prohibitively expensive, the introduction of next-generation sequencing (NGS) in the mid-2000s led to a 50 000-fold decrease in the cost of human genome sequencing [20]. Since then, further technological advances have meant that the sequencing of specifically targeted genes or an individual’s entire exome or genome can be performed efficiently and affordably. As we move towards a new era of genomic medicine, we are faced with many new opportunities for diagnosis and management, and equally many new challenges on how to integrate these new data safely and effectively into our existing clinical practice.
This review examines new opportunities and challenges in the treatment in diseases of the kidney and, in particular, how advances in genomic medicine can affect the utility of the percutaneous renal biopsy, how these two modalities interact with one another and how they can work in synthesis with one another to provide patient care.
INHERITED KIDNEY DISEASE
Inherited kidney disease is an umbrella term that encompasses many different disease states with a variety of different presentations. Given the broad variation of phenotypes, inherited kidney disease may cause disease that ranges in severity from extremely mild to incompatible with life and can present at any time from antenatally to the last decades of life. There are many kidney diseases that are inherited in a monogenic fashion due to a variant in a single gene, but there are equally as many kidney diseases that are influenced in a polygenic fashion. Even the term ‘inherited’ is imprecise, as many disease-causing variants arise de novo in the proband, although once they arise they are hereditary thereafter.
Most Mendelian inherited kidney diseases are rare, affecting only a tiny portion of the population. However, as a group they represent a significant burden of disease. They are the main cause of CKD and ESRD in the paediatric population [21, 22]. In an adult population, inherited kidney disease is thought to account for at least 10% of the CKD population [23–25].
Polycystic kidney disease (PKD) remains the most common inherited cause of CKD. It is thought to account for 7% of the UK incident ESRD population and has a lifetime prevalence of at least 9.3 cases per 10 000 genes sequenced [26]. However, the improving cost-effectiveness and speed of genomic testing has led to recognition that other forms of inherited kidney disease are more common than previously assumed. Groopman et al. [27] reported that whole-exome sequencing (WES) of 3000 adult patients with CKD revealed a monogenic cause of disease in 10%, equivalent to rates of genetic diagnosis achieved in cancer, and detected 66 separate monogenic disorders. Genomic testing was particularly effective in diagnosing those with kidney disease of unknown origin, in which they were able to make a diagnosis in 18% of patients.
Even in those where a clear monogenic form of kidney disease has not been identified, there is strong evidence of heritability within families. In a survey of 1840 patients attending nephrology outpatient clinics or dialysis in Ireland in 2014, 34% self-reported a family history of kidney disease [28]. Those with an unknown underlying aetiology were 3 times more likely to report a family history of inherited kidney disease, while those with an unspecified tubulointerstitial kidney disease were 8 times more likely to report a family history. Other studies in different populations have shown similar results [29, 30]. There is evidence that physiological parameters of the kidney are at least partially heritable. Glomerular filtration rate (GFR) is thought to be 30–60% heritable and handling of urinary calcium is thought to be 45% heritable [31–33].
As of 2019, >600 genes have been implicated in monogenic causes of kidney disease [34]. Testing and identification of many of these conditions may work in concert with, or in some cases replace, renal biopsy as the first-line diagnostic modality in some renal diseases.
POLYGENIC KIDNEY DISEASE
While monogenic causes contribute to a small fraction of all CKD, many more kidney diseases are influenced by a mixture of genetic, epigenetic and environmental risk factors. These kidney diseases are not inherited in a Mendelian fashion. Correlation between phenotype and genotype is weak, but genetic factors may nevertheless play an important role in developing these diseases by conveying an increased relative risk.
Genome-wide association studies (GWASs) are a tool used to look for associations between traits, including human disease and single-nucleotide polymorphisms (SNPs). These are observational studies and pool the results of many individuals to look for common links between SNPs and traits. However, as most GWAS loci tend to explain only a relatively small proportion of overall risk, translation of these findings into concrete clinical benefit has proven a challenge. This has been compounded by other limits, including technological challenges, small sample size and allelic heterogeneity. Genome-wide polygenic risk scores (GPSs) are designed to address these challenges by aggregating the effects of millions of genetic loci across the genome, including those that do not reach individual statistical significance. GPSs have now been shown to be able to predict risk in well-studied polygenic conditions such as coronary artery disease, type 2 diabetes mellitus and atrial fibrillation [35]. However, given the heterogenicity of CKD, the relative complexity of CKD diagnosis and the small numbers of patients in GWASs focused on specific diseases like IgA nephropathy (IgAN) and membranous nephropathy, GPSs may not be useful for the diagnosis of CKD [36].
Digenic inheritance is the simplest form of oligogenic inheritance. It may be mistaken for polygenic disease, but in this case only two separate loci are present that influence the expression and severity of a phenotype. Digenic inheritance has been seen in conditions including PKD and nephronophthisis [37, 38].
DIAGNOSTIC GENETIC TESTING
The aim of diagnostic genetic testing is to identify disease-causing variants in an individual or family. The vastness of the human genome makes this challenging. The human genome contains 3.2 billion base pairs and the average genome differs from the reference genome at 4–5 million sites [39]. There are 22 000 known genes in the human genome, of which 4000 are known to cause phenotypes that cause disease, susceptibility to disease or benign changes in laboratory values [40].
The aim of diagnostic testing is not only to identify variants but also to determine which among the many variants detected are predicted to be damaging. Several methods are used to try and determine this, including Sanger sequencing and NGS, each of which has specific advantages and disadvantages (Table 1).
Advantages and disadvantages of forms of genetic testing
| Technique | Sanger sequencing | Gene panel testing | WES | WGS |
|---|---|---|---|---|
| Tests | Single gene | Multiple known genes | Whole exome | Whole genome |
| Cost | Cost-effective | Cost-effective when testing known genes | Moderately expensive | Most expensive |
| Advantages |
| Can test a large range of known genes with high degree of accuracy |
|
|
| Disadvantages | Only tests one gene at a time |
|
|
|
| Uses | Cascade testing of family members. Testing of single-gene disorders, e.g. cystic fibrosis | Testing disorders with a specific phenotype and multiple known genes | Examine all CDS of the genome | Examines all regions of the genome |
| Technique | Sanger sequencing | Gene panel testing | WES | WGS |
|---|---|---|---|---|
| Tests | Single gene | Multiple known genes | Whole exome | Whole genome |
| Cost | Cost-effective | Cost-effective when testing known genes | Moderately expensive | Most expensive |
| Advantages |
| Can test a large range of known genes with high degree of accuracy |
|
|
| Disadvantages | Only tests one gene at a time |
|
|
|
| Uses | Cascade testing of family members. Testing of single-gene disorders, e.g. cystic fibrosis | Testing disorders with a specific phenotype and multiple known genes | Examine all CDS of the genome | Examines all regions of the genome |
Advantages and disadvantages of forms of genetic testing
| Technique | Sanger sequencing | Gene panel testing | WES | WGS |
|---|---|---|---|---|
| Tests | Single gene | Multiple known genes | Whole exome | Whole genome |
| Cost | Cost-effective | Cost-effective when testing known genes | Moderately expensive | Most expensive |
| Advantages |
| Can test a large range of known genes with high degree of accuracy |
|
|
| Disadvantages | Only tests one gene at a time |
|
|
|
| Uses | Cascade testing of family members. Testing of single-gene disorders, e.g. cystic fibrosis | Testing disorders with a specific phenotype and multiple known genes | Examine all CDS of the genome | Examines all regions of the genome |
| Technique | Sanger sequencing | Gene panel testing | WES | WGS |
|---|---|---|---|---|
| Tests | Single gene | Multiple known genes | Whole exome | Whole genome |
| Cost | Cost-effective | Cost-effective when testing known genes | Moderately expensive | Most expensive |
| Advantages |
| Can test a large range of known genes with high degree of accuracy |
|
|
| Disadvantages | Only tests one gene at a time |
|
|
|
| Uses | Cascade testing of family members. Testing of single-gene disorders, e.g. cystic fibrosis | Testing disorders with a specific phenotype and multiple known genes | Examine all CDS of the genome | Examines all regions of the genome |
SANGER SEQUENCING
Developed in 1977 by Frederick Sanger, Sanger sequencing was the primary method of gene sequencing for >40 years. While in many scenarios Sanger sequencing has been succeeded by NGS due to enhanced speed and efficiency, it remains a useful and cost-effective tool in diagnostic testing. It provides targeted sequencing with high analytical validity in detecting causal single-nucleotide variations (SNVs) and insertions and deletions (indels) <10 base pairs in length and is therefore the gold standard for confirmatory testing in single-gene disorders detected by NGS [41]. In practice, it is used in cascade testing of affected or at-risk family members once a pathogenic or likely pathogenic variant has been identified in a proband. It is also used in segregation analysis where a variant of uncertain significance (VUS) is identified, in an attempt to clarify pathogenicity.
NGS
NGS refers to high-throughput, in-parallel sequencing and has the ability to generate a very large volume of data very cheaply [42]. It can be used to code the entire exome or genome or to code only areas of specific interest.
There are numerous different NGS platforms that use a variety of sequencing technologies. However, all function using the core principle of the simultaneous parallel sequencing of millions of small fragments of DNA, ∼1000–10 000 base pairs in length. Each fragment is sequenced multiple times (30 times is the industry standard) to ensure a great depth of coverage and accurate data when detecting unexpected DNA variations [43].
The fragments are then pieced together using bioinformatics to map the individual reads to a reference human genome [44]. Algorithms are used to detect the best match between two or more fragments of genetic sequencing [45]. Another set of bioinformatic algorithms detects mismatches between the reference genome and the mapped fragments to detect SNVs and indels, in a process called variant calling [46]. Variant calling algorithms use statistical models of potential errors to detect which mismatches represent a true change and which are sequencing errors. The variants are then annotated with the addition of functional information such as the frequency of the variant in human population databases and the predicted function of the variant upon the protein product of the gene [47]. Databases such as ClinVar collate and publish information about previously interpreted variants. Large-scale genomic sequencing databases, such as the Genome Aggregation Database, publish data on normal population variation and can be used to help distinguish common from rare variants and thus aid in determination of pathogenicity [48]. The American College of Medical Genetics (ACMG) standards and guidelines are widely used criteria that assess variants into one of five categories: pathogenic, likely pathogenic, VUS, likely benign and benign [49]. These data then need to be interpreted in a clinical context with the input and expertise of clinicians [50].
There are many different methods of NGS. These may aim to sequence the entire exome or the entire genome or may focus on specific gene sequences, as is the case in gene panels.
WES AND WHOLE GENOME SEQUENCING (WGS)
The exome, that part of the genome that consists of exons, accounts for only 1–2% of the entire genome. WES targets ∼22 000 known genes, or roughly 95% of human exons. It examines only the protein-coding regions [CoDing Sequence (CDS)]. WGS sequences 50–100 times more of the genome than WES. It is untargeted and sequences exons, but also regulatory, intronic and intergenic regions.
The advantage of WES over WGS is that it is less expensive and the bioinformatic tools associated with it are more advanced. It significantly reduces the number of variants one has to interpret (22 000 versus 2–3 million), and as 75% of pathogenic variants lie within the exome, it is a valuable approach. However, there are emerging whole-genome approaches that detect structural variants and expansions of nucleotide repeats associated with disease that cannot be detected by WES. In time, once the analysis of WGS is more robust, it is likely that this will become the test of choice.
WES and WGS may be used de novo or as a second-line test when gene panel testing does not detect a disease-causing variant [51]. Success rates are variable and depend on the disorder being sequenced, but have been reported to be >25% in consecutive patients referred for genetic testing [52]. WES was used by Lata et al. [53] to sequence 3067 patients with CKD, giving a diagnostic yield of 9.3%, encompassing 66 separate monogenic disorders, of which 39 were found in only a single patient. Diagnostic rates are enhanced if the proband’s biological parents are also tested in so-called trio testing [54]. This is particularly true if it is a de novo disorder; in contrast, where a parent is also affected, triotesting will result in numerous inherited variants that can be difficult to filter.
Some laboratories offer homozygous exome testing on affected children of consanguineous relationships or if there are two affected siblings in a non-consanguineous relationship. Filtering is performed looking for homozygous or compound heterozygous variants common to both affected siblings and then segregation is completed on the parents using Sanger sequencing. Indeed, diagnoses have been made in some lethal foetal disorders by testing parents from consanguineous unions for common homozygous variants whose phenotype fits with the findings in the foetus [55].
There are fewer data on the use of WGS in the diagnosis of kidney disease [56]. The majority of disease-causing variants have been detected in the exon coding regions, although pathogenic variants have been detected in intronic regions [21, 57].
As massive parallel sequencing becomes less expensive, it has been argued that WES and WGS should replace targeted NGS as the first line in genetic sequencing [34]. WES and WGS massively expand the search window for potential causative variants. They may aid in detecting new variants and new genotype–phenotype correlations and may be more sensitive in detecting patients with more than one monogenic disorder causing their disease [58].
TARGETED GENE PANELS
Targeted gene panels are a form of NGS that targets only a specific set of curated genes. While WES and WGS look at the entirety of the exome and genome, respectively, targeted panels must be designed. Curated lists of genes are chosen by the testing geneticist or through the use of a resource such as PanelApp, a curated crowdsourcing tool that allows gene panels to be shared and evaluated by the scientific community (https://panelapp.genomicsengland.co.uk/). Because genes have to be selected, it means that panels are restricted by our current knowledge of the genes that cause disease, and must be updated frequently to ensure that they remain current. Unlike WES and WGS, it is not possible to retrospectively review the data panels provides, as our knowledge of the genes that cause disease constantly expands. As WES and WGS become more cost effective, targeted NGS has less of a role.
Nevertheless, panels have a role in testing homogenous conditions caused by a small number of known genes, such as testing for COL4A3, COL4A4 and COL4A5 in suspected Alport syndrome. The advantages to gene panels over WES or WGS are the reduced likelihood of finding an incidental finding or a VUS. While VUSs still occur in panel testing, they are less common. In addition, when a VUS occurs in a targeted gene test it is more likely to be pathogenic than benign, as the more genes one tests the more likely the variant is benign.
Panel-based sequencing has been used with success in renal disease. The Australian Renal Gene Panels service at the Children’s Hospital, Brisbane, QLD, Australia uses 10 targeted gene panels covering a curated range of 207 genes and was able to achieve a 43% diagnosis rate in patients referred over a 3-year period [59]. This was effective in homogeneous conditions such as Alport syndrome, but much less effective in heterogeneous conditions such as congenital abnormalities of the kidney and urinary tract (CAKUT).
BENEFITS AND RISKS OF GENETIC TESTING
Individuals undergoing genetic testing require pre- and post-testing counselling to ensure they understand the risks as well as the benefits of genetic testing [60]. Clinicians should document that informed consent has been given and that the patient is prepared for the possibility of uncertain and/or unexpected findings [60]. A multidisciplinary approach with the availability of nephrologists, clinical geneticists and genetic counsellors will aid in safeguarding this process. Our team has developed short patient animation videos on variants of uncertain significance to aid in the consent process (http://bit.ly/VariantUS). Genetic testing can be slow, often taking months or even years, thus it may not be possible to make real-time clinical decisions based on a genetic diagnosis.
In general, genetic diagnosis has the potential to help answer the questions that are most important to the patient: What is the diagnosis? Why did it occur? What will be the future course? Will it reoccur post-transplant? Are other organs affected? Is there effective treatment? Are family members at risk and can they consider becoming renal donors? [61].
The benefits of a genetic diagnosis include the ability to enhance the specificity of diagnosis, allowing guidance of the prognosis and management. It allows for genetic counselling and family planning and can provide reassurance for unaffected family members. On a population level, it allows us to understand the disease process and its prevalence and provides opportunities to develop future therapies. It offers opportunities for diagnosis in those who, by traditional methods, would not have a diagnosis. It can potentially help risk stratification in clinical trials and identify patients unlikely to respond to treatment, such as avoiding steroid therapy in nephrotic syndrome [62, 63].
However, there are concerns regarding clinicians’ understanding of genetic test reports, particularly the limitations of our knowledge when a VUS is identified. Some laboratories have questioned whether non-clinical geneticists should be informed of VUSs. The concern being that without a background understanding of normal human variation, someone will assume any variant is significant no matter how the report is worded.
There have been a number of high-profile legal cases where clinicians have misinterpreted DNA variants with serious consequences. If one overinterprets a gene variant and determines it is pathogenic (when in fact it is not), then it can have consequences not only to the proband, but also their at-risk relatives who might act on a result if they test positive. There have been cases where BRCA1 and BRCA2 were overinterpreted, leading to prophylactic surgery such as mastectomy, oophorectomy and colorectal surgery. Potentially misinterpretation could lead to a termination of a healthy pregnancy. Alternatively, if one underinterprets a variant, it can mean a patient misses vital opportunities for treatment, an opportunity for pre-natal testing or pre-implantation genetic diagnosis or avoidance of drugs contraindicated in that specific disorder. There are legal actions pending for all these scenarios.
Non-targeted NGS, which examines the whole exome or genome, may reveal unsolicited genetic findings, including some that may have a significant clinical impact on the patient. This may include actionable variants that put the individual at risk of developing cancer or cardiovascular disease [64]. It is important to appreciate that guidance from the European Society of Human Genetics states that the utility of the test and the diagnostic yield are considered before offering testing and whether testing will rule out or rule in a diagnosis. In contrast, the ACMG lists 59 clinically actionable variants that should be reported, regardless of the initial indication for sequencing, and this policy is observed throughout the USA [64, 65].
There have been reports of people experiencing genetic discrimination as a result of their genetic diagnosis [66]. In Europe, the European Union Charter of Fundamental Rights, Article 21.1 prohibits discrimination based on ‘genetic features’, but in practice the EU lacks a concrete legislative position and legislation is left to member states. In the USA, the Genetic Information Nondiscrimination Act protects the right to health insurance and employment [67]. However, more subtle discrimination may still occur.
The scope of some genetic counselling may be outside a nephrologist’s usual practice and there is a need for specialized genetic counsellors to work alongside physicians. These counsellors can support physicians in helping to interpret data, answer scientific queries and provide emotional support [68].
DISEASES OF THE KIDNEY
Advances in genomic medicine have affected diagnosis and treatment in several kidney diseases that previously relied solely on renal biopsy as the primary diagnostic modality. These include ciliopathies, autosomal dominant tubulointerstitial kidney disease (ADTKD), Alport syndrome and thin basement membrane nephropathy, FSGS and IgA nephropathy.
CILIOPATHIES
Ciliopathies are a group of multisystem, inherited disorders that cause functional or structural deficits in the cilia or the cilia anchoring structures. Cilia are cell organelles present in almost every cell with signalling or sensory functions [69, 70]. They may be motile or non-motile. Those that are affected in renal disease tend to be primary or non-motile cilia [71, 72]. Primary cilia are present on the apical surface of the tubule and collecting ducts. They act as a sensor that modulates urine osmolality and composition via activation of intracellular signalling pathways [73].
Primary cilia are present in cell types across the body, so ciliopathies may affect any organ system in the body and extrarenal features relating to cilia dysfunction are commonly associated with the renal phenotype. However, in practice, they primarily cause renal disease, retinal deterioration and cerebral abnormalities [74]. Overlapping clinical features can make the diagnosis of ciliopathies challenging.
Most ciliopathies are single-gene disorders that alter the function of the cilium–centrosome complex [71]. Autosomal dominant PKD (ADPKD) and Von Hippel–Lindau disease are the main autosomal dominant ciliopathies that affect the kidney. Others are primarily autosomal recessive.
Nephronophthisis, meaning wasting of the nephron, is an autosomal recessive ciliopathy. It is thought to be the most common inherited kidney disease to cause ESRD in the paediatric and adolescent population [75, 76]. Initial symptoms include polyuria, polydipsia and failure of urinary concentrating ability, progressing insidiously to CKD and eventually ESRD, which may be recognized late due to the insidious nature of its onset. It is distinguished in three forms: infantile, juvenile and adolescent, based on the age of onset of ESRD. These three forms manifest ESRD at the median ages of 1, 13 and 19 years, respectively [77–79].
It is characterized by renal cysts that occur at the corticomedullary junction. Kidneys are usually normal or reduced in size, though in the infantile form they may show a moderate increase in size [80–82]. Renal histology shows a characteristic triad of tubulointerstitial nephropathy, tubular basement membrane disruption and corticomedullary cysts [83]. Nephronophthisis may be isolated to the kidneys, however, it is often a component part of syndromes with other extrarenal features. Nearly 100 genes have been associated with nephronophthisis-related ciliopathies to date and as our knowledge of the genetic basis of nephronophthisis-related ciliopathies expands, it becomes clear that there is significant phenotypic overlap [84, 85]. Many multisystem ciliopathies may be caused by defects in multiple proteins with roles in ciliary structure, function and biogenesis [86]. Some heterozygous disorders such as Joubert syndrome have >30 known causative genes [87]. Our understanding of these disorders and the overlap between them continues to shift rapidly. Some of the most common ciliopathies associated with renal disease are summarized in Table 2. However, it is advised that all patients who present with nephronophthisis should undergo screening for ophthalmological and hepatic abnormalities at a minimum.
Summary of ciliopathies associated with renal disease
| Syndrome | Inheritance | Gene | Renal features | Extrarenal features | Renal histology |
|---|---|---|---|---|---|
| ADPKD | Autosomal dominant | PKD1, PKD2, GANAB and DNAJB11 | Polycystic kidneys, progression to ESKD in third to fifth decade of life | Polycystic liver and pancreas, increased risk of intracerebral aneurysm | Saccular expansions or diverticula of all portions of renal tubule, secondary glomerulosclerosis |
| ARPKD | Autosomal recessive | PKHD1 and DZIP1L | Polycystic liver, progressive CKD in the first decade of life | Polycystic liver, liver fibrosis, | Elongated, radially arranged cysts formed from the collecting tubules |
| Von Hippel–Lindau syndrome | Autosomal dominant | VHL | Clear cell carcinoma of the kidney | Haemangioblastomas in the spine, brain, pancreas and retina, pheochromocytoma, pancreatic neuroendocrine tumours | Benign and malignant cystic lesions, clear cell carcinoma |
| Nephronophthisis | Autosomal recessive | NPHP1-NPHP6, TMEM67, ANKS6, TTC21B, GLIS2, CEP164, INVS, MAPKB1, NEK8, CEP83, WDR19, ZNF423 and others | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | None | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Senior–Løken syndrome | Autosomal recessive | NPHP1, NPHP4, CEP290, IQCB1, SDCCAG8 and WDR19 | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | Retinal degeneration, retinitis pigmentosa and leber congenital amaurosis | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Joubert syndrome | Autosomal recessive | INPP5E, TMEM216, CC2D2A, AHI1, CEP41, RPGRIP1L and others | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life or multicystic kidneys in first year of life | Nephronophthisis, ataxia, hypotonia, learning difficulties and retinal dystrophy | Cystic dysplasia or cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Bardet–Biedl syndrome | Autosomal recessive | BBS1-BBS12, NPHP6, CEP290, MKS1, SDCCAG8 and SEPT7 | Structural and urinary tract abnormalities, tubulointerstitial disease | Obesity, learning difficulties, retinitis pigmentosa, post-axial polydactyly, behavioural difficulties, hypogonadism and nephronophthisis | Cystic lobulation of the kidney |
| Meckel syndrome | Autosomal recessive | B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TMEM67 and TMEM216 | Enlarged cyst filled kidneys | Occipital encephalocele, polydactyly and perinatal death | Little corticomedullary differentiation and deficient nephrons |
| Alström syndrome | Autosomal recessive | ALMS1 | Progressive CKD in third decade of life, nephrocalcinosis | Sensorineural hearing loss, nystagmus, dilated cardiomyopathy, asthma, insulin-dependent diabetes hypercholesterolaemia and obesity short stature | Mixed tubulointerstitial and glomerular disease |
| RHYNS syndrome | Autosomal recessive | TMEM67 | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis, ESKD in adolescence | Gaze palsy, sensorineural hearing loss, retinitis pigmentosa and skeletal dysplasia | Cysts at the corticomedullary junction, severe tubular atrophy and interstitial fibrosis |
| Oro-facial-digital syndrome | X-linked, Autosomal recessive | OFD1 and CPLANE1 | Polycystic kidneys, ESRD in third to fifth decade of life | Facial asymmetry, alae nasi hypoplasia, tongue abnormalities and cleft lip | Cysts of the glomeruli and distal tubules |
| Syndrome | Inheritance | Gene | Renal features | Extrarenal features | Renal histology |
|---|---|---|---|---|---|
| ADPKD | Autosomal dominant | PKD1, PKD2, GANAB and DNAJB11 | Polycystic kidneys, progression to ESKD in third to fifth decade of life | Polycystic liver and pancreas, increased risk of intracerebral aneurysm | Saccular expansions or diverticula of all portions of renal tubule, secondary glomerulosclerosis |
| ARPKD | Autosomal recessive | PKHD1 and DZIP1L | Polycystic liver, progressive CKD in the first decade of life | Polycystic liver, liver fibrosis, | Elongated, radially arranged cysts formed from the collecting tubules |
| Von Hippel–Lindau syndrome | Autosomal dominant | VHL | Clear cell carcinoma of the kidney | Haemangioblastomas in the spine, brain, pancreas and retina, pheochromocytoma, pancreatic neuroendocrine tumours | Benign and malignant cystic lesions, clear cell carcinoma |
| Nephronophthisis | Autosomal recessive | NPHP1-NPHP6, TMEM67, ANKS6, TTC21B, GLIS2, CEP164, INVS, MAPKB1, NEK8, CEP83, WDR19, ZNF423 and others | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | None | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Senior–Løken syndrome | Autosomal recessive | NPHP1, NPHP4, CEP290, IQCB1, SDCCAG8 and WDR19 | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | Retinal degeneration, retinitis pigmentosa and leber congenital amaurosis | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Joubert syndrome | Autosomal recessive | INPP5E, TMEM216, CC2D2A, AHI1, CEP41, RPGRIP1L and others | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life or multicystic kidneys in first year of life | Nephronophthisis, ataxia, hypotonia, learning difficulties and retinal dystrophy | Cystic dysplasia or cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Bardet–Biedl syndrome | Autosomal recessive | BBS1-BBS12, NPHP6, CEP290, MKS1, SDCCAG8 and SEPT7 | Structural and urinary tract abnormalities, tubulointerstitial disease | Obesity, learning difficulties, retinitis pigmentosa, post-axial polydactyly, behavioural difficulties, hypogonadism and nephronophthisis | Cystic lobulation of the kidney |
| Meckel syndrome | Autosomal recessive | B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TMEM67 and TMEM216 | Enlarged cyst filled kidneys | Occipital encephalocele, polydactyly and perinatal death | Little corticomedullary differentiation and deficient nephrons |
| Alström syndrome | Autosomal recessive | ALMS1 | Progressive CKD in third decade of life, nephrocalcinosis | Sensorineural hearing loss, nystagmus, dilated cardiomyopathy, asthma, insulin-dependent diabetes hypercholesterolaemia and obesity short stature | Mixed tubulointerstitial and glomerular disease |
| RHYNS syndrome | Autosomal recessive | TMEM67 | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis, ESKD in adolescence | Gaze palsy, sensorineural hearing loss, retinitis pigmentosa and skeletal dysplasia | Cysts at the corticomedullary junction, severe tubular atrophy and interstitial fibrosis |
| Oro-facial-digital syndrome | X-linked, Autosomal recessive | OFD1 and CPLANE1 | Polycystic kidneys, ESRD in third to fifth decade of life | Facial asymmetry, alae nasi hypoplasia, tongue abnormalities and cleft lip | Cysts of the glomeruli and distal tubules |
Summary of ciliopathies associated with renal disease
| Syndrome | Inheritance | Gene | Renal features | Extrarenal features | Renal histology |
|---|---|---|---|---|---|
| ADPKD | Autosomal dominant | PKD1, PKD2, GANAB and DNAJB11 | Polycystic kidneys, progression to ESKD in third to fifth decade of life | Polycystic liver and pancreas, increased risk of intracerebral aneurysm | Saccular expansions or diverticula of all portions of renal tubule, secondary glomerulosclerosis |
| ARPKD | Autosomal recessive | PKHD1 and DZIP1L | Polycystic liver, progressive CKD in the first decade of life | Polycystic liver, liver fibrosis, | Elongated, radially arranged cysts formed from the collecting tubules |
| Von Hippel–Lindau syndrome | Autosomal dominant | VHL | Clear cell carcinoma of the kidney | Haemangioblastomas in the spine, brain, pancreas and retina, pheochromocytoma, pancreatic neuroendocrine tumours | Benign and malignant cystic lesions, clear cell carcinoma |
| Nephronophthisis | Autosomal recessive | NPHP1-NPHP6, TMEM67, ANKS6, TTC21B, GLIS2, CEP164, INVS, MAPKB1, NEK8, CEP83, WDR19, ZNF423 and others | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | None | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Senior–Løken syndrome | Autosomal recessive | NPHP1, NPHP4, CEP290, IQCB1, SDCCAG8 and WDR19 | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | Retinal degeneration, retinitis pigmentosa and leber congenital amaurosis | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Joubert syndrome | Autosomal recessive | INPP5E, TMEM216, CC2D2A, AHI1, CEP41, RPGRIP1L and others | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life or multicystic kidneys in first year of life | Nephronophthisis, ataxia, hypotonia, learning difficulties and retinal dystrophy | Cystic dysplasia or cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Bardet–Biedl syndrome | Autosomal recessive | BBS1-BBS12, NPHP6, CEP290, MKS1, SDCCAG8 and SEPT7 | Structural and urinary tract abnormalities, tubulointerstitial disease | Obesity, learning difficulties, retinitis pigmentosa, post-axial polydactyly, behavioural difficulties, hypogonadism and nephronophthisis | Cystic lobulation of the kidney |
| Meckel syndrome | Autosomal recessive | B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TMEM67 and TMEM216 | Enlarged cyst filled kidneys | Occipital encephalocele, polydactyly and perinatal death | Little corticomedullary differentiation and deficient nephrons |
| Alström syndrome | Autosomal recessive | ALMS1 | Progressive CKD in third decade of life, nephrocalcinosis | Sensorineural hearing loss, nystagmus, dilated cardiomyopathy, asthma, insulin-dependent diabetes hypercholesterolaemia and obesity short stature | Mixed tubulointerstitial and glomerular disease |
| RHYNS syndrome | Autosomal recessive | TMEM67 | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis, ESKD in adolescence | Gaze palsy, sensorineural hearing loss, retinitis pigmentosa and skeletal dysplasia | Cysts at the corticomedullary junction, severe tubular atrophy and interstitial fibrosis |
| Oro-facial-digital syndrome | X-linked, Autosomal recessive | OFD1 and CPLANE1 | Polycystic kidneys, ESRD in third to fifth decade of life | Facial asymmetry, alae nasi hypoplasia, tongue abnormalities and cleft lip | Cysts of the glomeruli and distal tubules |
| Syndrome | Inheritance | Gene | Renal features | Extrarenal features | Renal histology |
|---|---|---|---|---|---|
| ADPKD | Autosomal dominant | PKD1, PKD2, GANAB and DNAJB11 | Polycystic kidneys, progression to ESKD in third to fifth decade of life | Polycystic liver and pancreas, increased risk of intracerebral aneurysm | Saccular expansions or diverticula of all portions of renal tubule, secondary glomerulosclerosis |
| ARPKD | Autosomal recessive | PKHD1 and DZIP1L | Polycystic liver, progressive CKD in the first decade of life | Polycystic liver, liver fibrosis, | Elongated, radially arranged cysts formed from the collecting tubules |
| Von Hippel–Lindau syndrome | Autosomal dominant | VHL | Clear cell carcinoma of the kidney | Haemangioblastomas in the spine, brain, pancreas and retina, pheochromocytoma, pancreatic neuroendocrine tumours | Benign and malignant cystic lesions, clear cell carcinoma |
| Nephronophthisis | Autosomal recessive | NPHP1-NPHP6, TMEM67, ANKS6, TTC21B, GLIS2, CEP164, INVS, MAPKB1, NEK8, CEP83, WDR19, ZNF423 and others | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | None | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Senior–Løken syndrome | Autosomal recessive | NPHP1, NPHP4, CEP290, IQCB1, SDCCAG8 and WDR19 | Polydipsia, polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life | Retinal degeneration, retinitis pigmentosa and leber congenital amaurosis | Cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Joubert syndrome | Autosomal recessive | INPP5E, TMEM216, CC2D2A, AHI1, CEP41, RPGRIP1L and others | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis and ESRD by second to third decade of life or multicystic kidneys in first year of life | Nephronophthisis, ataxia, hypotonia, learning difficulties and retinal dystrophy | Cystic dysplasia or cysts at the corticomedullary junction, severe tubular atrophy, interstitial fibrosis and chronic inflammation |
| Bardet–Biedl syndrome | Autosomal recessive | BBS1-BBS12, NPHP6, CEP290, MKS1, SDCCAG8 and SEPT7 | Structural and urinary tract abnormalities, tubulointerstitial disease | Obesity, learning difficulties, retinitis pigmentosa, post-axial polydactyly, behavioural difficulties, hypogonadism and nephronophthisis | Cystic lobulation of the kidney |
| Meckel syndrome | Autosomal recessive | B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TMEM67 and TMEM216 | Enlarged cyst filled kidneys | Occipital encephalocele, polydactyly and perinatal death | Little corticomedullary differentiation and deficient nephrons |
| Alström syndrome | Autosomal recessive | ALMS1 | Progressive CKD in third decade of life, nephrocalcinosis | Sensorineural hearing loss, nystagmus, dilated cardiomyopathy, asthma, insulin-dependent diabetes hypercholesterolaemia and obesity short stature | Mixed tubulointerstitial and glomerular disease |
| RHYNS syndrome | Autosomal recessive | TMEM67 | Polyuria, failure of concentrating ability, tubulointerstitial fibrosis, ESKD in adolescence | Gaze palsy, sensorineural hearing loss, retinitis pigmentosa and skeletal dysplasia | Cysts at the corticomedullary junction, severe tubular atrophy and interstitial fibrosis |
| Oro-facial-digital syndrome | X-linked, Autosomal recessive | OFD1 and CPLANE1 | Polycystic kidneys, ESRD in third to fifth decade of life | Facial asymmetry, alae nasi hypoplasia, tongue abnormalities and cleft lip | Cysts of the glomeruli and distal tubules |
Autosomal recessive PKD (ARPKD) is caused by variants in PKHD1, which encodes the transmembrane protein fibrocystin [88]. Less common and more severe than ADPKD, it tends to present in the first decade of life and is often detected prenatally [89, 90]. Up to 30% of patients may present with oligohydramnios, Potter’s sequence and pulmonary hypoplasia, leading to a significant risk of neonatal death [91, 92].
Although not a true ciliopathy, disease-causing variants in FAN1 may cause a rare autosomal recessive form of tubulointerstitial kidney disease called karyomegalic interstitial nephritis (KIN) [18, 93]. This is characterized by slowly progressive renal failure, progressing to ESRD in the fifth decade of life, bronchiectasis and deranged liver function tests. Karyomegaly describes the presence of an enlarged cell nucleus, and in KIN the characteristic renal pathology is tubulointerstitial fibrosis and atrophy and the presence of enlarged, hyperchromatic nuclei in the tubular epithelial cells. Karyomegaly can also be seen in other organs, including the brain and prostate [94].
ADTKD
ADTKD can prove a diagnostic challenge for physicians. It is notable for an autosomal dominant inheritance pattern, but is otherwise characterized by a lack of distinctive clinical or histological features, particularly in the case of ADTKD-MUC1. Previously known by a number of names, but particularly by medullary cystic kidney disease, a 2015 Kidney Disease: Improving Global Outcomes consensus report recommended the change to ADTKD, as cysts are not pathognomonic of the disease nor are they confined to the medulla when they do appear [95, 96].
ADTKD is characterized by nonspecific renal features (Figure 1A–C). It features progressive loss of kidney function, but the rate of decline is very variable and ESRD can occur at any time between the ages of 17 and 75 years [97]. Urinalysis is bland, with mild or absent proteinuria, and there may be nocturia or enuresis in children as concentrating ability is lost. Histological features are equally non-specific. A kidney biopsy may show interstitial scarring, but has no distinctive characteristic features [98]. Usual histological findings include interstitial fibrosis, tubular atrophy, microcyst formation and thickening and lamellation of the tubular basement membranes. There will be no evidence of glomerular disease and negative IF for immunoglobins and complement [95, 97].
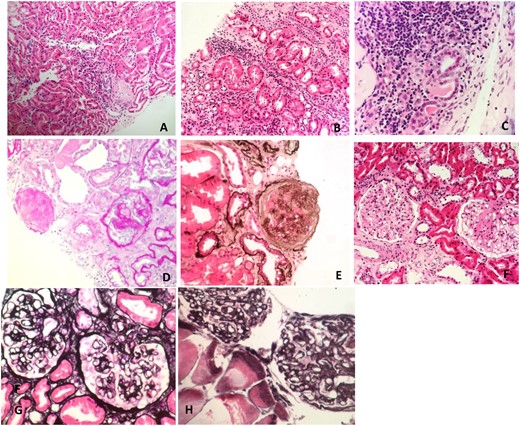
Renal tissue under LM. (A) Interstitial inflammatory infiltrate and tubulitis on haematoxylin and eosin (H&E) stain in a patient with ADTKD-HNF1B. (B) Interstitial inflammatory infiltrate and tubulitis on H&E stain in a patient with ADTKD-UMOD. (C) Interstitial inflammatory infiltrate and tubulitis on H&E stain in a patient with ADTKD-unknown. (D) Global and segmental glomerulosclerosis on perisodic acid–schiff stain in a patient with FSGS due to autosomal dominant INF2 mutation. (E) Glomerulosclerosis on silver stain in a patient with a COL4A4 mutation. (F) Segmental glomerulosclerosis on H&E stain in a patient with primary FSGS, genetics unknown. (G) Capillary loop thickening and double contour formation on silver stain in a patient with hereditary C3 nephropathy. (H) Capillary loop thickening and double contour formation on silver stain in a patient with hereditary C3 nephropathy.
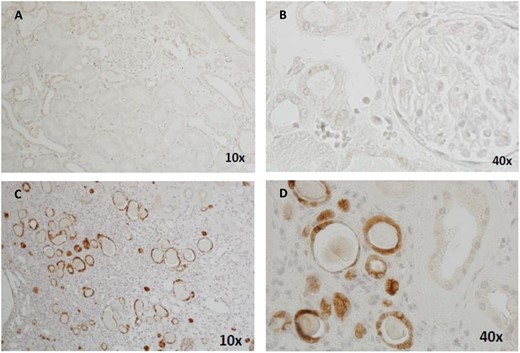
Immunohistochemical staining of renal biopsy tissue samples showing positive MUC1fs staining in affected individuals with ADTKD-MUC1 and negative MUC1fs staining in controls. (A) Kidney section from a negative control at 10× magnification. (B) Negative control at ×40 magnification. (C) Kidney section from an individual affected by ADTKD-MUC1 at ×10 magnification, showing positive staining of the distal tubules and collecting ducts at ×20 magnification. (D) Kidney section from an individual affected by ADTKD-MUC1 at ×20 magnification, showing positive staining of the distal tubules and collecting ducts at ×20 magnification.
Five genes are known to cause ADTKD: UMOD (ADTKD-UMOD), MUC1 (ADTKD-MUC1), REN (ADTKD-REN), HNF1B (ADTKD-HNF1B) and, more rarely, SEC61A1 (ADTKD-SEC61A1) [99–102]. ADTKD-UMOD, HNF1B, REN and SEC61A1 may have extrarenal features, while ADTKD-MUC1 does not (Table 3). Another category, ADT not otherwise specified (ADT-NOS) is used to refer to those who have not undergone genetic testing or in whom testing is inconclusive.
Characteristics of the four major causes of ADTKD
| Condition | ADTKD-MUC1 | ADTKD-UMOD | ADTKD-REN | ADTKD-HNF1B | ADTKD-SEC61A1 |
|---|---|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant |
| Age of onset of CKD (years) | 18–50 | 18–50 | First year of life | Variable, may be antenatal | 10–50 |
| Onset in childhood | No | Gout rarely | Frequent | May have prenatal findings | Variable |
| Renal features | Progressive renal disease with bland urinary sediment | Progressive renal disease with bland urinary sediment |
|
| Progressive renal disease, small dysplastic kidneys without cysts |
| Gout | In advanced CKD | Early onset, frequent | Early onset, frequent | Early onset, frequent | In second decade of life |
| Other electrolyte abnormalities | None identified | Low fractional excretion of urate, low urinary excretion of uromodulin | Hyperkalaemia,low urinary excretion of uromodulin | Hypomagnesaemia, hypokalaemia | None identified |
| Other clinical features | None | None |
|
| Congenital anaemia, intrauterine and postnatal growth retardation, polydactyly, mild mental retardation |
| Histopathology | Non-specific tublointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Small foci of tubulointerstitial lesions |
| Condition | ADTKD-MUC1 | ADTKD-UMOD | ADTKD-REN | ADTKD-HNF1B | ADTKD-SEC61A1 |
|---|---|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant |
| Age of onset of CKD (years) | 18–50 | 18–50 | First year of life | Variable, may be antenatal | 10–50 |
| Onset in childhood | No | Gout rarely | Frequent | May have prenatal findings | Variable |
| Renal features | Progressive renal disease with bland urinary sediment | Progressive renal disease with bland urinary sediment |
|
| Progressive renal disease, small dysplastic kidneys without cysts |
| Gout | In advanced CKD | Early onset, frequent | Early onset, frequent | Early onset, frequent | In second decade of life |
| Other electrolyte abnormalities | None identified | Low fractional excretion of urate, low urinary excretion of uromodulin | Hyperkalaemia,low urinary excretion of uromodulin | Hypomagnesaemia, hypokalaemia | None identified |
| Other clinical features | None | None |
|
| Congenital anaemia, intrauterine and postnatal growth retardation, polydactyly, mild mental retardation |
| Histopathology | Non-specific tublointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Small foci of tubulointerstitial lesions |
Characteristics of the four major causes of ADTKD
| Condition | ADTKD-MUC1 | ADTKD-UMOD | ADTKD-REN | ADTKD-HNF1B | ADTKD-SEC61A1 |
|---|---|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant |
| Age of onset of CKD (years) | 18–50 | 18–50 | First year of life | Variable, may be antenatal | 10–50 |
| Onset in childhood | No | Gout rarely | Frequent | May have prenatal findings | Variable |
| Renal features | Progressive renal disease with bland urinary sediment | Progressive renal disease with bland urinary sediment |
|
| Progressive renal disease, small dysplastic kidneys without cysts |
| Gout | In advanced CKD | Early onset, frequent | Early onset, frequent | Early onset, frequent | In second decade of life |
| Other electrolyte abnormalities | None identified | Low fractional excretion of urate, low urinary excretion of uromodulin | Hyperkalaemia,low urinary excretion of uromodulin | Hypomagnesaemia, hypokalaemia | None identified |
| Other clinical features | None | None |
|
| Congenital anaemia, intrauterine and postnatal growth retardation, polydactyly, mild mental retardation |
| Histopathology | Non-specific tublointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Small foci of tubulointerstitial lesions |
| Condition | ADTKD-MUC1 | ADTKD-UMOD | ADTKD-REN | ADTKD-HNF1B | ADTKD-SEC61A1 |
|---|---|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant |
| Age of onset of CKD (years) | 18–50 | 18–50 | First year of life | Variable, may be antenatal | 10–50 |
| Onset in childhood | No | Gout rarely | Frequent | May have prenatal findings | Variable |
| Renal features | Progressive renal disease with bland urinary sediment | Progressive renal disease with bland urinary sediment |
|
| Progressive renal disease, small dysplastic kidneys without cysts |
| Gout | In advanced CKD | Early onset, frequent | Early onset, frequent | Early onset, frequent | In second decade of life |
| Other electrolyte abnormalities | None identified | Low fractional excretion of urate, low urinary excretion of uromodulin | Hyperkalaemia,low urinary excretion of uromodulin | Hypomagnesaemia, hypokalaemia | None identified |
| Other clinical features | None | None |
|
| Congenital anaemia, intrauterine and postnatal growth retardation, polydactyly, mild mental retardation |
| Histopathology | Non-specific tublointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Non-specific tubulointerstitial atrophy | Small foci of tubulointerstitial lesions |
Since renal biopsy will not provide a precise diagnosis in ADTKD, genetic testing is a viable alternative, particularly as the autosomal dominant pattern means that multiple family members may be affected, and the variable age of onset and bland radiological and urinary sediment findings mean it may be difficult to distinguish the affected from the unaffected through clinical screening alone. Histology alone will not distinguish the various pathologies. Commercial genetic testing is available to look for pathogenic variants in UMOD, REN and HNF1B. However, MUC1 is an extraordinarily difficult gene to sequence and cannot currently be sequenced through conventional means, therefore testing is conducted in a very small selection of centres worldwide.
ADTKD-MUC1 is most commonly caused by a frameshift insertion in MUC1 that leads to the formation of a mutant protein, MUC1-fs [100]. The cause of this frameshift is the insertion of a single cytosine (c) into a sequence of seven c base pairs in a variable nucleotide tandem repeat (VNTR) in the MUC1 gene. A VNTR is a short sequence of nucleotides organized into a tandem repeat. In MUC1, the VNTR codes for a serine-, proline- and threonine-rich sequence of 20 amino acids that make up the extracellular part of the Mucin-1 protein. The VNTR is highly polymorphic and because of the high number of cytosine and guanine residues combined with the architecture of the VNTR, direct sequencing is largely ineffective [103]. For these reasons, MUC1 is refractory to many NGS methods and a novel mass spectrometer–based method has instead been developed to detect the C+ insertion in the VNTR [104]. More recently, single-model real-time sequencing has been used to assemble the VNTR in affected and non-affected families and identify the exact positions of causative changes, providing the structural and positional information that cannot be captured by this mass spectrometry method [103]. No truncating or missense variants have been found that cause ADTKD-MUC1.
The pathogenesis of ADTKD-MUC1 is thought to be due to the accumulation of MUC1-fs protein within the cell. However, recent evidence suggests that the accumulation of MUC1-fs protein alone is not immediately toxic and it is only when the unfolded protein response pathway—a homoeostatic mechanism that guards against the accumulation of unfolded proteins—becomes inactivated that it becomes harmful to the cell [105]. This may help explain the variable age of onset. Because of the unique structure of MUC1-fs, it is possible to develop antibodies that detect MUC1-fs but not wild-type MUC1. This has allowed for successful immunohistochemical staining of the MUC1-fs protein on epithelial tissues and in urinary smears [106]. Živná et al. [101] were able to show the presence of MUC1-fs protein in 11 of 12 renal biopsies of patients with ADTKD-MUC1 (Figure 2). Immunostaining of the kidney was 92% sensitive and 81% specific. Skin biopsy was offered as a less invasive alternative, but MUC1-fs was only present in the sebaceous glands, which are not consistently obtained on skin biopsy. However, staining for MUC1-fs on urinary smears was shown to be very effective. There was strong intracellular staining in urothelial cells in affected individuals, regardless of the stage of kidney disease. Staining was shown to be as effective in those with normal renal function, advanced CKD and those on dialysis. In 173 patients tested for MUC1-fs, sensitivity was 94% and specificity was 89%. Seventeen families were identified who had MUC1-fs protein present in urinary cells. Variants other than the C+ insertion into the MUC1 VNTR were identified. All encoded the same MUC1-fs protein. While still only available on a research basis, urinary staining may become a useful non-invasive test. This may be of increasing importance as treatments become available for ADTKD-MUC1. Currently there is no effective treatment for the condition, but Dvela-Levitt et al. [105] recently reported promising results in animal studies and organoids with the small molecule transmebrane P24 trafficking protein 9, which has been shown to promote lysosomal degradation of the toxic MUC1-fs from cells and reverse proteinopathy. The same therapy may have potential treatment implications for ADTKD-UMOD and other proteinopathies.
Alport syndrome
Alport syndrome is a disorder of type IV collagen production. Type IV collagen exists primarily in the basal lamina and is the major collagenous component of the basement membranes of the cochlea of the ear, the glomerulus of the kidney and the cornea, lens and retina of the eye [107]. Alport syndrome is classically characterized by a triad of renal dysfunction, variable sensorineural deafness and ocular abnormalities, including anterior dislocation of the lens, corneal opacities and temporal retinal thinning [108–110]. While an individual must have persistent haematuria to meet the diagnostic criteria for Alport syndrome, deafness and ocular abnormalities are not consistently found in patients with Alport syndrome and are no longer considered necessary for diagnosis in the presence of evidence of disease-causing variants in the type IV collagen genes or characteristic abnormalities of the glomerular basement membrane (GBM) [109].
Alport syndrome occurs due to variants in COL4A3, COL4A4 and COL4A5, all of which affect the expression of the α chains of type IV collagen, α3, α4 and α5 [110]. Approximately 85% of all Alport syndrome cases affect the gene COL4A5 and are inherited in an X-linked fashion, while 15% affect COL4A3 and COL4A4 and are inherited in an autosomal recessive fashion. Autosomal dominant inheritance of Alport syndrome, previously thought to be rare, is now believed to be more common than previously recognized [111]. Alport syndrome may also be inherited in a digenic fashion and patients with disease-causing variants in both COL4A5 and COL4A4 have been identified [112].
The earliest ultrastructural lesion seen in Alport syndrome is thinning of the GBM seen on EM [113]. In established Alport syndrome, changes on LM tend to be non-specific, with glomerulosclerosis, interstitial fibrosis and interstitial foam cells [114]. Differences in collagen immunostaining can be detected between X-linked and recessive Alport syndrome. In X-linked Alport syndrome there is often an absence of α5 type IV collagen in males and patchy or reduced α5 type IV collagen in females. Reduced levels of α5 type IV collagen may still be visible in males with features of Alport syndrome and missense variants in COL4A5 [115]. In autosomal recessive Alport syndrome, α5 type IV collagen is visible in Bowman’s capsule and the distal tubules, but not the GBM.
X-linked Alport syndrome tends to affect males more severely than females. Males tend to have progressive CKD and generally progress to ESRD between the second and third decades of life. Until recently, X-linked Alport syndrome in women was considered a benign status and women were considered only carriers. However, if a carrier is an individual or organism who carriers a single copy or a recessive allele but does not display the trait, then only 5% of women who are heterozygous for X-linked Alport syndrome can be said to be true carriers. The other 95% are affected by Alport syndrome. A study of 195 families with Alport syndrome and a proven COL4A5 disease-causing variant showed that 75% of women developed proteinuria, 28% developed hearing loss and 15% developed ocular defects. While the phenotype is usually milder than in males, 12% of women developed ESRD and 105 women developed deafness by the age of 40 years [116]. For this reason, in 2017, the Alport Syndrome Classification Working Group dispensed with the term ‘carrier’. X-linked Alport syndrome should now be considered a dominant disease, one in which women have a lower, but not trivial, risk of progressing to serious kidney disease [109]. Even within families, the severity of presentation may vary among women. This is likely due to inactivation of the X-chromosome in affected females [117, 118]. This may lead to a mosaic pattern seen on the GBM [119].
Autosomal recessive Alport syndrome affects both sexes equally and causes deafness and ESRD at a young age. Heterozygous individuals tend to have milder renal involvement. However, they have a higher lifetime risk of ESRD than the general population and a retrospective study suggests they benefit from renin–angiotensin–aldosterone system blockade [120]. It may be that the clinical significance of being a ‘carrier’ of a COL4A3, COL4A4 or COL4A5 disease-causing variant has been grossly underestimated.
Increasingly, it is recognized that Alport syndrome represents a broader spectrum of disease than previously thought. There is increasing recognition that variants in COL4A3, COL4A4 and COL4A5 are a cause of FSGS in adults and children and may be among the most common causes of hereditary FSGS in children [121–123].
Currently treatment for Alport syndrome is limited to renin–angiotensin blockade, although bardoxalone is now in phase 3 trials as a potential treatment for Alport syndrome, and some promise has also been seen in treatments with microRNA-21 [124, 125].
THIN BASEMENT MEMBRANE NEPHROPATHY (TBMN)
TBMN is characterized by the uniform thinning of the basement membrane seen on EM. A normal basement membrane is ∼350 nm, while a thin basement membrane is <250 nm [126, 127].
TBMN presents with normal renal function, dysmorphic haematuria and <500 mg/day of proteinuria [128, 129]. The most common presentation is that of microscopic haematuria, and proteinuria is not usually present. Because of this, the incidence of TBMN has not been described accurately, but some studies have suggested incidences as high as 1% of the population based on the percentage of children and adults who present with haematuria and no urological cause [130]. It tends to occur more commonly in women and this is thought to be because the GBM in men is naturally thicker [131]. At least two-thirds of those affected will have another affected relative [129].
At least 40% of patients with TBMN have a heterozygous disease-causing variant in the COL4A4 or COL4A3 gene, which in homozygous patients or compound heterozygotes causes autosomal recessive Alport syndrome [119]. Recent studies suggest that variants in other collagen genes, such as COL4A1, may also be implicated in TBMN [132].
There is ongoing debate as to whether TBMN is a term that should be discarded completely in favour of autospmal dominant Alport syndrome in patients with an identified COL4A3, COL4A4 or COL4A5 variant and ‘recurrent and persistent haematuria with other morphologic changes’ in those with evidence of a thin GBM on biopsy and negative or no genetic studies [109]. Proponents advise that this will help to avoid the erroneous assumption that TBMN is an entirely benign condition, while opponents believe it will create unwarranted fear, when only a small proportion progress to ESRD [133]. It is probably advisable that all those with TBMN have lifelong yearly assessment.
FSGS
FSGS has been thought of until recently as a single disease process. However, more accurately it should be considered as a histological description that describes the underlying disease pathology of multiple genetically and clinically distinct conditions. There have been recent efforts to stratify FSGS and nephrotic syndrome into hereditary, immune-based and circulating factor–based disease [134, 135]. FSGS is characterized by hyalinosis, mesangial sclerosis, capillary destruction, foam cells and adhesions between the glomerular tuft and Bowman’s capsule (Figure 1E–F) [136]. The underlying cause of this histological appearance is often not clinically apparent from histology alone and no non-genetic biomarker currently exists that can differentiate the forms of FSGS. Therefore molecular diagnosis is likely key to understanding the underlying pathogenesis and treatment.
The prevalence of FSGS varies widely with the population and is thought to be responsible for 5–20% of ESRD [137, 138]. It was previously thought that genetic testing was only of use in a paediatric population, as the likelihood of identifying a pathogenic variant correlated inversely with age. Disease-causing variants are identified in 60–100% of those diagnosed during the first year of life, 40–60% of young children, 25–40% of older children and 10–25% of adolescents [139–142]. Hereditary forms were long thought to account for only 1% of FSGS in adults [143]. However, recent literature has challenged this, with Yao et al. [144] achieving a genetic diagnosis rate of 11% in a recent study of 193 individuals with biopsy-proven FSGS. Some of these variants will present with steroid-resistant nephrotic syndrome (SRNS), while others will have significantly less proteinuria. Of the pathogenic variants detected, 55% were in COL4A3, COL4A4 and COL4A5, 40% were in podocyte genes and 5% were in CAKUT genes. More than 40 genes have now been identified that present with both syndromic and non-syndromic forms of hereditary FSGS (Table 4) and new FSGS-causing genes continue to be discovered [145, 146]. Disease-causing variants in COL4A3, COL4A4 and COL4A5 are more classically associated with Alport syndrome but are now thought to be one of the most common causes of hereditary FSGS in adults [119, 145]. Our rapidly expanding knowledge of the genes associated with FSGS makes WES the preferred choice for those with FSGS.
Genes causing syndromic and non-syndrome FSGS
| Gene | Protein | Inheritance | Syndrome | Renal phenotype |
|---|---|---|---|---|
| Non-syndromic forms of FSGS | ||||
| Cell signalling | ||||
| PLCE1 | 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase epsilon-1 | AR | No | Steroid-resistant nephrotic syndrome |
| KANK2 | KN motif and ankyrin repeat domain-containing protein 2 | AR | No | Early onset, steroid-resistant nephrotic syndrome, haematuria |
| KANK4 | KN motif and ankyrin repeat domains 4 | AD | No | VUS, may contribute to FSGS |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 | AD | No | FSGS |
| Slit diaphragm-associated proteins | ||||
| NPHS1 | Nephrin | AR | No | Congenital nephrotic syndrome or FSGS |
| NPHS2 | Podocin | AR | No | Early-onset FSGS |
| CD2AP | CD2-associated protein | AD/AR | No | Early-onset FSGS |
| MYO1E | Myosin 1E | AR | No | Early-onset FSGS |
| MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domain-containing 2 | AR | No | Steroid-resistant congenital nephrotic syndrome |
| Nuclear pore complex proteins | ||||
| XPO5 | Exportin 5 | AR | No | Childhood-onset steroid nephrotic syndrome |
| NUP85 | Nucleoporin 85 kDa | AR | No | Childhood-onset FSGS |
| NUP93 | Nucleoporin 93 kDa | AR | No | Childhood-onset FSGS |
| NUP205 | Nucleoporin 205 kDa | AR | Childhood-onset FSGS | |
| NUP160 | Nucleoporin 160 kDa | AR | No | Steroid-resistant nephrotic syndrome in the second decade |
| Cell membrane-associated proteins | ||||
| PTPRO | Protein tyrosine phosphatase receptor-type, O | AR | No | Steroid resistant nephrotic syndrome in childhood |
| MP2 | Epithelial membrane protein 2 | AR | No | Childhood-onset steroid-resistant nephrotic syndrome |
| Mitochondrial function | ||||
| COQ8B | Coenzyme Q8B | AR | No | Steroid-resistant nephrotic syndrome |
| Cilia | ||||
| TTC21B | Tetratricopeptide repeat domain-containing protein 21B | AR | No | Nephronophthisis, adolescent-onset FSGS with tubulointerstitial lesions |
| AVIL | Advillin | AR | No | Childhood-onset FSGS |
| Cytoskeleton, cell polarity adhesion | ||||
| INF2 | Inverted foramin 2 | AD | No | FSGS, other, severe histological appearances |
| ACTN4 | Actinin-α-4 | AD | No | Congenital steroid-resistant nephrotic syndrome |
| ARHGAP24 | Rho GTPase activating protein 24 | AD | No | Adolescent-onset FSGS |
| ANLN | Actin binding protein anillin | AD | No | FSGS with variable age of onset |
| CRB2 | Crumbs cell polarity complex component 2 | AR | No | Childhood-onset FSGS, congenital steroid-resistant nephrotic syndrome |
| ARHGDIA | Rho GDP dissociation inhibitor α | AR | No | Congenital or early-onset steroid-resistant nephrotic syndrome |
| FAT1 | Fat atypical cadherin | AR | No | VUS, may contribute to steroid-resistant nephrotic syndrome |
| DNA repair, transcription | ||||
| NXF5 | Nuclear RNA export factor 5 | X-linked | No | Adult-onset nephrotic syndrome |
| Cell vesicles | ||||
| TBC1D8B | TBC1 domain family protein 8 | X-linked | No | Early-onset steroid-resistant nephrotic syndrome |
| Syndromic forms of FSGS | ||||
| Lysosome | ||||
| SCARB2 | Lysosomal integral membrane protein 2 | AR | Action myoclonus-renal failure syndrome (ataxia, myoclonus, collapsing FSGS) | FSGS |
| DNA repair, transcription | ||||
| WT1 | Wilm’s tumour 1 | AD | Denys–Drash syndrome (Wilms’ tumour, male pseudohermaphroditism, FSGS), Frasier syndrome (FSGS, gonadoblastoma, male pseudohermaphroditism isolated congenital or childhood-onset diffuse mesangial sclerosis) | Childhood-onset FSGS |
| PAX2 | Paired box 2 | AD | Renal coloboma syndrome (renal hypoplasia, childhood-onset FSGS, optic nerve colobomas) | Adult-onset FSGS |
| EYA1 | Eyes absent homolog 1 | AD | Branchio-oto-renal syndrome (abnormalities in branchial, ear and renal development) | Adult-onset FSGS |
| LMX1B | LIM homoeobox transcription factor 1β | AD | Nail–Patella syndrome (hypoplastic or absent patella, dysplasia of elbows, short stature dystrophic nails, frequently glaucoma) | FSGS |
| SMARCAL1 | SMARCA-like protein 1 | AR | Schimke immuno-osseous dysplasia (immunodeficiency, skeletal dysplasia, childhood-onset FSGS) | Childhood-onset FSGS |
| LMNA | Lamin A | AD | Familial partial lipodystrophy with adult-onset FSGS, other syndromes | Adult-onset FSGS |
| WDR73 | WD repeat-containing protein 73 | AR | Galloway–Mowat syndrome (microcephaly joint contractures and developmental delay) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cell matrix | ||||
| COL4A3 | α3 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A4 | α4 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A5 | α5 Type 4 collagen | X-linked | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| ITGB4 | Integrin-β4 | AR | Epidermolysis bullosa | Steroid-resistant nephrotic syndrome |
| LAMB2 | Laminin-β2 | AR | Pierson syndrome (microcoria, neuromuscular junction defects, early-onset FSGS or diffuse mesangial sclerosis) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cytoskeleton, cell polarity adhesion | ||||
| MYH9 | Myosin heavy chain 9 | AD | Epstein–Fechtner syndrome (FSGS, cataracts, sensorineural deafness macrothrombocytopaenia leucocyte inclusions) | Childhood-onset FSGS |
| Mitochondrial function | ||||
| MT-TL1 | mitochondrial tRNA for leucine 1 | Mitochondrial | mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) | Secondary FSGS |
| MT-TL2 | Mitochondrially encoded tRNA leucine 2 | Mitochondrial | MELAS | Secondary FSGS |
| MT-TY | Mitochondrially encoded tRNA tyrosine | Mitochondrial | Ophthalmoplaegia, dilated cardiomyopathy, FSGS | Secondary FSGS |
| COQ2 | Coenzyme Q2 polyprenyltransferase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| COQ6 | Coenzyme Q6 monooxygenase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| Cell membrane | ||||
| CUBN | Cubilin | AR | Imerslund–Gräsbeck syndrome (childhood-onset nephrotic syndrome, and megaloblastic anaemia, peripheral neuropathy) | Steroid resistant nephrotic syndrome |
| SGPL1 | Sphingosine-1-phosphate lyase-1 | AR | Adrenal insufficiency, ichthyosis, immunodeficiency and neurological defects | Steroid-resistant nephrotic syndrome |
| PODXL | Podocalyxin-like protein | AR | Microcoria, omphalocele, steroid-resistant nephrotic syndrome | Childhood and adult-onset FSGS |
| Nuclear pore complex protein | ||||
| NUP107 | Nucleoporin 107 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| NUP133 | Nucleoporin 133 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| Gene | Protein | Inheritance | Syndrome | Renal phenotype |
|---|---|---|---|---|
| Non-syndromic forms of FSGS | ||||
| Cell signalling | ||||
| PLCE1 | 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase epsilon-1 | AR | No | Steroid-resistant nephrotic syndrome |
| KANK2 | KN motif and ankyrin repeat domain-containing protein 2 | AR | No | Early onset, steroid-resistant nephrotic syndrome, haematuria |
| KANK4 | KN motif and ankyrin repeat domains 4 | AD | No | VUS, may contribute to FSGS |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 | AD | No | FSGS |
| Slit diaphragm-associated proteins | ||||
| NPHS1 | Nephrin | AR | No | Congenital nephrotic syndrome or FSGS |
| NPHS2 | Podocin | AR | No | Early-onset FSGS |
| CD2AP | CD2-associated protein | AD/AR | No | Early-onset FSGS |
| MYO1E | Myosin 1E | AR | No | Early-onset FSGS |
| MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domain-containing 2 | AR | No | Steroid-resistant congenital nephrotic syndrome |
| Nuclear pore complex proteins | ||||
| XPO5 | Exportin 5 | AR | No | Childhood-onset steroid nephrotic syndrome |
| NUP85 | Nucleoporin 85 kDa | AR | No | Childhood-onset FSGS |
| NUP93 | Nucleoporin 93 kDa | AR | No | Childhood-onset FSGS |
| NUP205 | Nucleoporin 205 kDa | AR | Childhood-onset FSGS | |
| NUP160 | Nucleoporin 160 kDa | AR | No | Steroid-resistant nephrotic syndrome in the second decade |
| Cell membrane-associated proteins | ||||
| PTPRO | Protein tyrosine phosphatase receptor-type, O | AR | No | Steroid resistant nephrotic syndrome in childhood |
| MP2 | Epithelial membrane protein 2 | AR | No | Childhood-onset steroid-resistant nephrotic syndrome |
| Mitochondrial function | ||||
| COQ8B | Coenzyme Q8B | AR | No | Steroid-resistant nephrotic syndrome |
| Cilia | ||||
| TTC21B | Tetratricopeptide repeat domain-containing protein 21B | AR | No | Nephronophthisis, adolescent-onset FSGS with tubulointerstitial lesions |
| AVIL | Advillin | AR | No | Childhood-onset FSGS |
| Cytoskeleton, cell polarity adhesion | ||||
| INF2 | Inverted foramin 2 | AD | No | FSGS, other, severe histological appearances |
| ACTN4 | Actinin-α-4 | AD | No | Congenital steroid-resistant nephrotic syndrome |
| ARHGAP24 | Rho GTPase activating protein 24 | AD | No | Adolescent-onset FSGS |
| ANLN | Actin binding protein anillin | AD | No | FSGS with variable age of onset |
| CRB2 | Crumbs cell polarity complex component 2 | AR | No | Childhood-onset FSGS, congenital steroid-resistant nephrotic syndrome |
| ARHGDIA | Rho GDP dissociation inhibitor α | AR | No | Congenital or early-onset steroid-resistant nephrotic syndrome |
| FAT1 | Fat atypical cadherin | AR | No | VUS, may contribute to steroid-resistant nephrotic syndrome |
| DNA repair, transcription | ||||
| NXF5 | Nuclear RNA export factor 5 | X-linked | No | Adult-onset nephrotic syndrome |
| Cell vesicles | ||||
| TBC1D8B | TBC1 domain family protein 8 | X-linked | No | Early-onset steroid-resistant nephrotic syndrome |
| Syndromic forms of FSGS | ||||
| Lysosome | ||||
| SCARB2 | Lysosomal integral membrane protein 2 | AR | Action myoclonus-renal failure syndrome (ataxia, myoclonus, collapsing FSGS) | FSGS |
| DNA repair, transcription | ||||
| WT1 | Wilm’s tumour 1 | AD | Denys–Drash syndrome (Wilms’ tumour, male pseudohermaphroditism, FSGS), Frasier syndrome (FSGS, gonadoblastoma, male pseudohermaphroditism isolated congenital or childhood-onset diffuse mesangial sclerosis) | Childhood-onset FSGS |
| PAX2 | Paired box 2 | AD | Renal coloboma syndrome (renal hypoplasia, childhood-onset FSGS, optic nerve colobomas) | Adult-onset FSGS |
| EYA1 | Eyes absent homolog 1 | AD | Branchio-oto-renal syndrome (abnormalities in branchial, ear and renal development) | Adult-onset FSGS |
| LMX1B | LIM homoeobox transcription factor 1β | AD | Nail–Patella syndrome (hypoplastic or absent patella, dysplasia of elbows, short stature dystrophic nails, frequently glaucoma) | FSGS |
| SMARCAL1 | SMARCA-like protein 1 | AR | Schimke immuno-osseous dysplasia (immunodeficiency, skeletal dysplasia, childhood-onset FSGS) | Childhood-onset FSGS |
| LMNA | Lamin A | AD | Familial partial lipodystrophy with adult-onset FSGS, other syndromes | Adult-onset FSGS |
| WDR73 | WD repeat-containing protein 73 | AR | Galloway–Mowat syndrome (microcephaly joint contractures and developmental delay) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cell matrix | ||||
| COL4A3 | α3 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A4 | α4 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A5 | α5 Type 4 collagen | X-linked | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| ITGB4 | Integrin-β4 | AR | Epidermolysis bullosa | Steroid-resistant nephrotic syndrome |
| LAMB2 | Laminin-β2 | AR | Pierson syndrome (microcoria, neuromuscular junction defects, early-onset FSGS or diffuse mesangial sclerosis) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cytoskeleton, cell polarity adhesion | ||||
| MYH9 | Myosin heavy chain 9 | AD | Epstein–Fechtner syndrome (FSGS, cataracts, sensorineural deafness macrothrombocytopaenia leucocyte inclusions) | Childhood-onset FSGS |
| Mitochondrial function | ||||
| MT-TL1 | mitochondrial tRNA for leucine 1 | Mitochondrial | mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) | Secondary FSGS |
| MT-TL2 | Mitochondrially encoded tRNA leucine 2 | Mitochondrial | MELAS | Secondary FSGS |
| MT-TY | Mitochondrially encoded tRNA tyrosine | Mitochondrial | Ophthalmoplaegia, dilated cardiomyopathy, FSGS | Secondary FSGS |
| COQ2 | Coenzyme Q2 polyprenyltransferase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| COQ6 | Coenzyme Q6 monooxygenase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| Cell membrane | ||||
| CUBN | Cubilin | AR | Imerslund–Gräsbeck syndrome (childhood-onset nephrotic syndrome, and megaloblastic anaemia, peripheral neuropathy) | Steroid resistant nephrotic syndrome |
| SGPL1 | Sphingosine-1-phosphate lyase-1 | AR | Adrenal insufficiency, ichthyosis, immunodeficiency and neurological defects | Steroid-resistant nephrotic syndrome |
| PODXL | Podocalyxin-like protein | AR | Microcoria, omphalocele, steroid-resistant nephrotic syndrome | Childhood and adult-onset FSGS |
| Nuclear pore complex protein | ||||
| NUP107 | Nucleoporin 107 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| NUP133 | Nucleoporin 133 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
Genes causing syndromic and non-syndrome FSGS
| Gene | Protein | Inheritance | Syndrome | Renal phenotype |
|---|---|---|---|---|
| Non-syndromic forms of FSGS | ||||
| Cell signalling | ||||
| PLCE1 | 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase epsilon-1 | AR | No | Steroid-resistant nephrotic syndrome |
| KANK2 | KN motif and ankyrin repeat domain-containing protein 2 | AR | No | Early onset, steroid-resistant nephrotic syndrome, haematuria |
| KANK4 | KN motif and ankyrin repeat domains 4 | AD | No | VUS, may contribute to FSGS |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 | AD | No | FSGS |
| Slit diaphragm-associated proteins | ||||
| NPHS1 | Nephrin | AR | No | Congenital nephrotic syndrome or FSGS |
| NPHS2 | Podocin | AR | No | Early-onset FSGS |
| CD2AP | CD2-associated protein | AD/AR | No | Early-onset FSGS |
| MYO1E | Myosin 1E | AR | No | Early-onset FSGS |
| MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domain-containing 2 | AR | No | Steroid-resistant congenital nephrotic syndrome |
| Nuclear pore complex proteins | ||||
| XPO5 | Exportin 5 | AR | No | Childhood-onset steroid nephrotic syndrome |
| NUP85 | Nucleoporin 85 kDa | AR | No | Childhood-onset FSGS |
| NUP93 | Nucleoporin 93 kDa | AR | No | Childhood-onset FSGS |
| NUP205 | Nucleoporin 205 kDa | AR | Childhood-onset FSGS | |
| NUP160 | Nucleoporin 160 kDa | AR | No | Steroid-resistant nephrotic syndrome in the second decade |
| Cell membrane-associated proteins | ||||
| PTPRO | Protein tyrosine phosphatase receptor-type, O | AR | No | Steroid resistant nephrotic syndrome in childhood |
| MP2 | Epithelial membrane protein 2 | AR | No | Childhood-onset steroid-resistant nephrotic syndrome |
| Mitochondrial function | ||||
| COQ8B | Coenzyme Q8B | AR | No | Steroid-resistant nephrotic syndrome |
| Cilia | ||||
| TTC21B | Tetratricopeptide repeat domain-containing protein 21B | AR | No | Nephronophthisis, adolescent-onset FSGS with tubulointerstitial lesions |
| AVIL | Advillin | AR | No | Childhood-onset FSGS |
| Cytoskeleton, cell polarity adhesion | ||||
| INF2 | Inverted foramin 2 | AD | No | FSGS, other, severe histological appearances |
| ACTN4 | Actinin-α-4 | AD | No | Congenital steroid-resistant nephrotic syndrome |
| ARHGAP24 | Rho GTPase activating protein 24 | AD | No | Adolescent-onset FSGS |
| ANLN | Actin binding protein anillin | AD | No | FSGS with variable age of onset |
| CRB2 | Crumbs cell polarity complex component 2 | AR | No | Childhood-onset FSGS, congenital steroid-resistant nephrotic syndrome |
| ARHGDIA | Rho GDP dissociation inhibitor α | AR | No | Congenital or early-onset steroid-resistant nephrotic syndrome |
| FAT1 | Fat atypical cadherin | AR | No | VUS, may contribute to steroid-resistant nephrotic syndrome |
| DNA repair, transcription | ||||
| NXF5 | Nuclear RNA export factor 5 | X-linked | No | Adult-onset nephrotic syndrome |
| Cell vesicles | ||||
| TBC1D8B | TBC1 domain family protein 8 | X-linked | No | Early-onset steroid-resistant nephrotic syndrome |
| Syndromic forms of FSGS | ||||
| Lysosome | ||||
| SCARB2 | Lysosomal integral membrane protein 2 | AR | Action myoclonus-renal failure syndrome (ataxia, myoclonus, collapsing FSGS) | FSGS |
| DNA repair, transcription | ||||
| WT1 | Wilm’s tumour 1 | AD | Denys–Drash syndrome (Wilms’ tumour, male pseudohermaphroditism, FSGS), Frasier syndrome (FSGS, gonadoblastoma, male pseudohermaphroditism isolated congenital or childhood-onset diffuse mesangial sclerosis) | Childhood-onset FSGS |
| PAX2 | Paired box 2 | AD | Renal coloboma syndrome (renal hypoplasia, childhood-onset FSGS, optic nerve colobomas) | Adult-onset FSGS |
| EYA1 | Eyes absent homolog 1 | AD | Branchio-oto-renal syndrome (abnormalities in branchial, ear and renal development) | Adult-onset FSGS |
| LMX1B | LIM homoeobox transcription factor 1β | AD | Nail–Patella syndrome (hypoplastic or absent patella, dysplasia of elbows, short stature dystrophic nails, frequently glaucoma) | FSGS |
| SMARCAL1 | SMARCA-like protein 1 | AR | Schimke immuno-osseous dysplasia (immunodeficiency, skeletal dysplasia, childhood-onset FSGS) | Childhood-onset FSGS |
| LMNA | Lamin A | AD | Familial partial lipodystrophy with adult-onset FSGS, other syndromes | Adult-onset FSGS |
| WDR73 | WD repeat-containing protein 73 | AR | Galloway–Mowat syndrome (microcephaly joint contractures and developmental delay) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cell matrix | ||||
| COL4A3 | α3 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A4 | α4 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A5 | α5 Type 4 collagen | X-linked | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| ITGB4 | Integrin-β4 | AR | Epidermolysis bullosa | Steroid-resistant nephrotic syndrome |
| LAMB2 | Laminin-β2 | AR | Pierson syndrome (microcoria, neuromuscular junction defects, early-onset FSGS or diffuse mesangial sclerosis) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cytoskeleton, cell polarity adhesion | ||||
| MYH9 | Myosin heavy chain 9 | AD | Epstein–Fechtner syndrome (FSGS, cataracts, sensorineural deafness macrothrombocytopaenia leucocyte inclusions) | Childhood-onset FSGS |
| Mitochondrial function | ||||
| MT-TL1 | mitochondrial tRNA for leucine 1 | Mitochondrial | mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) | Secondary FSGS |
| MT-TL2 | Mitochondrially encoded tRNA leucine 2 | Mitochondrial | MELAS | Secondary FSGS |
| MT-TY | Mitochondrially encoded tRNA tyrosine | Mitochondrial | Ophthalmoplaegia, dilated cardiomyopathy, FSGS | Secondary FSGS |
| COQ2 | Coenzyme Q2 polyprenyltransferase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| COQ6 | Coenzyme Q6 monooxygenase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| Cell membrane | ||||
| CUBN | Cubilin | AR | Imerslund–Gräsbeck syndrome (childhood-onset nephrotic syndrome, and megaloblastic anaemia, peripheral neuropathy) | Steroid resistant nephrotic syndrome |
| SGPL1 | Sphingosine-1-phosphate lyase-1 | AR | Adrenal insufficiency, ichthyosis, immunodeficiency and neurological defects | Steroid-resistant nephrotic syndrome |
| PODXL | Podocalyxin-like protein | AR | Microcoria, omphalocele, steroid-resistant nephrotic syndrome | Childhood and adult-onset FSGS |
| Nuclear pore complex protein | ||||
| NUP107 | Nucleoporin 107 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| NUP133 | Nucleoporin 133 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| Gene | Protein | Inheritance | Syndrome | Renal phenotype |
|---|---|---|---|---|
| Non-syndromic forms of FSGS | ||||
| Cell signalling | ||||
| PLCE1 | 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase epsilon-1 | AR | No | Steroid-resistant nephrotic syndrome |
| KANK2 | KN motif and ankyrin repeat domain-containing protein 2 | AR | No | Early onset, steroid-resistant nephrotic syndrome, haematuria |
| KANK4 | KN motif and ankyrin repeat domains 4 | AD | No | VUS, may contribute to FSGS |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 | AD | No | FSGS |
| Slit diaphragm-associated proteins | ||||
| NPHS1 | Nephrin | AR | No | Congenital nephrotic syndrome or FSGS |
| NPHS2 | Podocin | AR | No | Early-onset FSGS |
| CD2AP | CD2-associated protein | AD/AR | No | Early-onset FSGS |
| MYO1E | Myosin 1E | AR | No | Early-onset FSGS |
| MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domain-containing 2 | AR | No | Steroid-resistant congenital nephrotic syndrome |
| Nuclear pore complex proteins | ||||
| XPO5 | Exportin 5 | AR | No | Childhood-onset steroid nephrotic syndrome |
| NUP85 | Nucleoporin 85 kDa | AR | No | Childhood-onset FSGS |
| NUP93 | Nucleoporin 93 kDa | AR | No | Childhood-onset FSGS |
| NUP205 | Nucleoporin 205 kDa | AR | Childhood-onset FSGS | |
| NUP160 | Nucleoporin 160 kDa | AR | No | Steroid-resistant nephrotic syndrome in the second decade |
| Cell membrane-associated proteins | ||||
| PTPRO | Protein tyrosine phosphatase receptor-type, O | AR | No | Steroid resistant nephrotic syndrome in childhood |
| MP2 | Epithelial membrane protein 2 | AR | No | Childhood-onset steroid-resistant nephrotic syndrome |
| Mitochondrial function | ||||
| COQ8B | Coenzyme Q8B | AR | No | Steroid-resistant nephrotic syndrome |
| Cilia | ||||
| TTC21B | Tetratricopeptide repeat domain-containing protein 21B | AR | No | Nephronophthisis, adolescent-onset FSGS with tubulointerstitial lesions |
| AVIL | Advillin | AR | No | Childhood-onset FSGS |
| Cytoskeleton, cell polarity adhesion | ||||
| INF2 | Inverted foramin 2 | AD | No | FSGS, other, severe histological appearances |
| ACTN4 | Actinin-α-4 | AD | No | Congenital steroid-resistant nephrotic syndrome |
| ARHGAP24 | Rho GTPase activating protein 24 | AD | No | Adolescent-onset FSGS |
| ANLN | Actin binding protein anillin | AD | No | FSGS with variable age of onset |
| CRB2 | Crumbs cell polarity complex component 2 | AR | No | Childhood-onset FSGS, congenital steroid-resistant nephrotic syndrome |
| ARHGDIA | Rho GDP dissociation inhibitor α | AR | No | Congenital or early-onset steroid-resistant nephrotic syndrome |
| FAT1 | Fat atypical cadherin | AR | No | VUS, may contribute to steroid-resistant nephrotic syndrome |
| DNA repair, transcription | ||||
| NXF5 | Nuclear RNA export factor 5 | X-linked | No | Adult-onset nephrotic syndrome |
| Cell vesicles | ||||
| TBC1D8B | TBC1 domain family protein 8 | X-linked | No | Early-onset steroid-resistant nephrotic syndrome |
| Syndromic forms of FSGS | ||||
| Lysosome | ||||
| SCARB2 | Lysosomal integral membrane protein 2 | AR | Action myoclonus-renal failure syndrome (ataxia, myoclonus, collapsing FSGS) | FSGS |
| DNA repair, transcription | ||||
| WT1 | Wilm’s tumour 1 | AD | Denys–Drash syndrome (Wilms’ tumour, male pseudohermaphroditism, FSGS), Frasier syndrome (FSGS, gonadoblastoma, male pseudohermaphroditism isolated congenital or childhood-onset diffuse mesangial sclerosis) | Childhood-onset FSGS |
| PAX2 | Paired box 2 | AD | Renal coloboma syndrome (renal hypoplasia, childhood-onset FSGS, optic nerve colobomas) | Adult-onset FSGS |
| EYA1 | Eyes absent homolog 1 | AD | Branchio-oto-renal syndrome (abnormalities in branchial, ear and renal development) | Adult-onset FSGS |
| LMX1B | LIM homoeobox transcription factor 1β | AD | Nail–Patella syndrome (hypoplastic or absent patella, dysplasia of elbows, short stature dystrophic nails, frequently glaucoma) | FSGS |
| SMARCAL1 | SMARCA-like protein 1 | AR | Schimke immuno-osseous dysplasia (immunodeficiency, skeletal dysplasia, childhood-onset FSGS) | Childhood-onset FSGS |
| LMNA | Lamin A | AD | Familial partial lipodystrophy with adult-onset FSGS, other syndromes | Adult-onset FSGS |
| WDR73 | WD repeat-containing protein 73 | AR | Galloway–Mowat syndrome (microcephaly joint contractures and developmental delay) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cell matrix | ||||
| COL4A3 | α3 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A4 | α4 Type 4 collagen | AR, AD | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| COL4A5 | α5 Type 4 collagen | X-linked | ATS (deafness, renal failure, ocular abnormalities) | FSGS, ATS |
| ITGB4 | Integrin-β4 | AR | Epidermolysis bullosa | Steroid-resistant nephrotic syndrome |
| LAMB2 | Laminin-β2 | AR | Pierson syndrome (microcoria, neuromuscular junction defects, early-onset FSGS or diffuse mesangial sclerosis) | Childhood-onset steroid-resistant nephrotic syndrome |
| Cytoskeleton, cell polarity adhesion | ||||
| MYH9 | Myosin heavy chain 9 | AD | Epstein–Fechtner syndrome (FSGS, cataracts, sensorineural deafness macrothrombocytopaenia leucocyte inclusions) | Childhood-onset FSGS |
| Mitochondrial function | ||||
| MT-TL1 | mitochondrial tRNA for leucine 1 | Mitochondrial | mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) | Secondary FSGS |
| MT-TL2 | Mitochondrially encoded tRNA leucine 2 | Mitochondrial | MELAS | Secondary FSGS |
| MT-TY | Mitochondrially encoded tRNA tyrosine | Mitochondrial | Ophthalmoplaegia, dilated cardiomyopathy, FSGS | Secondary FSGS |
| COQ2 | Coenzyme Q2 polyprenyltransferase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| COQ6 | Coenzyme Q6 monooxygenase | AR | Sensorineural deafness, childhood FSGS, coenzyme Q10 deficiency | Childhood-onset FSGS |
| Cell membrane | ||||
| CUBN | Cubilin | AR | Imerslund–Gräsbeck syndrome (childhood-onset nephrotic syndrome, and megaloblastic anaemia, peripheral neuropathy) | Steroid resistant nephrotic syndrome |
| SGPL1 | Sphingosine-1-phosphate lyase-1 | AR | Adrenal insufficiency, ichthyosis, immunodeficiency and neurological defects | Steroid-resistant nephrotic syndrome |
| PODXL | Podocalyxin-like protein | AR | Microcoria, omphalocele, steroid-resistant nephrotic syndrome | Childhood and adult-onset FSGS |
| Nuclear pore complex protein | ||||
| NUP107 | Nucleoporin 107 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
| NUP133 | Nucleoporin 133 kDa | AR | Galloway–Mowat syndrome (developmental delay, microcephaly and early onset FSGS) | Childhood-onset FSGS |
Inheritance is variable and can be autosomal recessive, autosomal dominant or X-linked. The genes effected often produce proteins that localize to the podocyte and its slit diaphragm, such as nephrin (NPHS1), podocin (NPHS2), inverted formin 2 (INF2), α-actinin-4 (ACTN4), CD2-associated protein (CD2AP) and laminin-beta2 (LAMB2) [147–151].
The syndromic forms of inherited FSGS tend to encode proteins that are expressed not only in the podocyte, but in other tissue types. They include WT1, LAMB2, ITGB4 and LMX1B [152]. Major syndromes associated with hereditary FSGS include Denys–Drash syndrome (WT1), nail–patella syndrome (LXM1B) and Pierson syndrome (LAMB2). Certain variants in genes thought to be syndromic may also cause only isolated renal disease—or perhaps cause extrarenal features so subtle that the clinical syndrome is not detectable—such as has been seen in patients with LXM1B variants that cause autosomal dominant FSGS without extrarenal manifestations [153]. Genetic variants may also cause an increased susceptibility to FSGS but may not present unless a second, environmental ‘hit’ occurs, as is the case with APOL1 risk variants. Determining the significance of detecting rare variants can also be challenging. A recent study reported a 5.5% rate of rare, disease-causing variants associated with FSGS in an asymptomatic control population [154].
Identification of the cause of FSGS may have a significant impact on diagnosis and management. The majority of monogenic forms of FSGS do not respond to corticosteroids and have a very low risk of recurrence. Identifying an underlying genetic cause may avoid unnecessary exposure to toxic steroid-containing treatment regimens. There is also increased interest in therapies including ciclosporin, coenzyme Q10 and vitamin B12 to treat certain forms of hereditary FSGS [155, 156]. While previously genetic testing was only recommended for the paediatric population, it has now been suggested that genetic testing be considered in any case where the cause of FSGS cannot be classified by clinicopathological assessment [156].
APOL1
APOL1 is a gene that codes for apolipoprotein L1. Variants in these genes have been identified as initiating factors in the increased risk for hypertension and kidney disease found in those of sub-Saharan African ancestry. The discovery of APOL1 has been instrumental in understanding the 4-fold risk of CKD in the population with African ancestry [157].
There are two known risk alleles in APOL1, G1 and G2, both of which are only known to be present in those of African ancestry [158]. Much as the sickle cell trait is protective of malaria, a single copy of an APOL1 risk allele is thought to be protective of African sleeping sickness, a parasitic infection transmitted by Trypanosoma brucei rhodesiense that is endemic in sub-Saharan Africa [159]. However, the presence of two risk alleles (G1/G1, G1/G2 or G2/G2) leads to an increased risk of FSGS and of human immunodeficiency virus–associated nephropathy (HIVAN).
APOL1 nephropathy may present with a number of histopathological presentations, including tubulointerstitial injury and arteriosclerosis, FSGS, HIVAN, lupus nephritis, solidified glomerulosclerosis and sickle cell nephritis [158, 160, 161]. Individuals are often labelled as having hypertensive or non-specific forms of CKD, leading to disease misclassification [162].
In a study of 196 non-diabetic, non-nephrotic, African American patients with progressive CKD, characterized by chronic tubulointerstitial injury and arteriosclerosis, those with two APOL1 risk variants tended to be younger and display a greater degree of interstitial fibrosis and microcystic tubular dilatation. They displayed solidified fibrosis and thyroidization-type tubular atrophy [160]. In a study of 138 children and young adults with FSGS, those with two APOL1 risk variants had more segmental glomerulosclerosis, more total glomerulosclerosis and more tubular atrophy and interstitial fibrosis. They were more likely to develop the collapsing variant of FSGS [163].
The lifetime risk of FSGS in patients of African ancestry is thought to be 0.2, 0.3 and 4.25% in those with none, one or two APOL1 risk alleles, respectively [157]. In those who developed FSGS, progression to ESRD was also faster, with a hazard ratio of 2.3. Those who develop APOL1-related FSGS seem to have similar sensitivity to corticosteroids as the general population, with ∼30% responding to steroids [157].
There is little prospective data on the outcomes of recipients or donors with two APOL1 risk alleles. Survival of deceased donor renal allografts with two APOL1 risk alleles was significantly shorter than those without [164]. Transplantation of a kidney from a healthy living donor with two APOL1 risk alleles has been associated with an increased rate of decline in GFR, with ESRD occurring 10 years post-donation, however, further research into this area is required [165].
FABRY DISEASE
Fabry disease is an X-linked, multisystem disorder. A disorder of glycosphingolipid metabolism, it is caused by disease-causing variants in the GLA gene on chromosome X [166]. This leads to a deficiency in lysosomal α-galactosidase A, which causes an accumulation of globotriaosylceramide (GL-3) within cell lysosomes [167].
Accumulation of GL-3 affects the heart, causing arrhythmia and cardiomyopathy; the peripheral nervous system, causing pain and burning; the skin, causing angiokeratomas; and the kidney, causing proteinuria and nephropathy [168]. It affects males more severely, but may also cause significant symptoms in heterozygous females. It is often not detected before adulthood.
Nephropathy in Fabry disease is because of the accumulation of GL-3 in the podocytes, which leads to their destruction [169]. Podocytes have a limited ability to regenerate, leading to segmental or global glomerulosclerosis and to CKD [170]. The characteristic pathological features are myelin-like inclusions in the podocyte cytoplasm that are seen on EM [171]. These inclusions may also be seen less prominently in glomerular endothelial cells, tubular epithelial cells and mesangial cells [172, 173]. LM shows vacuolization of the podocyte cytoplasm and glomerular sclerosis [171]. IF is unremarkable, although Fabry disease has been reported in conjunction with IgAN and pauci-immune glomerulonephritis [174, 175].
An enzymatic blood test of α-galactosidase A activity can be used to confirm the diagnosis and genomic testing can be used to identify the culprit variant [176]. Enzyme replacement therapy with recombinant galactosidase can stabilize disease, with enzyme therapy ameliorating the decrease in GFR but not entirely halting it [177]. Even with enzyme therapy, nephropathy often progresses over time to ESRD.
IgAN
IgAN is the most common form of glomerulonephritis in the world [178]. It is characterized by haematuria, proteinuria and considerable heterogenicity in the progression of CKD. Its histological appearance is characterized by a variable appearance on LM and IgA-dominant deposits on IF [179].
The causes of IgAN are poorly defined and it is generally not considered to be hereditary. However, there have been multiple reports of families presenting with biopsy-proven IgAN and a Mendelian inheritance, although penetrance is often incomplete [180]. There is also evidence of geographic and ethnic clustering, with the prevalence of IgAN being high within Asian and Native American ethnic groups and low in those with African ancestry [181]. An Italian study examined 54 unaffected family members of patients with IgAN and found microscopic haematuria present in 24%. The same group found a 16-fold increased risk of IgAN in first-degree relatives of those with IgAN in a cohort of northern Italians [182]. However, even among families who display familial clustering, there is significant heterogenicity, with variable penetrance and no specific pattern of inheritance [183].
The genetic locus of IgAN remains elusive, made more complex by the heterogenicity of the disease. More than 130 candidate gene association studies for IgAN have been conducted without success [180]. GWASs in European, Chinese and Japanese cohorts have shown associations at the 6p21 human leucocyte antigen and 22q12, but a definitive risk allele or a monogenic form of IgAN has not yet been discovered [184]. Most susceptibility loci that have been identified via GWAS encode genes involved in the mucosal pathogen response, suggesting the intestinal immune network may be involved in the pathogenesis of IgAN [185]. This supports the notion that IgAN occurs due to a complex interplay of genetic and environmental factors.
Other hereditary forms of kidney disease, such as Alport syndrome may masquerade as hereditary IgAN [186, 187].
GENETIC TESTING IN RENAL TRANSPLANTATION
Genetic testing offers many benefits to individuals opting to undergo it. It may provide reassurance, have diagnostic and therapeutic benefits and allow for genetic counselling and family planning. These benefits are valuable but not unique to patients with renal disease, and they apply across the spectrum of organ systems and diseases. However, there is one potential benefit of genetic testing that is peculiar to renal disease, and this lies in the field of renal transplantation.
Allogenic renal transplantation remains the gold standard for management of ESRD, conveying increased survival and an improved quality of life compared with dialysis as well as improved cost-effectiveness [188–191]. Long-term mortality in patients who have undergone transplant is 50% lower than in those still on dialysis.
Organs are a vital and scarce resource and living donor transplants are an essential part of a successful renal transplant programme. They offer a significant number of benefits to the transplant recipient. Individuals who receive a living transplant enjoy a lower risk of rejection, better allograft function and longer life than those who receive a deceased donor transplant [192]. They enjoy less fatigue and better quality of life [193]. They spend less time on dialysis than those awaiting deceased donor transplants and avoid the attendant risks [194].
Donor nephrectomy is not without risk and potential donors should undergo rigorous screening prior to donation to ensure that no harm comes to the donor. The worst-case scenario is for a donor to go on to develop significant CKD or even ESRD themselves following donation. For many years it was thought that donation was a benign procedure, as the rate of decline in GFR in donors was comparable to that of the general population [195]. However, because of the intense screening they undergo, donors are a rarefied patient group, and when compared with a group of well-selected, non-donor controls, the risk of ESRD is 8–11 times higher [196, 197].
Many living donors are first or second-degree relatives of the kidney recipient. It is known that those with a first- or second-degree relative are at higher risk of developing ESRD even in the absence of known Mendelian disorders [198, 199].
Approximately 15% of those who reach ESRD currently do so without a primary renal diagnosis. If a genetic cause of disease can be identified in any of these cases, it will not only give the patient a diagnosis, it will allow for potential testing of any living donors and appropriate counselling as to the risks of living donation.
This is also true in those who may have a higher lifetime risk of developing kidney disease due to genetic factors, for instance, those who have two APOL1 risk alleles. Renal donors with two risk alleles are at much higher risk for progressing to ESRD than those without [165]. A study of African American kidney donors says that 96% of donors would want transplant centres to offer routine APOL1 testing to donors, but even in the event they had two risk alleles, 61% would still wish to donate [200]. A survey of 383 American nephrologists and transplant surgeons report that only 4% routinely send APOL1 testing, but 87% thought it would help donors make an informed decision and 67% would begin sending the test in the next year if available [201]. Identification of at-risk individuals would help in choosing appropriate candidates and stratifying risk.
INDICATIONS FOR GENOMIC ASSESSMENT
Genetic testing is now increasingly available in the clinical setting and availability is only likely to increase. Therefore it is important that clinicians know how and when to apply it as a diagnostic modality.
When assessing a patient for a possible inherited cause of disease, a rigorous baseline assessment is essential. Genetic testing is rarely successful without a detailed and complete phenotype. Therefore initial assessment including a detailed history, pedigree and examination, as well as first-line tests including biochemistry and radiology are essential. A review of previous pathology, if available, is useful.
Genetic testing is recommended in all children with SRNS and in any child where the clinical phenotype does not give a clear diagnosis [202, 203]. In adults, the role of genetic testing is less clear. It is currently recommended in patients who have a known or suspected hereditary form of nephropathy; in those who have a phenotype with a strong hereditary basis, such as tubulointerstitial kidney disease or CAKUT; in those with extrarenal features compatible with a syndromic form; in potential living renal donors and in those with nephropathy of uncertain aetiology.
FUTURE DIRECTIONS
Percutaneous renal biopsy continues to be extremely relevant to our understanding of the kidney, both in individuals and across populations. Ongoing clinical studies are collecting renal biopsy tissue samples for the purposes of studying acute kidney injury and CKD and to help identify novel biomarkers. Single-cell RNA sequencing has been used on biopsy samples to investigate lupus nephritis and investigate early kidney development [204, 205]. Further integration of genomics, transcriptomics, proteomics and biopsy will enhance precision medicine and provide a more targeted approach to treating patients.
This review was written in collaboration with NDT Educational.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the kind support of Martina Zivna and Veronika Baresova, who provided images for this article.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
REFERENCES


{kind=link}
{kind=link}
Comments