





Abstract
Renal tubulointerstitial lesions (TILs), a key pathological hallmark for chronic kidney disease to progress to end-stage renal disease, feature renal tubular atrophy, interstitial mononuclear leukocyte infiltration and fibrosis in the kidney. Our study tested the renoprotective and therapeutic effects of compound K (CK), as described in our US patent (US7932057B2), on renal TILs using a mouse unilateral ureteral obstruction (UUO) model.
Renal pathology was performed and renal draining lymph nodes were subjected to flow cytometry analysis. Mechanism-based experiments included the analysis of mitochondrial dysfunction, a model of tubular epithelial cells (TECs) under mechanically induced constant pressure (MICP) and tandem mass tags (TMT)-based proteomics analysis.
Administration of CK ameliorated renal TILs by reducing urine levels of proinflammatory cytokines, and preventing mononuclear leukocyte infiltration and fibrosis in the kidney. The beneficial effects clearly correlated with its inhibition of: (i) NF-κB-associated priming and the mitochondria-associated activating signals of the NLRP3 inflammasome; (ii) STAT3 signalling, which in part prevents NLRP3 inflammasome activation; and (iii) the TGF-β-dependent Smad2/Smad3 fibrotic pathway, in renal tissues, renal TECs under MICP and/or activated macrophages, the latter as a major inflammatory player contributing to renal TILs. Meanwhile, TMT-based proteomics analysis revealed downregulated renal NLRP3 inflammasome activation-associated signalling pathways in CK-treated UUO mice.
The present study, for the first time, presents the potent renoprotective and therapeutic effects of CK on renal TILs by targeting the NLRP3 inflammasome and STAT3 signalling.
INTRODUCTION
Renal tubulointerstitial lesions (TILs) are a key pathological hallmark for chronic kidney disease (CKD) to progress to end-stage renal disease (ESRD) and uraemia [1–4]. We and others [5–9] have reported that NLRP3 inflammasome-associated molecular mechanisms are involved in the development of renal inflammation and fibrosis.
Compound K (CK) is a major absorbable intestinal bacterial metabolite of ginsenosides, which are the major bioactive components from ginseng [10, 11]. CK inhibits proinflammatory cytokine production via downregulation of the NF-κB signalling pathway [12, 13], and has been shown to inhibit ER stress and NLRP3 inflammasomes in adipose tissue [14]. Whether CK has therapeutic effects on renal inflammation and progression to diffuse fibrosis in the kidney, the latter as a late stage of TILs, remains unknown.
The present study demonstrates that CK markedly inhibits the activation of NLRP3 inflammasomes and mononuclear leukocyte infiltration, and negatively regulates STAT3 signalling in renal tissues, particularly renal tubular epithelial cells (TECs), under mechanically induced constant pressure (MICP) and/or activated macrophages. In agreement with these findings, tandem mass tags (TMT)-based proteomics analysis showed that downregulated renal NLRP3 inflammasome activation-associated signalling pathways were seen in CK-treated unilateral ureteral obstruction (UUO) mice. Our results suggest CK as a potential drug candidate for the treatment of renal TILs and the NLRP3 inflammasome as a therapeutic target for CKD.
MATERIALS AND METHODS
Preparation of CK
The pure compound 20-O-[β-d-glucopyranosyl]-20[S]-protopanaxa-diol was prepared as described in our US patent (US7932057B2) for the generation of CK [15].
Animal experiments
Male 8-week-old C57BL/6 mice were used for the induction of UUO [5] and ischaemia reperfusion (I/R) injury with unilateral nephrectomy (NX) (NX + I/R) [16], as described previously. Sham control mice underwent an identical procedure without ureteric ligation for the UUO model or without clamping of renal vessels for the NX + I/R model. Briefly, CK (30 mg/kg body weight) was administered intraperitoneally 1 day before (preventive group) or 3 days after (therapeutic group) the induction of UUO, or 1 day after the ligation of renal vessels in the NX + I/R model. Diseased mice that received only 3% cremophor (Sigma-Aldrich, St Louis, MO, USA) daily via intraperitoneal injections were used as the vehicle control group. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the National Defense Medical Center, Taiwan, in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Urine collection and pathological evaluation
Because of the ligation of only a single ureter in the UUO mice, urine samples were collected from the dilated pelvises of the obstructed (left) sides of the kidneys using needles and syringes, as described previously [5]. Urine samples from sham control mice were collected in metabolic cages, as described previously [6–8]. For the assessment of renal function in NX + I/R mice, serum levels of BUN and Cr were determined as described previously [6]. Renal tissues were processed for light microscopy as described previously [5, 17].
Isolation of lymphoid cells from renal draining lymph nodes
Renal draining lymph nodes were dissected from the same side as the ligated kidneys (left) of UUO mice or the sham control mice as described previously [5], and lymphoid cells were harvested for flow cytometry analysis.
In vitro experiments
An MICP on renal TECs (M-1) (Bioresource Collection and Research Center, Hsinchu, Taiwan) was adopted to simulate the microenvironments inside the renal pelvises of the diseased kidneys of UUO mice, as described previously [5]. A murine cell line of macrophages (J774A.1) was purchased from the American Type Culture Collection.
Immunohistochemistry
Formalin-fixed and paraffin-embedded renal tissue sections for immunohistochemistry (IHC) were incubated with collagen-III (Southern Biotech, Birmingham, AL, USA), p-NF-κB p65 (Abcam, Cambridge, UK), F4/80 (monocytes/macrophages; Bio-Rad, Hercules, CA, USA) or CD3 (DAKO, Carpentaria, CA, USA) antibodies, as described previously [6–8].
Total soluble collagen analysis
The concentration of total soluble collagen in renal tissues was measured using a Sircol collagen assay kit (Biocolor Ltd, Antrim, UK) according to the manufacturer’s instructions.
ELISA and transcription factor activity assay
Levels of IL-1β, TNF-α, MCP-1 and IL-6 in the urine or supernatants of cultured cells, and IL-17A and TGF-β in renal tissues were determined using commercial ELISA kits (R&D Systems, Minneapolis, MN, USA). Caspase-1 activity (R&D Systems) was determined in cytosolic proteins extracted from renal tissues, and NF-κB p65 activity (Active Motif, Shinjuku-Ku, Tokyo, Japan) in nuclear proteins extracted from renal tissues or cultured cells was measured using an ELISA-based transcription factor activity assay according to the manufacturer’s instructions.
Flow cytometry analysis and mitochondrial dysfunction
Mitochondrial membrane potential was detected using MitoTracker Deep Red and MitoTracker Green (Thermo Fisher Scientific, Waltham, MA, USA), and mitochondrial ROS production was assayed using MitoSOX (Invitrogen, Waltham, MA, USA) using a FACSCalibur as described previously [18, 19].
Reporter assay for NF-κB and STAT3
J774A.1 macrophages stably expressing a secreted embryonic alkaline phosphatase (SEAP) reporter gene that could be induced by the activation of NF-κB (J-Blue) were established and the SEAP activity was determined using QUANTI-BlueTM reagent (InvivoGen) according to the manufacturer’s instructions. For the STAT3 luciferase reporter assay, J774A.1 macrophages were subjected to infection with lentiviral particles expressing the STAT3 reporter gene according to the manufacturer’s instructions (QIAGEN, Hilden, Germany).
Western blot analysis
Antibodies against NLRP3, caspase-1 (AdipoGen, San Diego, CA, USA), IL-1β (R&D Systems), p-STAT3, p-Smad2 or p-Smad3 (Cell Signalling Technology, Danvers, MA, USA) were used.
Real-time PCR assay
RNA of tissues or cells was extracted using REzol (Protech Technology, Taipei, Taiwan) and cDNA was prepared as described previously [6]. The measurement of mitochondrial DNA or extraction of cytosolic DNA was performed as described previously [19].
Statistical analysis
Data are presented as the means±SEM. Data obtained from in vitro and in vivo experiments were analysed using one-way ANOVA and a subsequent Scheffe’s test. A P < 0.05 was considered statistically significant for each of the experiments.
RESULTS
Downregulated renal NLRP3 inflammasome activation-associated signalling pathways with TMT-based proteomics analysis in UUO mice
In order to systematically understand the effects of CK on the UUO mice, TMT-based proteomics analysis was conducted; in total, 4758 proteins were identified in renal tissues of sham control mice, UUO mice treated with vehicle (Vehicle + UUO mice; disease control group) and UUO mice treated with CK (CK + UUO mice) at Day 14. Among the proteins, the TMT ratio values were calculated based on reporter ion intensities of 126 N and 128 N (sham control mice), 127 C and 128 C (Vehicle + UUO mice), and 127 N and 130 N (CK + UUO mice). Replicate samples for each group and no analytical replicates were included in the TMT-based quantitative proteomics analysis (Supplementary data, Table S1). Next, GSEA was used to evaluate the biologically enriched pathways/networks in the comparisons of CK + UUO mice versus Vehicle + UUO mice. The data suggested that signalling pathways associated with NLRP3 inflammasome activation correlated with the mechanisms of action of the potential renoprotective effects of CK in the treated UUO mice. Twenty-nine of the 186 Kyoto Encyclopedia of Genes and Genomes gene sets were significantly enriched, and CK administration resulted in the downregulation of 26 and the upregulation of 3 signalling pathways (Supplementary data, Table S2). Five of these 29 pathways, which positively regulate the NLRP3 inflammasome, including lysosome [20, 21], oxidative phosphorylation [22, 23], fatty acid metabolism [24, 25], sphingolipid metabolism [26] and the renin–angiotensin system [27, 28], were downregulated in renal cortical tissues from CK-treated UUO mice. Additionally, CK upregulated a ribosome-mediated pathway, which has been shown to negatively regulate the NLRP3 inflammasome [29].
CK prevents renal TILs in UUO mice
First, a preventive treatment with CK was performed. As shown in Figure 1A and B, Vehicle + UUO mice exhibited renal pathological changes mimicking TILs on both Days 7 and 14, but this effect was greatly inhibited in CK + UUO mice at both time points. Consistent with this, the level of neutrophil gelatinase-associated lipocalin (NGAL), a marker of renal TILs, was significantly lowered in both renal tissues and urine samples collected from the renal pelvic cavity of CK + UUO mice compared with those of Vehicle + UUO mice (Supplementary data, Figure S1A–C). Furthermore, although Vehicle + UUO mice exhibited increased renal infiltration of macrophages (F4/80) and T cells (CD3) at Days 7 and 14, compared with sham control mice, this effect was significantly inhibited in CK + UUO mice (Figure 1C–F).
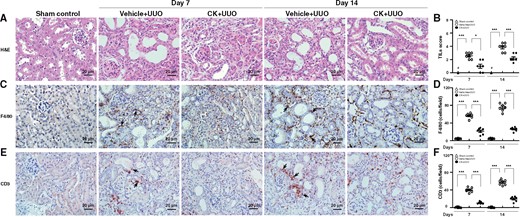
CK prevented renal TILs and mononuclear lymphocyte infiltration. Renal histopathological evaluation using (A) light microscopy, showing dilatation or atrophy of renal tubules, renal interstitial mononuclear leukocyte infiltration and fibrosis, and ischaemic collapse of the glomerular tufts with dilated Bowman’s space at Day 7 compared with sham control mice. These histopathological changes became relatively diffuse in distribution and more intense in severity in untreated mice at Day 14. (B) Scoring of TILs. IHC for (C and D) macrophages (F4/80) and (E and F) pan T cells (CD3). The quantitative results are reported as the number of (D) macrophages and (F) pan T cells in the renal tubulointerstitial compartment. Arrows indicate positive staining. H&E, hematoxylin and eosin. Original magnification was 400×. n = 7 mice per group. #Not detectable. *P < 0.05, ***P < 0.005.
UUO mice indeed displayed diffuse fibrosis, revealed by Masson’s trichrome staining of kidney sections (Figure 2A and B), and this effect was also markedly suppressed in UUO mice treated with CK. Consistent with these findings, increased protein expression and mRNA levels of collagen-III (Col-III) were detected by IHC and real-time PCR, respectively, in the Vehicle + UUO mice compared with those of sham control mice at Days 7 and 14, but these changes were inhibited in CK + UUO mice (Figure 2C–E). Although the amounts of total renal soluble collagen were increased in renal tissues of Vehicle + UUO mice compared with sham control mice, these changes were suppressed in CK + UUO mice at Days 7 and 14 (Figure 2F).
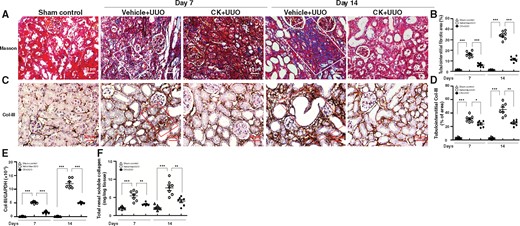
CK reduced renal fibrosis-related gene/protein expression. (A) Masson's trichrome. (B) Scoring of tubulointerstitial fibrotic area. White solid frame indicates positively stained renal tubulointerstitial fibrotic area. IHC for (C and D) Col-III. (D) Quantitative results reported as the positive area of Col-III in the tubulointerstitial compartment. Red solid frame indicates positively stained area. Original magnification was 400×. (E) Real-time PCR of renal mRNA levels of Col-III. (F) Total renal soluble collagen contents determined by ELISA. n = 7 mice per group. *P < 0.05, **P < 0.01, ***P < 0.005.
CK reduces the levels of proinflammatory cytokines in renal tissues and urine collected from the dilated renal pelvis
CK administration significantly decreased urine levels of IL-1β, TNF-α, MCP-1 and IL-6 in CK + UUO mice compared with Vehicle + UUO mice at Day 14 (Figure 3A–D). Similarly, the analysis of renal mRNA expression of IL-1β, TNF-α, MCP-1 or IL-6 revealed that CK suppressed the production of these proinflammatory cytokines within the diseased kidney (Figure 3E–H).

CK reduced proinflammatory cytokine levels in urine and renal tissues. ELISA of urine protein levels of (A) IL-1β, (B) TNF-α, (C) MCP-1 and (D) IL-6 collected from the dilated pelvis. Real-time PCR of renal mRNA levels of (E) IL-1β, (F) TNF-α, (G) MCP-1 and (H) IL-6. n = 7 mice per group. ns, no significant difference. *P < 0.05, **P < 0.01, ***P < 0.005.
CK inhibits the activation of T cells and the differentiation into Th17 cells in renal draining lymph nodes
Significantly increased percentages of activated CD4+ CD69+ T cells were observed in Vehicle + UUO mice compared with those of sham control mice at Days 7 and 14, while the activation of T cells was inhibited in CK + UUO mice at Day 14 (Figure 4A). A similar profile was noted in the percentages of activated CD8+CD69+ T cells of the renal draining lymph nodes in Vehicle + UUO and CK + UUO mice (Figure 4B) at Day 14. In addition, consistent with a pathogenic role of Th17 cells in TILs of the UUO model [5], we observed greatly increased mRNA levels of IL-17A and ROR-γ in the renal draining lymph nodes of Vehicle + UUO mice at Days 7 and 14, while these increases were hardly seen in CK + UUO mice (Figure 4C and D).
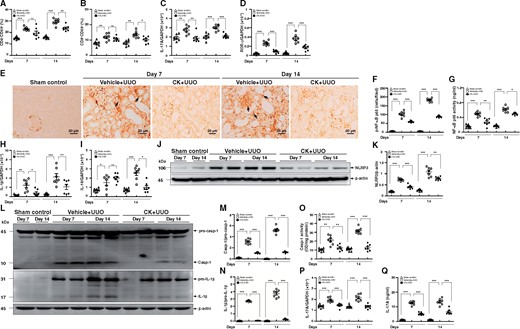
CK inhibited T cell activation in renal draining lymph nodes and NF-κB/NLRP3 inflammasome activation in renal tissues. Percentages of (A) CD4+CD69+ and (B) CD8+CD69+ cells determined by flow cytometry analysis, and real-time PCR of mRNA levels of (C) IL-17A and (D) ROR-γ in renal draining lymph nodes. IHC for (E and F) p-NF-κB p65. (F) Quantitative data as the number of p-NF-κB p65 cells in the tubulointerstitial compartment. Arrows indicate positive staining. Original magnification was 400×. (G) NF-κB p65 activity determined by an ELISA-based transcription factor activity assay. Real-time PCR of renal mRNA levels of (H) IL-1β and (I) IL-18. Western blot analysis for (J) NLRP3 and (K) semi-quantitative analysis, (L) caspase-1 and IL-1β, and semi-quantitative analysis of (M) caspase-1 and (N) IL-1β. (O) Caspase-1 activity determined by an enzyme activity assay. (P) Real-time PCR of renal mRNA levels of IL-17A. (Q) ELISA of renal protein levels of IL-17A. n = 7 mice per group. ns, no significant difference. *P < 0.05, **P < 0.01, ***P < 0.005.
CK inhibits the activation of renal NF-κB/the NLRP3 inflammasome
As shown in Figure 4E and F, greatly increased numbers of renal TECs and mononuclear leukocytes, and occasionally glomerular intrinsic cells, were positive for p-NF-κB p65 in Vehicle + UUO mice as demonstrated by IHC, but these changes were markedly inhibited in CK + UUO mice at Days 7 and 14. Consistent with this, CK + UUO mice exhibited significantly lowered NF-κB p65 activity in renal cortical tissues compared with Vehicle + UUO mice at Days 7 and 14 (Figure 4G). Moreover, the assessment of the activation of the NLRP3 inflammasome in renal tissues revealed significantly reduced renal mRNA levels of IL-1β (Figure 4H) and IL-18 (Figure 4I) in CK + UUO mice at both Days 7 and/or 14 compared with Vehicle + UUO mice. Furthermore, Vehicle + UUO mice exhibited increased expression levels of renal NLRP3 compared with sham control mice, whereas these increases were greatly inhibited in CK + UUO mice (Figure 4J and K). In parallel, CK + UUO mice showed markedly decreased protein levels of active caspase-1 and IL-1β, compared with those of Vehicle + UUO mice at Days 7 and 14, as demonstrated by western blot analysis (Figure 4L–N). As expected, the activity of caspase-1 in CK + UUO mice was markedly reduced, at levels comparable to those of the sham control mice (Figure 4O). In addition, renal mRNA (Figure 4P) and protein (Figure 4Q) levels of IL-17A were significantly reduced in CK + UUO mice compared with Vehicle + UUO mice.
CK inhibits the activation of the NLRP3 inflammasome in cultured renal TECs
As shown in Figure 5A, the activation levels of NF-κB p65 in the nuclei of renal TECs under MICP treated with saline only were increased compared with those of the normal controls, but these increases was greatly blocked in CK-treated renal TECs under MICP. Also, compared with normal controls, the renal TECs under MICP exhibited significantly increased mRNA levels of NLRP3 (Figure 5B), caspase-1 (Figure 5C) and IL-1β (Figure 5D), and protein levels of pro-IL-1β and NLRP3 (Figure 5E), suggesting that MICP serves as a primary signal of the NLRP3 inflammasome in renal TECs. However, these effects were nearly completely inhibited by CK treatment (Figure 5A–E). In addition, we observed that ATP-induced increases in caspase-1 activity, as well as protein levels of active caspase-1 and mature IL-1β in renal TECs under MICP, were again suppressed by CK treatment (Figure 5F and G). Together, these results indicate that CK inhibits the NLRP3 inflammasome by reducing both priming and activation signals of the NLRP3 inflammasome in renal TECs.
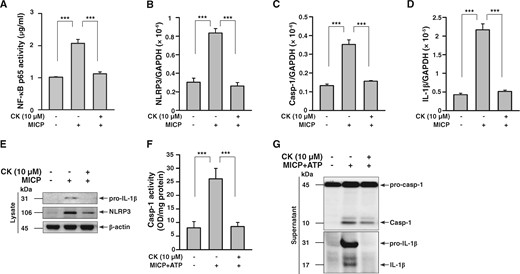
CK inhibited NLRP3 inflammasome activation in cultured renal TECs under MICP. To validate the effect of CK on NLRP3 inflammasome activation in renal TECs, an MICP system that mimics stimulation by a constant, high pressure within the renal pelvis as a result of obstruction of the ureter in UUO mice was employed. Renal TECs (murine renal tubular epithelial cell line M-1) were incubated for 30 min with or without CK, and were cultured in an MICP system at 60 mmHg for 24 h and/or with or without 5 mM ATP (30 min). (A) NF-κB p65 activity by an ELISA-based transcription factor activity assay. Real-time PCR of mRNA levels of (B) NLRP3, (C) caspase-1 and (D) IL-1β. Western blot analysis for (E) pro-IL-1β and NLRP3. (F) Caspase-1 activity determined by an enzyme activity assay. Western blot analysis for (G) caspase-1 and IL-1β. The measurement of IL-1β and caspase-1 released into the culture medium was performed on extracted supernatants. The data represent three separate experiments. ***P < 0.005.
CK inhibits the activation of the NLRP3 inflammasome by reducing mitochondrial damage in cultured macrophages
As shown in Figure 6, CK treatment significantly reduces IL-1β secretion induced by NLRP3 inflammasome activators, including ATP, alum crystals and monosodium urate, in macrophages (Figure 6A–C). The inhibitory effect of CK on the NLRP3 inflammasome was confirmed by measuring the expression levels of IL-1β and active caspase-1 in the supernatants and cell lysates by western blot analysis (Figure 6D and E). Meanwhile, CK reduced NLRP3 expression in LPS-activated macrophages and these effects were associated with reduced NF-κB activation by CK treatment (Figure 6F and G). Moreover, while CK significantly reduced LPS- and ATP-mediated IL-1β secretion, and caspase-1 activation, in J774A.1 macrophages (Figure 6H), this effect was markedly abolished by MCC950, an NLRP3 inhibitor (Figure 6H). These results suggest that CK inhibits the NLRP3 inflammasome, through which this compound exerts its beneficial effects on UUO mice. In addition, CK significantly decreased the mitochondrial damage, mitochondrial ROS production, and mitochondrial DNA release in LPS- and ATP-activated macrophages (Figure 6I–K). These results indicate that CK inhibits the activation of the NLRP3 inflammasome in macrophages by reducing NF-κB-associated priming and mitochondria-associated activation signals of the inflammasome.

CK inhibited NLRP3 inflammasome activation and mitochondrial damage in cultured macrophages. Murine J774A.1 macrophages were incubated for 30 min with or without CK, then for 5.5 h with or without 1 µg/mL of LPS, and with or without (A) 5 mM ATP (30 min), (B) 500 μg/mL of alum (24 h) or (C) 100 μg/mL of MSU (24 h) for IL-1β levels in culture media measured using an ELISA. The cells were incubated for 30 min with or without CK, followed by incubation for 6 h with or without 1 µg/mL of LPS, and then with or without 5 mM ATP for (D) IL-1β, (E) caspase-1 and (F) NLRP3 determined by western blot analysis. The analysis of IL-1β and caspase-1 released into the culture medium was performed on the extracted supernatants. J-Blue cells were incubated for 30 min with or without CK, and 24 h with or without 1 µg/mL of LPS for (G) the activation levels of NF-κB determined by an NF-κB reporter assay. (H) Use of MCC950 (1 µM), an NLRP3 inhibitor, for IL-1β and caspase-1 by ELISA or western blot analysis. Macrophages were incubated for 30 min with or without CK, 5.5 h with or without 1 µg/mL of LPS, 15 min with or without 5 mM ATP, and 15 min with 25 nM MitoTracker Deep Red and MitoTracker Green for (I) the mitochondrial inner transmembrane potential by flow cytometry. Macrophages were incubated for 30 min with or without CK, 5.5 h with or without 1 µg/mL of LPS, 15 min 5 μM MitoSOX, and 10 min with or without 5 mM ATP for (J) mitochondrial ROS generation in the cells by flow cytometry. The cells were incubated for 30 min with or without CK or Mito-TEMPO, 5.5 h with or without 1 µg/mL of LPS, and 30 min with or without 5 mM ATP for (K) mtDNA copy number by real-time PCR. The data represent three separate experiments. Alum, alum crystals; MCC950, N-(1, 2, 3, 5, 6, 7-Hexahydro-s-indacen-4-ylcarbamoyl)-4-(2-hydroxy-2-propanyl)-2-furansulfonamide, an NLRP3 inhibitor; MFI, mean fluorescence intensity; MSU, monosodium urate; mtDNA, mitochondrial DNA; Mito-TEMPO, mitochondria-targeted antioxidant; NAC, N-acetyl-L-cysteine. #Not detectable. *P < 0.05, **P < 0.01, ***P < 0.005.
CK inhibits STAT3 activation in renal tissues and cultured macrophages
STAT3 signalling has also been shown to play a key role in renal inflammatory processes that involving NF-κB [30–32] or the NLRP3 inflammasome [32, 33]. As shown in Figure 7A and B, significantly increased levels of p-STAT3 were seen in the Vehicle + UUO mice, but were hardly increased in CK + UUO mice. Moreover, the effect of CK on the activation of STAT3 was detected using a J774A.1 macrophage cell line. As shown in Figure 7C and D, CK greatly reduced the phosphorylation level and activity of STAT3 in LPS-activated macrophages using a STAT3 reporter assay. In addition, treatment with CK resulted in significantly reduced LPS- and ATP-mediated IL-1β secretion and caspase-1 activation, as well as reduced LPS-mediated expression of IL-6 and TNF-α (Figure 7E–H). Furthermore, treatment with tofacitinib, a STAT3 inhibitor, significantly blocked the activation of the inflammasome in cells (Figure 7E and F). These results suggest that the inhibitory effect of CK on STAT3 signalling is also involved in its mechanism of action.
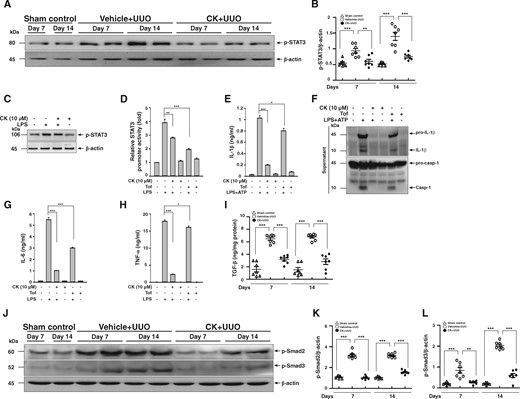
CK inhibited STAT3 activation in renal tissues and cultured macrophages. (A) p-STAT3 in renal tissues assessed by western blot analysis and (B) semi-quantitative analysis. J774A.1 macrophages were incubated for 30 min with or without CK, then for 1 h with or without 1 µg/mL of LPS for (C) p-STAT3 determined by western blot analysis. Macrophages stably expressing a STAT3 reporter gene incubated for 30 min with or without CK, or 1 µM Tof, an inhibitor of STAT3, and then 1 h with or without 1 µg/mL of LPS for (D) STAT3 promoter activity determined by luciferase reporter assay. J774A.1 macrophages were incubated for 30 min with or without CK or 1 µM Tof, then for 5.5 h with or without 1 µg/mL of LPS, and 30 min with or without 5 mM ATP, and (E) IL-1β levels in culture media measured using ELISA and (F) representative western blots of IL-1β and caspase-1. The measurement of IL-1β and caspase-1 released into the culture medium was performed on supernatants extracted. J774A.1 macrophages were incubated for 30 min with or without CK, or 1 µM Tof and 6 h with or without 1 µg/mL of LPS, for protein levels of (G) IL-6 and (H) TNF-α determined by ELISA. The data represent three separate experiments. (I) TGF-β levels in renal tissue measured using ELISA. (J) p-Smad2 and p-Smad3 in renal tissues assessed by western blot analysis and (K and L) semi-quantitative analysis. Tof, tofacitinib. n = 7 mice per group. *P < 0.05, **P < 0.01, ***P < 0.005.
STAT3 signalling has been shown to promote renal fibrosis leading to TILs through the expression of multiple fibrosis-related genes, including TGF-β1 in the renal setting [34, 35], and NLRP3 alone prompts the activation of TGF-β/Smad signalling in renal TECs [36]. As expected, renal protein levels of TGF-β were upregulated in Vehicle + UUO mice compared with those of sham control mice, while treatment with CK markedly downregulated the expression of TGF-β (Figure 7I). Moreover, analysis of the expression levels of p-Smad2 and p-Smad3 in renal tissues showed that both of the signalling molecules involved in the activation of the TGF-β/Smad signalling pathway were greatly reduced in CK + UUO mice (Figure 7J–L).
Validation of the therapeutic effects of CK on renal TILs in UUO mice
Based on the results above regarding favourable preventive treatment with CK of TILs in the mouse UUO model, we performed a therapeutic study (after the induction of the UUO model) to further validate its therapeutic effects. For this purpose, the administration of CK was initiated at an established stage of renal TILs (3 days after induction of the UUO model) to mimic clinical treatment in patients. CK exhibited remarkable therapeutic effects in UUO + CK mice compared with UUO + Vehicle mice (the disease control group), as demonstrated by significant improvements in renal histopathology (Figure 8A–D), and decreased renal infiltration of F4/80 macrophages (Figure 8E and F) and CD3 T cells (Figure 8G and H) in the treated mice. However, treatment with CK starting at Day 7 and Day 14, when diffuse and extensive renal fibrosis, respectively, had been well established, failed to show detectable improvement at a relatively late stage of the renal condition (data not shown).
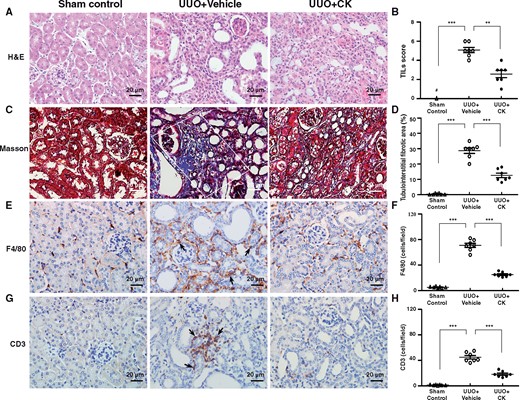
Therapeutic effects of CK on UUO mice. Renal histopathological evaluation using (A and B) hematoxylin and eosin (H&E) and (C and D) Masson’s trichrome staining. Scoring of (B) TILs and (D) renal tubulointerstitial fibrotic area. White solid frame indicates positively stained renal tubulointerstitial fibrotic area. IHC for (E and F) macrophages (F4/80) and (G and H) pan T cells (CD3). Quantitative results reported as the number of (F) macrophages and (H) pan T cells in the renal tubulointerstitial compartment. Arrows indicate positive staining. H&E, hematoxylin and eosin. Original magnification was 400×. n = 7 mice per group. #Not detectable. **P < 0.01, ***P < 0.005.
Validation of the therapeutic effects of CK on renal function and histopathology in NX + I/R mice
Since inflammation is an early event when acute renal injury progresses towards CKD, it was necessary to investigate the therapeutic efficacy of CK in another model of acute kidney injury-to-CKD progression in mice to assess its therapeutic implications. The NLRP3 inflammasome is a mediator in the ischaemic AKI [37]. As shown in Figure 9A and B, NX + I/R mice treated with CK (NX + I/R + CK mice) showed significantly improved renal function and renal pathology, including tubular necrosis, tubular regeneration and mononuclear leukocyte infiltration (Figure 9C–E) compared with NX + I/R mice treated with vehicle only (NX + I/R + Vehicle mice).
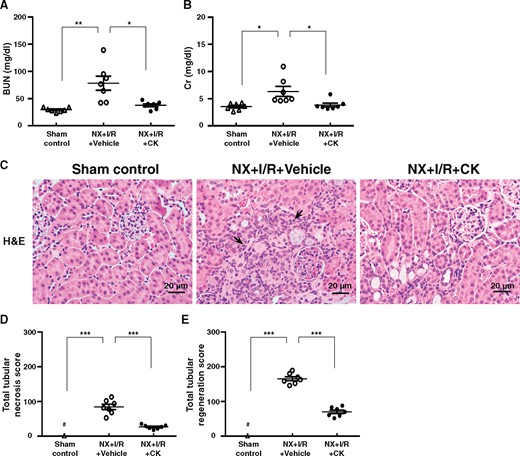
Therapeutic effects of CK on NX + I/R mice. Serum levels of (A) BUN and (B) Cr. Renal histopathological evaluation by (C) Light microscopy, showing tubular necrosis, tubular regeneration and mononuclear leukocyte infiltration. Scoring of (D) tubular necrosis and (E) tubular regeneration. Arrows indicate mononuclear leukocyte infiltration. Original magnification was 400×. NX + I/R, ischaemia–reperfusion injury with unilateral nephrectomy; H&E, hematoxylin and eosin. n = 7 mice per group. #Not detectable. *P < 0.05, **P < 0.01, ***P < 0.005.
DISCUSSION
In the present study, treatment with CK (our US patent: US7932057B2) dramatically improved renal TILs in UUO mice, involving the following: (i) priming and activating signals for NLRP3 inflammasome activation; (ii) STAT3 signalling, which in part prevents NLRP3 inflammasome activation; and (iii) the TGF-β-dependent fibrotic pathway in renal tissues, renal TECs under MICP and/or activated macrophages, the latter being part of a key molecular mechanism underlying renal TILs. Meanwhile, we found downregulated renal NLRP3 inflammasome activation-associated signalling pathways in UUO mice treated with the compound using TMT-based proteomics analysis. As illustrated in Figure 10, our findings support CK as a potential therapeutic candidate for the treatment of renal TILs. This ginsenoside-derived pure compound is effective in the early or developmental stages of renal inflammation and fibrosis, thus making it a suitable drug candidate for the effective prevention of CKD’s progression to ESRD and uraemia.
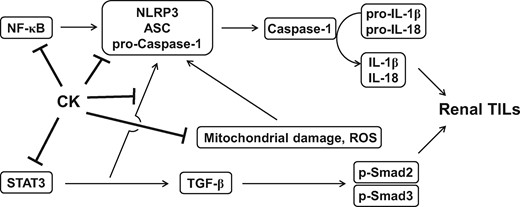
Schematic mechanism of action for the therapeutic effects of CK on renal TILs.
The NLRP3 inflammasome is implicated in renal inflammation and fibrosis of UUO mice [9]. Our data demonstrate that CK inhibits the activation of the NLRP3 inflammasome by greatly reducing renal levels of NLRP3, caspase-1 activity and IL-1β protein, and urine levels of IL-1β. Consistent with these findings, proteomics analysis disclosed several signalling pathways involved in the upregulation of the renal NLRP3 inflammasome (lysosome, oxidative phosphorylation, fatty acid metabolism and sphingolipid metabolism, and the renin–angiotensin system) that were downregulated in renal cortical tissues from CK-treated UUO mice. Moreover, CK upregulated a ribosome pathway, which inhibits the activation of the NLRP3 inflammasome (Supplementary data, Table S2). These results suggest that inhibition of the NLRP3 inflammasome underlie the renoprotective effects of CK on renal TILs in UUO mice (Figure 9). In addition, our demonstration of inhibition of the NLRP3 inflammasome in CK-treated renal TECs under an MICP system confirms the renoprotective effects of CK in this mouse UUO model, via negative regulation of both priming and activation signals of the NLRP3 inflammasome in renal TECs that are primarily involved in the renal TILs of UUO mice. Collectively, our results support a critical role for the NLRP3 inflammasome that underlies the pathogenesis of renal TILs, a key pathological lesion for the progression of CKD to ESRD and uraemia.
Renal fibrosis in UUO mice involves the activation of STAT3 signalling [38], which is implicated in NF-κB- [30–32] or NLRP3 inflammasome-mediated [32, 33] renal inflammatory processes. Notably, we demonstrated that CK inhibited: (i) renal p-STAT3 levels in UUO mice, and (ii) both p-STAT3 expression levels and activity in macrophages. Furthermore, the inhibitory effect rendered by CK on the NLRP3 inflammasome in macrophages is likely generated through the blockade of p-STAT3. Thus, our data suggest that the inhibition of STAT3 activation could be a mechanistic pathway of action for CK in the mouse UUO model.
Additionally, NLRP3 itself has been shown to trigger T cell differentiation [39, 40], while activation of NLRP3 inflammasomes of macrophages has also been shown to mediate the migration of T cells into lesion sites, which involved a receptor for MCP-1, CCR2, in an autoimmune disease model [41]. The present study demonstrates that CK treatment decreases renal infiltration of T cells when it is administered before or after the induction of UUO, and inhibits T cell activation in the renal draining lymph nodes of UUO mice. Whether NLRP3 itself exerts a chemotactic action in mediating the development of renal TILs before the inflammasome further activates Th cells in UUO mice, and the molecular mechanisms involved, require further investigation.
Of note, CK itself has been reported to have low oral bioavailability (5%) in rats [42, 43], as a result of its high polarity [44]. Instead, in a rat model of myoclonic seizures, intraperitoneal injections of CK were employed considering the poor water solubility and low bioavailability of the compound [45]. Therefore, in the present study, we employed an intraperitoneal route to administer CK to the mice. We demonstrated that this strategy also helped to limit variation in the uptake of the compound by the animals throughout the experiment.
CONCLUSION
Taken together, the present study validates the preventive and therapeutic effects of CK on renal TILs by modulating the NLRP3 inflammasome and STAT3 (Figure 10), and justifies the pure compound as a promising drug candidate for the common, progressive renal condition CKD.
FUNDING
This work was supported by grants from the Ministry of Science and Technology (MOST 106-2320-B-016-012-MY3), the Ministry of National Defense-Medical Affairs Bureau from National Defense Medical Center (MAB-106-007 and MAB-107-042), the Tri-Service General Hospital, Taipei, Taiwan (TSGH-C107-063) and the Molecular Medicine Research Center, Chang Gung University from The Featured Areas Research Center Program within the framework of the Higher ducation Sprout Project by the Ministry of Education, Taiwan.
AUTHORS’ CONTRIBUTIONS
W.-H.H.: Induction of mouse UUO model, experiments on preventive effects of CK on UUO mice and draft manuscript preparation. K.-F.H., W.-T.W. and H.-W.C.: NLRP3 inflammasome-related experiments and data analyses. L.-H.T. and Y.-L.T.: preparation of renal sections for histopathology and histochemistry. L.J.C.: proteomics analysis. Y.-C.L. and S.-L.L.: Preparation of CK and quality control of the compound. J.-H.L. and L.-J.H.: Flow cytometric analysis of immune cells. C.-L.C.: Interpretation of immune cells in kidneys and their draining lymph nodes. Y.-J.H. and C.-H.C.: interpretation of renal function and data from IHC. S.-M.K.: design of the study and in vitro experiments using cell models. A.C.: design of the study, renal pathology and finalizing the writing of the manuscript.
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments