





Abstract
Biology has taught us that a protein as abundantly made and conserved among species as Tamm–Horsfall protein (THP or uromodulin) cannot just be a waste product serving no particular purpose. However, for many researchers, THP is merely a nuisance during urine proteome profiling or exosome purification and for clinicians an enigmatic entity without clear disease implications. Thanks to recent human genetic and correlative studies and animal modeling, we now have a renewed appreciation of this highly prevalent protein in not only guarding urinary homeostasis, but also serving as a critical mediator in systemic inter-organ signaling. Beyond a mere barrier that lines the tubules, or a surrogate for nephron mass, mounting evidence suggests that THP is a multifunctional protein critical for modulating renal ion channel activity, salt/water balance, renal and systemic inflammatory response, intertubular communication, mineral crystallization and bacterial adhesion. Indeed, mutations in THP cause a group of inherited kidney diseases, and altered THP expression is associated with increased risks of urinary tract infection, kidney stone, hypertension, hyperuricemia and acute and chronic kidney diseases. Despite the recent surge of information surrounding THP’s physiological functions and disease involvement, our knowledge remains incomplete regarding how THP is normally regulated by external and intrinsic factors, how precisely THP deficiency leads to urinary and systemic pathophysiology and in what clinical settings THP can be used as a theranostic biomarker and a target for modulation to improve patient outcomes.
INTRODUCTION
Tamm–Horsfall protein (THP) or uromodulin (gene name UMOD) is a 90-kDa protein produced almost exclusively in the kidney by the epithelial cells lining the thick ascending limb (TAL) of the loop of Henle and early distal tubules [1–3]. This protein was initially discovered by Tamm and Horsfall in 1950 as an inhibitor of viral hemagglutination in the urine [4]. Although THP has since been assigned many more functions, few have been validated clinically or experimentally. Nonetheless, its highly restricted renal expression, extraordinary abundance and conservation among species support important biological functions and fuel a steady interest for both researchers and clinicians [1].
Initial biochemical characterization of THP was met with tremendous technical difficulties [3]. Due to its heavy glycosylation, which accounts for 30–40% of its total molecular mass and low isoelectric point (pI ∼5.00), this acidic protein is poorly stained by Coomassie Blue and hardly visible on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Because of its high propensity to self-aggregate and its susceptibility to proteases, measurements of THP in solutions and fluids such as the urine can be highly variable [5]. The TAL cells, where THP is synthesized, are terminally differentiated, polarized cells that are difficult to cultivate in vitro, and this has hampered in-depth investigation into the biosynthesis, posttranslational modification, intracellular trafficking, membrane targeting and release into the urine and bloodstream [6]. Notwithstanding these difficulties, the progress made in studying THP is quite substantial and is starting to fundamentally transform our understanding of the multifaceted role of THP in regulating kidney physiology, response to injury and cross talk of the kidney with other organ systems [1, 7–9].
In fact, THP has been assigned a role in the following functions: water impermeability of the thick ascending loop of Henle [2], ion-channel activity in TAL and early distal tubular cells [10–13], calcium and magnesium homeostasis [14, 15], cross talk with proximal tubules within the kidney [7, 16, 17], regulation of systemic granulopoiesis [9], homeostasis of mononuclear phagocytes in the kidney [18], susceptibility and response to kidney injury [17–19], susceptibility and response to urinary tract infection (UTI) [20–22] and kidney stone pathogenesis [23, 24]. In addition, mutations within the coding region of UMOD have been causally linked to a group of autosomal dominant, progressive kidney diseases collectively called uromodulin-associated kidney disease (UAKD) [25–27]. Recent genetic studies have identified single-nucleotide polymorphisms (SNPs) in the upstream regulatory region of THP that are associated with varying risk of hypertension and chronic kidney disease (CKD), suggesting that this protein plays an even broader role in regulating kidney function [28–30]. This review will synthesize key aspects of THP biology and focuses predominantly on several novel areas of advancements (in the context of our existing knowledge of THP) while pointing out some areas of controversies. We refer readers to previous and recent comprehensive reviews for the topics not covered in this review due to space constraints [1, 31]. For the sake of consistency, we will use ‘THP’ as an abbreviation for Tamm–Horsfall protein or uromodulin and reserve UMOD only when discussing the human gene.
THP BIOLOGY
Protein structure: sequence and domain composition
THP is a glycosylphosphatidylinositol (GPI) anchor-linked protein of remarkable structure and unique properties [3]. The precursor protein is composed of 640 amino acids based on the complementary DNA sequence (Figure 1A) [32]. Motifs identified in the THP primary sequence include a signal peptide directing its entry into the secretory pathway (residues 1–24), one epidermal growth factor-like (EGF-like) domains (residues 31–64), two calcium-binding EGF-like domains (residues 65–107 and 108–149), a D8C domain containing eight conserved cysteines (residues 199–287), a fourth EGF-like domain (residues 295–319) [33], one zona pellucida (ZP)-like domain (residues 334–585), eight potential N-linked glycosylation sites and a stretch of hydrophobic amino acids at the C-terminus that acts as a signal for GPI anchorage (residue 614). There are 48 cysteine residues involved in the formation of 24 disulfide bridges. The predicted EGF-like domains are believed to mediate THP protein interactions and the ZP-like domain promotes self-aggregation and polymerization.
![THP structure and expression. (A) Schematic representation of the primary amino acid structure of THP and its various domains. The top shows THP precursor with a signal peptide and 640 amino acids and a GPI anchoring site (614). The bottom shows mature urinary THP that is released after cleavage by hepsin. Two specific sequences (IHP and EHP) are important to prevent intracellular THP polymerization through the ZP domains. EHP is cleaved during THP release in the urine. See text for specific abbreviations. (B) In addition to its expression in the TALs, THP can be occasionally detected in the glomerulus by confocal immunofluorescence (top 2 panels are from a deceased donor nephrectomy) and immunohistochemistry [bottom panels are from the Human Protein Atlas (https://www.proteinatlas.org)]. The findings are reproduced in three different tissues using three different antibodies. Arrowheads in the images show THP staining in the glomerulus.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/35/1/10.1093_ndt_gfy394/2/m_gfy394f1.jpeg?Expires=1750042281&Signature=mR-mGQPaxTQ2kajNcgrY9fM1T2il78gvRoVVgS9KrefPgW~aenkfKwGx3SOOfXfuZqbqCM~zN6cm4pPeqxBggoPsLO4gKkK3WFDGUrxELZyPQX-2GFlt0g509PAX8xf3ruHdUdlNgvcnxjP1TUntglFRRds9We4oi3uDquPQjig~t8r~AJe2eTA48XvP2y8uagv8aAMvLjxGeHCXzqnXWCe8IQj7dDn3pfqM3QobrMRp7T7uRA9-7lTyFSxVUo86Sf-0KHg6DCW9kglNQQ4n0EZkR9iTcLWmP10Kcv3WAAVZFPDqJZKmG-kmOnhMWE5Zott2ICzhvZtSEQwj6JAVsQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
THP structure and expression. (A) Schematic representation of the primary amino acid structure of THP and its various domains. The top shows THP precursor with a signal peptide and 640 amino acids and a GPI anchoring site (614). The bottom shows mature urinary THP that is released after cleavage by hepsin. Two specific sequences (IHP and EHP) are important to prevent intracellular THP polymerization through the ZP domains. EHP is cleaved during THP release in the urine. See text for specific abbreviations. (B) In addition to its expression in the TALs, THP can be occasionally detected in the glomerulus by confocal immunofluorescence (top 2 panels are from a deceased donor nephrectomy) and immunohistochemistry [bottom panels are from the Human Protein Atlas (https://www.proteinatlas.org)]. The findings are reproduced in three different tissues using three different antibodies. Arrowheads in the images show THP staining in the glomerulus.
Biosynthesis and cellular trafficking
Since THP has a leader peptide and is a GPI-anchored plasma membrane protein, its synthesis and intracellular trafficking proceed through the well-established secretory pathway. During biosynthesis, the THP precursor is cotranslationally translocated into the endoplasmic reticulum (ER) where the signal peptide is removed and the protein is glycosylated on seven of its eight potential glycosylation sites [34], disulfide bonds are formed and a preformed GPI anchor is added on its C-terminus, most likely on S614 [35]. Following this addition, the membrane-bound THP is transported to the Golgi apparatus where the high mannose glycans are trimmed and complex glycans added. Both the mature glycans and GPI anchor are sorting signals, directing the protein predominantly to the apical membrane of the TAL epithelial cells [36, 37]. It is proposed that THP reaches the luminal side of the plasma membrane in a polymerization-incompetent conformation retained by the interaction of two hydrophobic motifs, one located within the ZP domain, called the internal hydrophobic patch (IHP, residues 430–462), and the other positioned between the ZP domain and the GPI anchor site, called the external hydrophobic patch (EHP, residues 598–607) [33, 38]. Proteolytic cleavage by a recently identified hepsin protease, at residue F587, disturbs the hydrophobic interaction between the IHP and EHP generating a polymerization-capable monomer that is subsequently assembled into polymeric filaments [39, 40]. THP is secreted in the urine as a high-molecular weight polymer (Mr = 1–10 × 106 Da) that under electron microscopy analysis appears as a fibril matrix that can assume a gel-like structure depending on the ionic strength [41]. Urinary THP monomer is composed of 563 amino acids and, depending on its glycosylation status, migrates as an 80–90 kDa band on reducing SDS-PAGE. Monomeric forms of THP in the urine have been reported after treatment with urea [42]. A truncated form lacking a portion of the ZP polymerization domain can be isolated from the urine [18]. This form is predominantly monomeric, although it can also form dimers, as it retains the N-terminal part of the ZP domain [43].
The rate limiting step in THP maturation is thought to be processing in the ER, due to its complex tertiary structure determined by its high number of cysteine residues (48.7% of amino acid content), all engaged in the formation of intramolecular disulfide bridges [44]. In the ER, glycosylation is initiated on seven of eight potential N-linked glycosylation sites. In the Golgi complex, all glycan chains are modified to complex polyantennary sialic acid–terminated carbohydrates, with the exception of N274, which retains a high-mannose moiety [34]. This unmodified high-mannose moiety is remarkably conserved and is present in THP of all the species examined (see further). Evidence for O-linked glycosylation has also been reported [45]. Thus the molecular weight of THP is significantly contributed (∼30%) by glycosylation and such high carbohydrate content is believed to be important for the physicochemical properties and function of THP.
In addition to this canonical apical targeting, THP also sorts, to a lesser degree, to the basolateral domain of the tubular epithelial cells, as demonstrated by confocal immunofluorescence and electron microscopy [16, 46, 47]. Basolaterally sorted THP is released in the interstitium where it enters circulation and becomes part of the serum proteome, where it can be easily measured by immunoassays [48–51]. The mechanisms that govern basolateral release of THP are not yet clear, but the circulating form of THP is predominantly monomeric, as shown recently by Micanovic et al. [18]. It is unclear why circulating THP, despite being full length, does not aggregate. This could be partially due to its low concentration in the serum compared with urine (20–50 ng/mL versus 20–50 µg/mL, respectively). Other explanations include an alternative cleavage site at the C-terminus that retains the EHP sequence and interferes with the aggregating property of the ZP domain, similar to what was shown in cell culture models [33, 38, 40]. Recent work by Tokonami et al. [52] showed a role for the calcium-sensing receptor in regulating THP trafficking within TAL cells and apical release in the urine. Whether these recent findings also translate into changes in basolateral release will need to be determined.
THP EXPRESSION, LOCALIZATION AND SECRETION
THP is primarily expressed in cells of the TALs of Henle and early distal tubules, but it is absent from macula densa cells [46, 53, 54]. This pattern of expression of THP has been primarily established in rodents and to a lesser extent in human kidneys. Interestingly, we consistently observe focal expression of THP in some glomeruli in humans (Figure 1B). This is also observed in a publically available atlas database using multiple antibodies applied to different sets of human kidney tissue [55] and was also reported by McGiven et al. [56] in 1978 in a series of human kidney biopsies. The significance of this occasional presence of THP in the Bowman space or in the glomerular tuft in human kidneys is unclear, but certainly deserves future investigation since glomerular function appeared to be somewhat affected by THP deficiency [8], and certain THP mutations can lead to glomerulocystic kidney disease, an inherited kidney disease characterized by cystic dilatation of the Bowman’s space and collapse of the glomerular tuft [26].
Regulation of THP expression is poorly understood. Recent in silico phylogenetic footprinting analysis showed that UMOD promoter is likely regulated by a large network of interacting transcription factors [57]. One of those transcription factors, hepatocyte nuclear factor 1 β has been shown to activate THP expression [58]. SNP variants in the upstream regulatory region of the UMOD gene also affect the expression of THP [50, 59, 60]. Other factors such as dietary salt and diuretics also affect the expression of THP [61, 62]. These latter effects are likely due to the interplay between THP and ion transporters targeted by diuretics. The synthesis of THP is dynamic and likely varies based on physiological and pathological stressors and nephron mass [63]. For example, the expression of THP increases in early diabetes even without evidence of kidney injury [64, 65]. THP expression can also fluctuate even when kidney function is normal, as demonstrated by varied THP measurements both in the serum and urine [50, 66, 67]. Although it has been suggested that the production of THP reaches a plateau beyond a glomerular filtration rate (GFR) of 90 mL [66], the variance of THP remains quite high, suggesting that with normal kidney function, the impact of GFR or nephron mass on THP levels is reduced compared with other regulatory factors that modulate THP expression and production.
While urinary THP is excreted at a rate of 30–60 mg/day with normal kidney function [1, 66], the range of THP in the serum is less well established (range 30–500 ng/mL) and several recent studies measured serum THP in a few cohorts [50, 51, 67–71]. Both urine and serum THP are directly associated with kidney function [49]. However, THP expression and secretion are very dynamic and can change rapidly in response to pathophysiological conditions, as we discuss below. Also, the fact that THP is regulated by a network of transcription factors [57] strongly suggests that this protein is under intricate regulation [72] and that the changes in its level result from complex interactions.
THE MULTIFACETED ROLE OF THP IN MAINTAINING HOMEOSTASIS
Under normal circumstances, THP appears to regulate various molecular targets within the cells where it is produced and on cells downstream of TAL through its release in the urine or in the interstitium and then the circulation (Figure 2, Homeostasis). Clearly, alterations in the functioning of THP, whether due to a decrease in its expression, mutations or impairment in its downstream targets, could underlie the pathogenesis of many kidney diseases (Figure 2, Disease). Based on studies of THP−/− mice, it is now established that THP deficiency, independent of kidney function, creates a state of inflammation, both systematically and in the kidney [1, 7–9]. This is now also supported by multiple human clinical studies that measured the level of serum and urine THP [67, 68, 72].
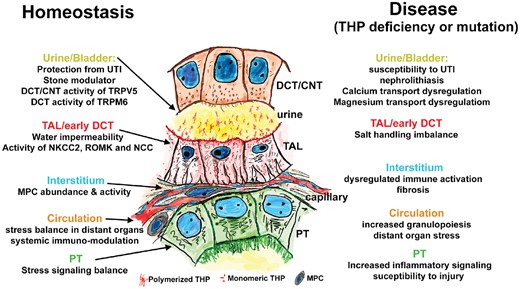
THP function in health and disease. This annotated drawing summarizes how THP is an important guardian of homeostasis (left side) both in the cells where it is produced (TALs and early DCTs) and also in target cells reached through the urine or via the interstitium and bloodstream. Disturbance in THP function either through deficiency or mutations can lead to disease by altering its function at various sites (right side). CNTs: connecting tubules; PTs: proximal tubules.
An interesting phenomenon is a concentration gradient of THP from the urine (highest) toward the serum (lowest). There is likely another concentration gradient of THP in the interstitium between the medulla (where TAL density is increased) and the cortex [7]. In addition to regulating intricate physiological functions (altering a gradient signals stress), gradients are essential for chemotaxis, and it is likely that such a THP gradient may be important in the homeostasis of mononuclear phagocytic cells (MPCs) in the inner stripe of the outer medulla, since THP knockout mice have a reduced MPC population in this region [18]. The various roles attributed to THP are summarized in Figure 2 and listed below.
Within the TAL and early distal convoluted tubule (DCT), THP regulates water permeability [2] and activity of the sodium potassium chloride cotransporter type 2 (NKCC2), renal outer medullary potassium and sodium chloride cotransporter channels [10–13]. Evidence for such regulation has been established experimentally, especially in knockout mice. This has important implications in disease associated with THP deficiency. For example, a recent study by Liu et al. [10] showed that chronic THP deficiency leads to a compensatory increase in proximal reabsorption of solutes accompanied by upregulation of urate transporters, initially triggered by reduced apical translocation of NKCC2 in the TAL cells. These changes lead to hypertension and hyperuricemia, which are phenotypic features consistently present in advanced CKD, a state of relative THP deficiency [67, 68, 71].
In the urine, THP can regulate calcium and magnesium reabsorption in DCT and connecting segment cells by modulating the abundance of transient receptor potential cation channel subfamily V member 5 (TRPV5) and transient receptor potential cation channel subfamily M member 6 channels on these cells [14, 15]. This has implications on divalent cation homeostasis and urolithiasis as discussed below.
In the renal interstitium, THP can target multiple cell types within that microenvironment. By accessing the basolateral domain of epithelial cells [16], THP inhibits pro-inflammatory signaling from the neighboring proximal tubules, such as neutrophil–chemokine release [17] and production of interleukin (IL)-23, which can stimulate the release of IL-17 systemically and enhancement of granulopoiesis through granulocyte colony-stimulating factor [9]. Therefore changes in THP levels in the interstitium could be considered as stress or danger signals that will trigger a response from the epithelium that cascades into a more systemic reaction [7].
THP will also interact with immune cells in the renal interstitium. In fact, THP appears to be a significant regulator of the abundance and phagocytic activity of resident MPCs [18]. It is likely that this role has important implications in the immune balance within the renal interstitium that can be perturbed during injury.
Since THP is released by the kidney in the circulation, do circulating THPs have a distant effect on other organs? This is a likely scenario because an effect of THP on epithelial and immune cells may have far-reaching impacts well beyond the kidneys. The extent that THP impacts nonkidney organ systems may be highly dependent on its levels in the circulation. Several recent studies have shown that THP levels in the serum are inversely correlated with markers of inflammation, such as C-reactive protein and IL-1β, independent of kidney function and even in patients with normal kidney function [67, 68, 72]. The fact that a high THP level in the serum is associated with protection from cardiovascular mortality and kidney injury, independent of kidney function, strongly supports that THP has a positive impact on the homeostasis of the entire organism [50].
THP IN SPECIFIC DISEASE STATES
The role of THP in specific disease states has been studied in several animal models and clinical settings and reviewing these data is important to understand the behavior of THP and interpret changes in its level during systemic and/or kidney disease. Since THP has pleiotropic effects both within the cells where it is produced and on distant targets, it is reasonable to anticipate that changes in THP production will depend on whether the kidneys, and specifically THP-producing cells, are affected by the disease. In addition, our current knowledge (including several unpublished observations) suggests that the production of THP is dynamic and is affected by pathophysiological stressors [53, 72, 73]. Therefore it is logical to interpret the changes in THP levels in the serum and/or urine depending on the presence of a systemic illness and whether the disease affects the kidney and causes acute kidney injury (AKI).
Systemic illness and inflammation
Because of its protective, counterinflammatory functions and its role in maintaining homeostasis, it is expected that THP production by the kidney will compensatorily increase in systemic illness without kidney injury [53]. Examples would be stress response to infections, transient hemodynamics changes and fight/flight responses [1, 7]. When THP production is increased in that setting, it is likely that the kidney is counteracting uncontrolled systemic inflammation through inhibition of cytokine/chemokine signaling or via activation and polarization of macrophages.
AKI
Studies using animal models of AKI, namely the ischemia–reperfusion injury (IRI) model, helped elucidate the role of THP in AKI. During AKI, THP production is significantly diminished both at the RNA and protein level, as shown by multiple groups and confirmed in few human studies [1]. Therefore AKI is a state of THP deficiency [74]. The mechanisms through which THP expression is regulated during AKI are not fully understood [74].
Renal IRI in THP knockout mice is significantly more severe than in wild-type animals [16, 17, 19]. The injury is associated with increased neutrophil-activating inflammatory signaling in S3 tubules, leading to tubular necrosis in these segments. Therefore THP produced in the TAL inhibits chemokine signaling and the susceptibility of neighboring proximal tubules to injury, supporting the presence of a THP-dependent TAL–S3 tubular cross-talk phenomenon [1, 75]. Translocation of THP from the basolateral surface of TAL toward the basolateral domain of neighboring S3 tubules, the interstitium and circulation is important to suppress subsequent inflammation and begin recovery [16]. The effect of THP on neutrophil infiltration is likely to also involve regulation of the IL-23/IL-17 axis, whereby THP deficiency seen in AKI will stimulate granulopoiesis [9]. Furthermore, THP is important in macrophage migration, phagocytic activity, alternative polarization and healing. The latter may be important in promoting a swift transition toward a recovery phase [18]. Therefore a prolonged state of THP deficiency during AKI will likely lead to a more severe and persistent inflammatory state. Indeed, administration of exogenous THP after AKI mitigates subsequent injury and hastens recovery [18].
CKD
Based on our understanding of the function of THP in maintaining homeostasis and regulating inflammation and immune cell activation, it is likely that THP is a defender of the kidney against CKD [1]. Indeed, high baseline THP, independent of kidney function, is protective from subsequent decline in GFR [67, 76]. In fact, the synthesis of THP per functioning nephron unit is increased in CKD [49] (THP per unit GFR, the uromodulin index as we proposed previously [1]), that is, when the kidney is under attack. This was elegantly demonstrated by Thornley et al. [49], who showed that THP excretion per milliliter of creatinine clearance had a higher range in patients with CKD compared with normal subjects. However, despite increased production by intact nephrons, fibrosis will cause additional nephrons to drop out, leading to an absolute decrease in THP production in progressive CKD. The results will be that total THP levels drop as CKD progresses and GFR declines [49, 51, 71]. Conversely, in patients with very early CKD or with kidney disease without an apparent decrease in kidney function, THP synthesis will be increased. This has been reported in early diabetes without GFR impairment (in human and animal models of diabetes) or in patients prior to the onset of CKD [59, 64, 65, 67, 77–79]. This effect was also demonstrated to be independent of renal mass [49, 77].
Interpreting changes in THP levels with progressive or advanced CKD can be challenging, as the surviving functional nephron units are under stress and will likely continue to over-produce THP. At the same time, the surviving nephron units are progressively diminished. The net sum of these two opposing tendencies and their effects on the levels of circulating THP are difficult to predict and likely to introduce variability. It may be more useful in these situations to follow 24-h urine THP measurements normalized by unit of clearance. In fact, significant tubular atrophy observed in kidney biopsies correlated with a decrease in urinary THP excretion per unit GFR, suggesting that with more advanced disease, the synthesis of THP per nephron unit will eventually decline [49]. In these instances, decreased 24-h THP excretion per GFR unit likely implies a depletion of tubular reserves and progression to end-stage disease [49].
THP in urinary homeostasis and disease
It is not entirely surprising that THP plays major roles in the urinary tract [80], given the fact that the bulk of THP made by the TAL cells is excreted in the urine. Thus urinary THP binds in a dose-dependent manner to the FimH adhesin of type 1-fimbriated uropathogenic Escherichia coli, which causes up to 85% of all uncomplicated UTIs [81, 82]. The binding is mediated specifically by the evolutionally conserved, unmodified high-mannose residues of the THP, resulting in competitive inhibition and preventing E. coli from adhering to their cognate urothelial receptor, that is, uroplakin Ia, which bears the identical high-mannose receptor. There is some evidence that the unmodified mannoses on the soluble receptor (THP) and the immobilized receptor (uroplakin Ia) are co-evolved during evolution [2, 42]. The same principle might apply to the complex sugars shared by THP and uroplakin III [83]. Some of these predictions have already panned out in THP−/− mice, which are significantly more prone to experimentally induced UTIs than wild-type [20–22]. A previously suspected role of THP in inhibiting urinary calcium crystallization has also been validated in THP−/− mice. Spontaneous intrarenal crystals comprised of hydroxyapatite have been detected in knockout mice by histochemical staining and transmission electron microscopy and confirmed by infrared spectroscopy [23, 24, 84]. These crystals are deposited on the basement membrane, concentrated in the papillary region and not associated with overt inflammation—features resembling early stages of nephrolithiasis in idiopathic calcium oxalate stone–forming patients. The THP−/− mice are also highly prone to renal calcium crystallization induced by experimental hyperoxaluria. Such crystals are primarily intratubular. Although the exact mechanisms for THP to inhibit renal calcification remain to be elucidated, THP likely interferes with multiple steps during urolithiasis, including crystal formation, growth, aggregation and epithelial attachment. Furthermore, the effect of THP on distal calcium reabsorption through its effect on TRPV5 will reduce urinary calcium and may mitigate the risk for supersaturation of calcium oxalate and calcium phosphate [15].
An immunomodulatory role for THP in the urine has also received experimental support. THP accomplishes this role, possibly by binding to various cytokines, serving as a trap or sink and hence reducing the urinary levels of cytokines [8]. Although the disease implications of THP-mediated cytokine modulation have yet to be defined, reduced levels of THP have already been associated with UTI and kidney stone risks in humans. In one cohort, kidney stone formers were found to contain 30% less urinary THP than the nonstone-forming controls [85]. In another cohort, a THP variant, that is, the rs4293393-T allele, which is associated with high THP, is protective of kidney stones [86]. Similar correlation between high urinary THP levels and low risks of UTI has also been demonstrated in older community-dwelling adults by one of the coauthors of this review [87]. Even in individuals with normal levels of THP, deficiency in high-mannose- and complex-type glycosylation may still play a role in the susceptibility to UTI and kidney stones, respectively [85, 88].
CLINICAL STUDIES USING THP AS A BIOMARKER
The last decade has seen a number of studies published that have evaluated the role of serum and urine THP as a potential biomarker. These have included its use in the prognostication of progressive GFR decline, incident ESRD, posttransplant graft loss, all-cause mortality, cardiovascular disease (CVD) events and UTIs.
Urine THP
Data from two large cohorts of older adults demonstrate that higher THP levels are associated with significantly lower odds of loss of kidney function over nearly a decade [76, 89]. Given that THP may serve as a marker of tubular reserve, its use as a risk stratification biomarker for AKI has been hypothesized. Not surprisingly, studies in both children and adults undergoing cardiac surgery show that those with low levels of THP preoperatively have the greatest risk of AKI postoperatively [90–92]. Studies are needed that utilize THP levels to risk-stratify persons at increased risk of AKI and target prevention strategies.
Elegant laboratory studies have demonstrated that increasing concentrations of THP inhibit bacterial growth and THP−/− mice have an increased risk for developing bladder infections with greater evidence of histological damage than THP+/+ mice [20, 21, 82, 93]. Supporting this is a large cohort study demonstrating that persons with high urinary THP have a significantly lower risk of UTI events compared with those in those with the lowest THP [94]. Furthermore, THP may also be important in preventing the development of bacteremia in those with UTI [95].
Serum THP
A number of studies have reported positive correlations between serum/plasma THP levels and kidney function assessed using GFR [51, 71]. In a cohort of 426 persons with different stages of CKD, plasma THP showed very good ability to identify those with at least CKD Stage 3 [71]. Lower levels of serum THP may identify early kidney function loss even when serum creatinine values are still within a seemingly normal range (71–106 µmol/L) [67]. Serum THP levels were lower in patients with CKD, albuminuria and hypertension [67] and were inversely associated with the development of incident CKD. Even in persons who have undergone kidney transplants, lower levels of serum THP were associated with double the risk of graft loss after multiple adjustments for confounders [96]. Higher THP levels in the serum are also associated with better CVD and mortality outcomes [50, 70]. Taken together, this growing body of compelling evidence suggests that the presence of high levels of THP in the urine and serum is associated with better kidney, CVD and mortality outcomes.
INTERPRETING THP LEVELS IN KIDNEY DISEASE
Based on this discussion, interpretation of the levels and changes in THP measurements is shown in Figure 3. Both urinary and serum/plasma THP can be measured using commercially available enzyme-linked immunosorbent assays (ELISAs). As discussed above, both measurements correlate with kidney function and could be useful. We advocate the use of plasma or serum THP since THP in the circulation is monomeric and less prone to degradation compared with urine. Both of these factors could reduce the variability in measurements. However, in advanced CKD, measuring 24-h urine THP may have some advantages, as discussed above. Of note, there are limited data on the level of THP in glomerulonephritis such as systemic lupus erythematosus [68]. The available evidence suggests that serum THP levels are lower in patients with kidney involvement and injury, but more data are needed to explain how measurements of THP could be used to monitor disease activity and kidney involvement.
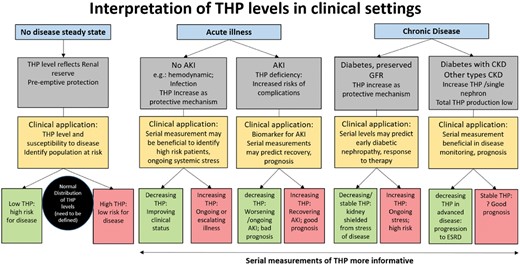
Interpretation of THP levels in clinical settings. THP can be measured either in the blood or in the serum. During acute illness or chronic disease, serial measurements of THP may be more informative than a single measurement. This figure does not account for the presence of minor risk genetic SNP variants in the promoter area of THP, which alter THP expression and have an independent effect on kidney disease risk.
OPPOSING EFFECTS BETWEEN UMOD PROMOTER REGION SNPs AND THE EXPRESSION OF THP ON KIDNEY FUNCTION: AN EXPRESSION QUANTITATIVE TRAIT LOCI EFFECT ON NEIGHBORING GENES?
One of the areas of debate recently has been the opposing effect on kidney function between the level of expression of THP and the presence of particular SNPs in the UMOD gene promoter area uncovered from genome wide association studies (GWAS) studies [97, 98]. We believe that the confusion developed because of data interpretation. The major alleles in variants rs12917707 and rs4293393, which are in linkage disequilibrium, are associated with an increased risk of kidney disease compared with the minor alleles. In addition, these upstream variants that increase the risk positively affect the level of THP expression [60]. By transitivity, it has been proposed that increased risk of the major alleles occurs through increased THP expression. However, what is strikingly inconsistent with this inference is that in the presence of the major allele, increased expression of THP is paradoxically associated with protection, independent of renal function and even in patients with normal kidney function [50, 67, 68, 70]. This strongly suggests that although risk variants can regulate THP expression, the effect of these SNPs on kidney risk is independent of THP expression and likely to offset the change in risk imparted by THP expression [50, 67]. Can SNPs upstream of the UMOD gene regulate other genes that are involved in CKD?
Indeed, emerging understanding of gene regulatory elements suggests that each gene could have multiple regulatory elements that explain a fraction of its expression [expression quantitative trait loci (eQTLs)] [99–102]. eQTLs can regulate multiple genes within a close distance within a chromosome (e.g. 1 megabase; cis-eQTL effect) or even more distantly (even on separate chromosomes; trans-eQTL). The variability of eQTL effects for a specific regulatory element is also likely to be tissue specific [99]. For the case of the UMOD gene, multiple loci have been implicated in its expression [103]. In addition to the variants rs12917707 upstream of UMOD on chromosome 16, variant rs12446492 in the neighboring PDILT gene or even variants in KCNJ1, SORL1 (both on chromosome 11) and CAB39 (chromosome 2) are associated with urinary THP levels [103]. Furthermore, multiple genes neighboring the UMOD gene (such as ACSM2A/2B) are also enriched in the kidney and display an expression profile that is tightly linked with kidney function, suggesting a role in regulating kidney disease [102]. Based on this evidence, it is very likely that risk variants in the UMOD promoter area regulate multiple neighboring or distal genes involved in kidney disease [57, 99, 102]. We and other groups proposed that this is likely to occur through the intricate network of transcription factors that bind these regulatory DNA motifs and are likely to coregulate gene expression [1, 57, 102]. Therefore a variant that alters the binding of certain transcription factors and hinders or potentiates initiation complex activation for one gene will concomitantly alter the expression of other genes by altering the availability of functionally linked transcription factors. A conceptual model of such regulation is shown in Figure 4. In summary, as scientific discoveries unravel the intricate and complex regulation of CKD risk-associated transcripts, we caution against a simplified or fragmented explanation of the mechanism underlying how risk variants in regulatory elements are linked with kidney disease.
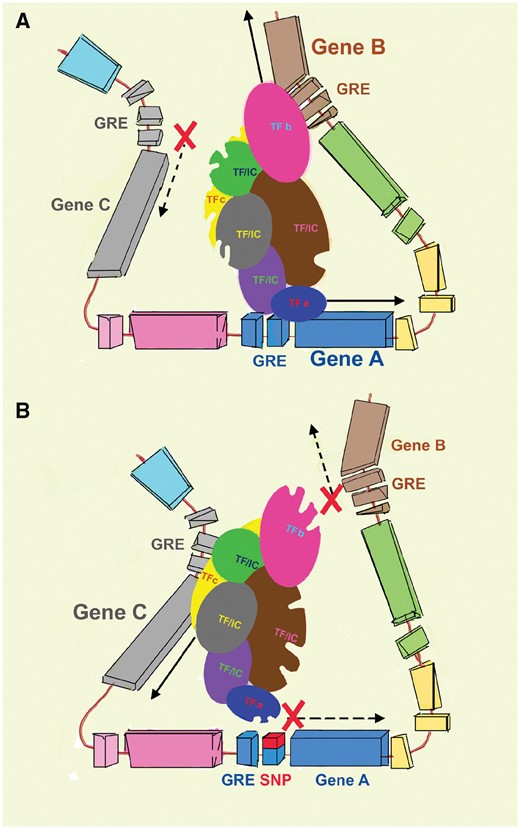
Model of eQTL interactions. (A) A network of transcription factors (TFs) and associated initiation complex (IC) molecules concomitantly regulate the expression of multiple neighboring genes through interactions with gene regulatory elements (GRE). Through this large network, the engagement of specific TFs with GREs (e.g. TFa for Gene A) alter the availability and/or conformation of other linked TFs (TFb and TFc for Genes B and C, respectively). This will cause either enhancement or inhibition of transcription in these neighboring genes. (B) Therefore, in the presence of a polymorphism within a specific GRE, gene expression will be altered not only in the area of immediate proximity (Gene A), but also in the other coregulated distant genes.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
As the most abundant protein in healthy human urine, THP has recently received the level of attention that it rightly deserves. The investigative efforts have been tremendously rewarding, as we now know with some certainty that THP is a multifunctional protein that plays indispensable roles in urinary and systemic homeostasis and whose structural and functional alterations can lead to a range of kidney and urinary diseases. Investigators in the field can take pride in their perseverance in studying a protein that was discovered almost 70 years ago and which many regard as no longer attractive. Looking forward, we must guard against the notion that there is not much else to learn about THP or that THP is only a surrogate marker for nephron mass. In fact, we are at the beginning stage of unraveling the systemic effects of THP through its release in the circulation. The kidney is known to produce erythropoietin, a hormone that will determine the production of red cells and oxygen delivery to the entire circulation. Therefore it is not unreasonable to propose, based on the mounting evidence, that THP is another hormone-like peptide produced by the kidney that shapes systemic immunity and inflammatory signaling balance.
Furthermore, many critically important questions remain unanswered. For instance, what is the functional significance of the high expression of THP in TAL but the absent expression of THP in the macula densa? What underlies the switch of THP from the default apical membrane targeting to the basolateral domain during renal ischemia and other conditions that predicate the release of THP into the circulation? Is it because of the intrinsic factors, that is, structural alterations in THP or changes in the intracellular environment of TAL cells? Exactly how is THP expression regulated by salt and water intake and how do the changes in THP levels alter the apical translocation and activity of NKCC2? How does THP expression within a specific nephron correlate with the functional status of that particular nephron? In what clinical settings can THP serve as a predictor of patient outcome? More importantly, can the levels of THP be upregulated to serve therapeutic purposes? Similarly, can structural defects caused by misfolding of mutated THP be corrected by chemical chaperones? Answers to some of these questions will undoubtedly have direct translational values in improving the clinical management of THP-related diseases.
FUNDING
This work was supported by funding from the National Institute for Diabetes and Digestive and Kidney Diseases (NIDDK-1R01DK111651) and a VA Merit review award to T.M.E.-A.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments