





Abstract
Magnesium is known to protect against phosphate-induced tubular cell injuries in vitro. We investigated in vivo effects of magnesium on kidney injuries and phosphate metabolism in mice exposed to a high phosphate diet.
Heminephrectomized mice were maintained on a high phosphate/normal magnesium diet or a high phosphate/low magnesium diet for 6 weeks. We compared renal histology, phosphaturic hormones and renal α-Klotho expression between the two diet groups.
High phosphate diet–induced tubular injuries and interstitial fibrosis were remarkably aggravated by the low-magnesium diet. At 1 week after high phosphate feeding when serum creatinine levels were similar between the two groups, the low magnesium diet suppressed not only fecal phosphate excretion but also urinary phosphate excretion, resulting in increased serum phosphate levels. Parathyroid hormone (PTH) levels were not appropriately elevated in the low magnesium diet group despite lower 1,25-dihydroxyvitamin D and serum calcium levels compared with the normal magnesium diet group. Although fibroblast growth factor 23 (FGF23) levels were lower in the low magnesium diet group, calcitriol-induced upregulation of FGF23 could not restore the impaired urinary phosphate excretion. The low magnesium diet markedly downregulated α-Klotho expression in the kidney. This downregulation of α-Klotho occurred even when mice were fed the low phosphate diet.
A low magnesium diet aggravated high phosphate diet–induced kidney injuries. Impaired PTH secretion and downregulation of renal α-Klotho were likely to be involved in the blunted urinary phosphate excretion by the low magnesium diet. Increasing dietary magnesium may be useful to attenuate phosphate-induced kidney injury.
INTRODUCTION
Hyperphosphatemia is highly prevalent among patients with chronic kidney disease (CKD). Phosphate overload accelerates vascular calcification and amplifies the cardiovascular risk of CKD patients [1–6]. Excess phosphate is also harmful to the kidney. Clinical studies have shown that higher serum phosphate levels are associated with an increased risk of CKD progression [7–13]. Furthermore, a high phosphate diet exacerbates kidney damage such as tubular injuries, interstitial fibrosis and inflammation in animal models of CKD [14–17]. Therefore, suppressing phosphate toxicity may improve not only cardiovascular outcomes, but also renal prognoses.
Magnesium has recently been focused on because of its protective effects on phosphate toxicity in terms of cardiovascular risk in CKD [18]. Experimental studies, both in vitro and in vivo, have shown that magnesium inhibits high phosphate–induced vascular calcification by preventing the formation and growth of calcium phosphate crystals and suppressing the differentiation of vascular smooth muscle cells to osteoblast-like cells [19–27]. Among patients undergoing hemodialysis, we have previously reported that the risk of cardiovascular death associated with hyperphosphatemia is attenuated with increasing serum magnesium levels [28].
Magnesium may also be protective against phosphate toxicity in the kidney [29]. In our previous cohort study, the association between serum phosphate levels and the risk of CKD progression was pronounced among patients with lower serum magnesium levels, but it disappeared among patients with higher serum magnesium levels [30]. Consistently an in vitro experiment showed an inhibitory effect of magnesium on high phosphate–induced proximal tubular cell death [30]. Therefore, magnesium may be a beneficial mineral that can counteract phosphate toxicity in the kidney. To date, however, in vivo evidence is lacking. The current study aimed to investigate whether dietary magnesium impacts on the high phosphate diet–induced kidney damage in a CKD mouse model.
MATERIALS AND METHODS
Animals
Seven-week-old male C57BL6J mice were purchased from CLEA Japan (Tokyo, Japan) and maintained under 12-h light–dark cycles and pathogen-free conditions with free access to food [AIN93G diet (CLEA Japan)] and water. Heminephrectomy was performed under anesthesia (butorphanol tartrate [5.0 mg/kg intaperitoneally (i.p.) (Meiji Seika Pharma, Tokyo, Japan)], midazolam [4.0 mg/kg i.p. (Astellas, Tokyo, Japan)] and medetomidine [0.3 mg/kg i.p. (Nippon Zenyaku Kogyo, Fukushima, Japan)]). A left flank incision was made and, after ligation of the renal pedicle, the left kidney was removed. One week later these mice were randomly divided into one of the following diet groups: a high phosphate (1.25%)/normal magnesium (0.05%) diet (n = 6), a high phosphate (1.25%)/low magnesium (0.005%) diet (n = 15), a low phosphate (0.25%)/normal magnesium (0.05%) diet (n = 6) and a low phosphate (0.25%)/low magnesium (0.005%) diet (n = 6). Mice were sacrificed at 6 weeks after the initiation of these experimental diets to collect serum and kidney samples (long-term experiments) (Figure 1A).
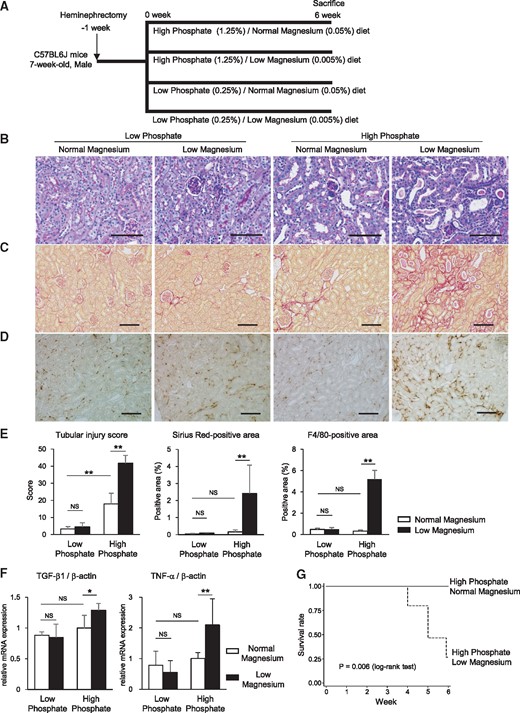
High phosphate diet–induced kidney injury is exacerbated by low magnesium feeding. (A) Heminephrectomized mice were administered either a high phosphate (1.25%)/normal magnesium (0.05%) diet (n = 6), a high phosphate (1.25%)/low magnesium (0.005%) diet (n = 15), a low phosphate (0.25%)/normal magnesium (0.05%) diet (n = 6) or a low phosphate (0.25%)/low magnesium (0.005%) diet (n = 6) for 6 weeks. (B–D) Representative micrographs showing (B) PAS-, (C) Sirius Red– and (D) F4/80-stained kidney sections. Scale bars: 100 µm for PAS staining, 200 µm for Sirius Red staining and F4/80 staining. (E) Tubular injury scores and Sirius Red– and F4/80-positive areas were significantly increased by the high phosphate/low magnesium diet. (F) TGF-β1 and TNF-α mRNA expression levels in the whole kidney were upregulated in the high phosphate/low magnesium diet group. (G) Kaplan–Meier curves showing the significantly reduced survival rate in mice maintained on the high phosphate/low magnesium diet compared with the high phosphate/normal magnesium diet (P = 0.006; log-rank test). At 6 weeks, all mice in the high phosphate/normal magnesium diet group (n = 6) were alive, whereas only 4 of 15 mice in the high phosphate/low magnesium diet group were alive. Data are presented as the mean ± standard deviation. Differences between the two groups were compared using a Student’s t-test with a Bonferroni correction (NS, not significant; *P<0.05, **P<0.01).
We also performed short-term experiments to examine renal histology, biochemical parameters and renal expression of α-Klotho. The heminephrectomized mice were divided into the high phosphate/normal magnesium diet group or the high phosphate/low magnesium diet group and were sacrificed at 1 and 2 weeks after the initiation of these diets (n = 4−8). The expression of α-Klotho was additionally examined after 1 week of the low phosphate/normal magnesium diet and low phosphate/low magnesium diet (n = 6 in each group).
During the 1-week experiment, mice were subcutaneously injected with low-dose calcitriol (30 pg/g body weight) (Sigma-Aldrich, St Louis, MO, USA) or vehicle (phosphate-buffered saline containing ethanol and Tween-20) on Days 1, 3, 5 and 6. We collected 24-h urine and fecal samples using metabolic cages.
All experiments were performed in accordance with the ethical guideline of the Animal Committee of Osaka University (approval no. 27-072-000).
Biochemical parameters
Serum and urine phosphate, magnesium and calcium levels as well as urinary N-acetyl-β-d-glucosaminidase (NAG) activity levels were measured using clinical diagnostic reagents (Wako Junyaku, Osaka, Japan). Serum creatinine levels were measured using the Kainos CRE reagent (Kainos Laboratories, Tokyo, Japan). We used an intact fibroblast growth factor 23 (FGF23) enzyme-linked immunosorbent assay (ELISA) kit (Kainos Laboratories) to measure serum intact FGF23 levels and a mouse parathyroid hormone (PTH) 1-84 ELISA kit (Immutopics, San Clemente, CA, USA) to measure plasma 1-84 PTH levels. Serum 1,25-dihydroxyvitamin D levels were measured using a 1,25-dihydroxy vitamin D radioimmunoassay kit (Immunodiagnostic Systems, Boldon, Tyne and Wear, UK).
Real-time polymerase chain reaction
Expression levels of messenger ribonucleic acid (mRNA) in the whole kidney were quantified by real-time SYBR-Green polymerase chain reaction analyses using an ABI PRISM 7900HT (Applied Biosystems, Foster City, CA, USA) with the specified primers (Supplementary data, Table S1). Total RNA was extracted from frozen kidney samples stored at −80°C using TRIzol (Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s instructions.
Histological analyses
Paraffin-embedded kidney sections were stained using the standard periodic acid–Schiff (PAS) and Sirius Red staining methods. Tubular injury scores were determined morphometrically as previously described [31]. Briefly, high-power magnification (×400) fields of the PAS-stained renal cortex were analysed using a BZ-9000 microscope (Keyence, Osaka, Japan). Each image was then divided into 96 (12×8) grids and those containing atrophic and/or necrotic tubular cells were counted. The tubular injury score was the average number of these grids per six randomly selected fields for one kidney. Immunohistochemical staining was performed as previously described [32], using rat anti-human Klotho monoclonal antibody (clone no. KM2076; Trans Genic, Kobe, Japan), rat anti-mouse F4/80antibody (Bio-Rad, Hercules, CA, USA) and rabbit anti-type IIa Na-Pi cotransporter (Npt2a) antibody (gift from Dr H. Segawa) [33]. The percentages of Sirius Red–positive, F4/80-positive, α-Klotho-positive and Npt2a-positive areas were measured in three to five randomly selected fields per kidney using ImageJ software (http://rsbweb.nih.gov/ij/index.html; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
A Student’s t-test with a Bonferroni correction was used to make multiple comparisons. Kaplan–Meier survival curves were compared using a log-rank test. All data are presented as the mean ± standard deviation. A two-sided P-value < 0.05 was considered to indicate statistical significance. All statistical analyses were performed using Stata 13 statistical software (StataCorp, College Station, TX, USA).
RESULTS
Low magnesium diet exacerbates high phosphate–induced kidney injuries
After 6 weeks of high phosphate diet feeding, heminephrectomized mice exhibited renal tubular injuries such as tubular dilation, loss of the brush border, and flattening and detachment of tubular epithelial cells (Figure 1B). These histological changes were markedly aggravated by the low magnesium diet (Figure 1B). Compared with the high phosphate diet alone, the high phosphate/low magnesium diet significantly exacerbated interstitial fibrosis as indicated by the Sirius Red staining and interstitial infiltration of F4/80-positive cells (Figure 1C–E). Renal mRNA expression levels of pro-fibrotic and pro-inflammatory cytokines, transforming growth factor (TGF)-β1 and tumor necrosis factor-α, were enhanced by the low magnesium diet (Figure 1F). Body weight was remarkably reduced by the high phosphate/low magnesium diet compared with the high phosphate/normal magnesium diet (Supplementary data, Table S2). Kaplan–Meier curves revealed a high mortality rate in the high phosphate/low magnesium diet group (Figure 1G). The low magnesium diet aggravated the kidney injuries even at 2 weeks after the high phosphate feeding (Supplementary data, Figure S1).
In contrast, no significant differences were found in these parameters between the low and normal magnesium diet groups under a low phosphate diet (Figure 1E and F).
Low magnesium diet suppresses urinary phosphate excretion
Next we examined how dietary magnesium affects phosphate metabolism. The high phosphate/low magnesium diet for 6 weeks significantly elevated serum creatinine levels (Supplementary data, Table S2), rendering it difficult to isolate the effect of dietary magnesium from that of the deteriorated glomerular filtration rate. Thus we performed a short-term (1 week) high phosphate diet feeding experiment since, at this time, both serum creatinine levels and urinary NAG activity levels were comparable between the two groups (Figure 2A and B).
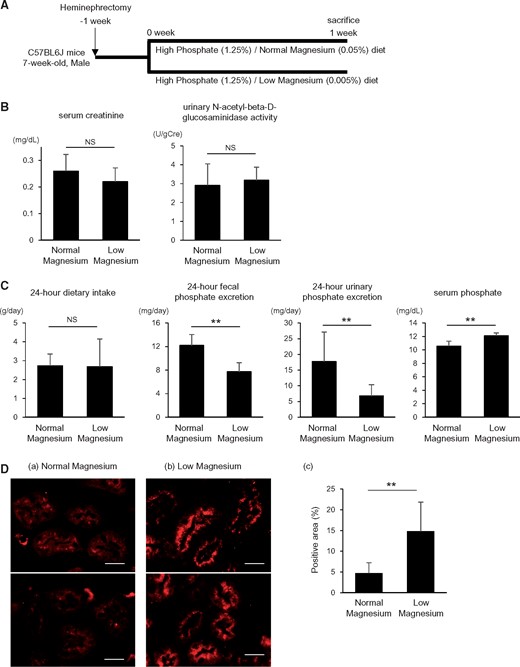
The low magnesium diet impairs urinary phosphate excretion. (A) Heminephrectomized mice were maintained on either a high phosphate (1.25%)/normal magnesium (0.05%) diet or a high phosphate (1.25%)/low magnesium (0.005%) diet for 1 week. (B) At 1 week after initiation of the high phosphate diet, no significant differences were found in serum creatinine levels and urinary NAG activity between the normal and low magnesium diet groups. (C) Despite reduced fecal phosphate excretion, urinary phosphate excretion was suppressed in the low magnesium diet group, resulting in elevated serum phosphate levels. Data are presented as the mean ± standard deviation. Differences between the two groups were compared using Student’s t-test. (D) Representative micrographs showing immunofluorescence staining for type IIa Na-Pi (Npt2a) cotransporter: (a) high phosphate (1.25%)/normal magnesium (0.05%) diet group and (b) high phosphate (1.25%)/low magnesium (0.005%) diet group. Scale bars: 20 µm. (c) The NPT2A-positive areas were increased by the low magnesium diet under high phosphate feeding conditions. Data are presented as the mean ± standard deviation. Differences between the two groups were compared using a Student’s t-test (n = 4–8 in each group; NS, not significant; **P<0.01).
Although the amount of dietary intake was similar, fecal phosphate excretion was reduced in the low magnesium diet group, indicating that the intestinal phosphate absorption was enhanced by the low magnesium intake (Figure 2C). Nevertheless, the 24-h urinary phosphate excretion was substantially decreased by the low magnesium diet (Figure 2C). As a result of the positive phosphate balance, serum phosphate levels were significantly elevated in the low magnesium diet group compared with the normal magnesium diet group (Figure 2C).
Effects of magnesium on phosphaturic hormones
To elucidate the mechanism underlying the impaired urinary phosphate excretion by the low magnesium diet, we measured two major phosphaturic hormones, FGF23 and PTH, after 1 week of feeding the high phosphate diet.
The 1-84 PTH levels were not appropriately elevated in the low magnesium diet group despite lower serum calcium and 1,25-dihydroxyvitamin D levels as compared with the normal magnesium diet group (Figure 3A and B). Serum magnesium levels were very low in the low magnesium diet group (Figure 3B), which could impair the secretion of PTH.
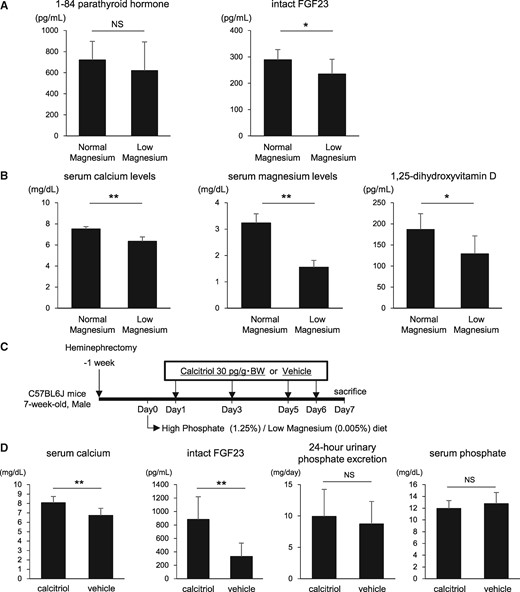
Calcitriol raises FGF23 levels but does not restore the impaired urinary phosphate excretion in the low magnesium diet group. (A) At 1 week after initiation of the high phosphate diet, plasma 1-84 PTH levels and serum intact FGF23 levels were measured using an ELISA. FGF23 levels were significantly reduced after the low magnesium feeding. (B) Serum levels of calcium, magnesium and 1,25-dihydroxyvitamin D were significantly lower in the low magnesium diet group than in the normal magnesium diet group. (C) Mice maintained on the high phosphate/low magnesium diet were injected with calcitriol (30 pg/g body weight) or vehicle on Days 1, 3, 5 and 6. (D) Calcitriol normalized serum calcium levels and markedly raised serum intact FGF23 levels but did not alter urinary phosphate excretion. Data are presented as the mean ± standard deviation. Differences between the two groups were compared using a Student’s t-test. (n = 6–8 in each group; NS, not significant; *P<0.05, **P<0.01).
Intact FGF23 levels were slightly lower in the low-magnesium diet group despite higher serum phosphate levels (Figure 3A). After administration of calcitriol to the low magnesium diet group, serum calcium levels were normalized and FGF23 was dramatically upregulated (Figure 3C and D). However, the calcitriol-induced FGF23 elevation did not restore the impaired urinary phosphate excretion by the low magnesium diet (Figure 3D).
Low magnesium diet downregulates renal α-Klotho expression
Finally, we examined renal expression of α-Klotho, a coreceptor of FGF23. After 1 week of high phosphate feeding, the low magnesium diet substantially reduced renal α-Klotho mRNA expression levels (Figure 4A). Consistently, the immunohistochemical analysis showed much weaker staining for α-Klotho in the low magnesium diet group than in the normal magnesium diet group (Figure 4B). NPT2A expression on proximal tubular brush border membranes was increased in the low magnesium diet group (Figure 2D). Calcitriol administration had little impact on the low magnesium diet–induced downregulation of α-Klotho (Figure 4C).
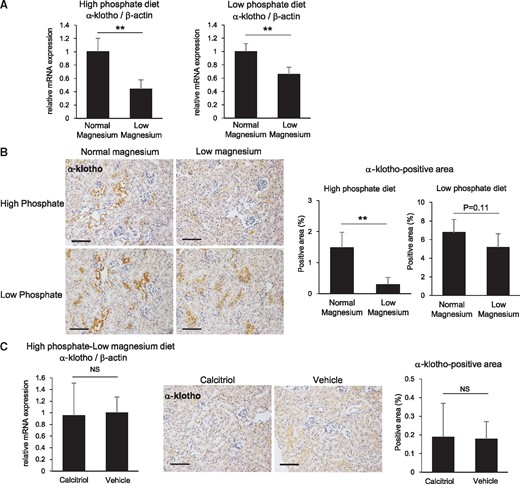
The low magnesium diet downregulates renal α-Klotho expression. (A) At 1 week after initiation of the high and low phosphate diets, the low magnesium diet significantly suppressed renal α-Klotho mRNA expression. (B) Representative immunohistochemical images for α-Klotho in the kidney (scale bar: 200 µm). The α-Klotho-positive areas were significantly reduced by the low magnesium diet under high phosphate feeding conditions and were nonsignificantly reduced under low phosphate feeding conditions. (C) Calcitriol administration did not restore the high phosphate/low magnesium diet–induced downregulation of α-Klotho. Data are presented as the mean ± standard deviation. Differences between the two groups were compared using a Student’s t-test. (n = 5–8 in each group; NS, not significant; **P<0.01).
The low magnesium diet suppressed renal α-Klotho mRNA expression even under the low phosphate diet condition (Figure 4A). The immunohistochemical analysis revealed a consistent result, although the difference did not reach statistical significance (Figure 4B).
DISCUSSION
In this study we found that the low magnesium diet exacerbated high phosphate diet–induced kidney injuries in heminephrectomized mice. Since magnesium is complexed with phosphate in the intestinal tract like a phosphate binder, the low magnesium diet decreased fecal phosphate excretion and enhanced intestinal phosphate absorption. In addition, we found that the low magnesium feeding upregulated NPT2A expression in the proximal tubules and suppressed urinary phosphate excretion, thereby leading to an excessive positive phosphate balance that could contribute to the aggravation of kidney injuries and high mortality rate. This is the first study to show that dietary magnesium deficiency hampers urinary phosphate excretion and downregulates renal α-Klotho expression in the context of CKD.
Under the low phosphate feeding condition, the low magnesium diet did not induce apparent kidney injuries and was not lethal. Therefore the severe kidney injuries and increased mortality rate in the high phosphate/low magnesium diet group were not likely to be due to a direct effect of magnesium deficiency itself, but due to the excessive positive phosphate balance caused by the low magnesium diet. In fact, mice in the high phosphate/low magnesium diet group were debilitated, as is obvious by their decreased body weights, like klotho mutant mice that are characterized by excess phosphate accumulation. Notably, the low magnesium diet aggravated the tubular injuries even at 2 weeks after the high phosphate diet when mice were not yet so weakened; therefore the aggravation of phosphate-induced kidney injuries caused by the low magnesium diet is considered to be independent of the systemic weakness and high lethality caused by this diet.
FGF23, the major phosphaturic hormone, was decreased by the low magnesium diet, presumably due to severe hypocalcemia and low 1,25-dihydroxyvitamin D levels [34]. The reduced FGF23 levels could have been partly involved in the impaired urinary phosphate excretion in the low magnesium diet group. However, the urinary phosphate excretion in the low magnesium diet group remained suppressed even after FGF23 levels were upregulated by calcitriol administration. This suggests that the decrease in FGF23 by the low magnesium diet was not mainly responsible for the impaired urinary phosphate excretion. α-Klotho functions as an FGF23 coreceptor that promotes the binding of FGF23 to its receptors and thereby inhibits phosphate reabsorption in the proximal tubules [35]. We found that the high phosphate diet–induced α-Klotho downregulation was further aggravated by the low magnesium diet, which could explain the blunted response of urinary phosphate excretion to calcitriol-induced FGF23 upregulation. Consistent with our finding, Rodríguez-Ortiz et al. [36] reported that α-Klotho expression in the rat parathyroid glands was suppressed under a low magnesium condition. The mechanism by which magnesium modulates α-Klotho expression is uncertain. It is known that vitamin D–responsive elements exist upstream of the Klotho gene and that calcitriol upregulates α-Klotho expression in the kidney of healthy mice [37]. However, a previous study using a CKD mouse model showed that calcitriol did not ameliorate renal α-Klotho downregulation caused by a high phosphate diet [38]. Similarly, we confirmed that calcitriol did not prevent the low magnesium diet–induced downregulation of α-Klotho. Therefore 1,25-dihydroxyvitamin D deficiency induced by the low magnesium diet is unlikely to be the primary cause of α-Klotho downregulation in our study. In the high phosphate diet experiment, it might be possible that the positive phosphate balance caused by the low magnesium diet, and its consequence of kidney injury aggravation, contributed to the downregulation of α-Klotho. However, renal histology was unremarkable at 1 week after the high phosphate diet when the expression of α-Klotho was severely reduced. Notably, the low magnesium–induced α-Klotho downregulation occurred even when mice were fed the low phosphate diet; thus the excess phosphate accumulation and kidney injuries seem not to be a prerequisite for the low magnesium–induced α-Klotho downregulation. Further studies are required to elucidate how magnesium regulates α-Klotho.
Apart from its impact on phosphate metabolism, α-Klotho downregulation might directly contribute to the aggravation of kidney damage. α-Klotho has been shown to increase resilience against various kinds of insults to the kidney, including phosphate overload [39–41]. This renoprotective action of α-Klotho is supposed to be derived from an antiapoptotic property, autophagy flux upregulation and the suppression of interstitial fibrosis via inhibiting TGF-β1, Wnt and angiotensin II signaling, as well as transient receptor potential canonical 6 channel [39–47]. Therefore the low magnesium diet–induced α-Klotho downregulation might have been directly involved in the exacerbation of phosphate-induced kidney injuries.
Our previous in vitro study showed that magnesium prevents proximal tubular cell death and mitochondrial membrane depolarization induced by phosphate load [30]. In line with this experimental evidence, a retrospective cohort study showed that the risk of CKD progression associated with higher serum phosphate levels was attenuated among patients with higher serum magnesium levels [30]. Therefore the low magnesium status itself may amplify the phosphate toxicity, thus contributing to the aggravation of phosphate-induced kidney injuries in a manner independent of its effects on phosphate metabolism. Unfortunately this direct action of magnesium on kidney injury cannot be proven by the current in vivo study, where many other important factors, such as phosphate, were substantially dysregulated.
In our study, 1-84 PTH levels were similar between the high and low magnesium diet groups. This is surprising given that PTH secretion in the low magnesium diet group should have been strongly stimulated by lower serum calcium and 1,25-dihydroxyvitamin D levels and higher serum phosphate levels. In other words, PTH secretion was inappropriately suppressed by the low magnesium diet. While extracellular magnesium is known to inhibit PTH secretion by acting on calcium-sensing receptors (CaSRs) [36], this mode of action is considered to be largely limited owing to the dysregulated calcium levels, a much stronger agonist of CaSRs. More importantly, intracellular magnesium deficiency is known to impair PTH secretion and its action on the target organs, causing hypoparathyroidism [48]. This phenomenon could have developed in mice fed with the low magnesium diet, partly contributing to the impaired urinary phosphate excretion. Furthermore, since PTH is known to stimulate FGF23 production [49], attenuated PTH signaling might be involved in the lower FGF23 levels of the low magnesium diet group and the further decrease in urinary phosphate excretion.
We have recently reported that hypomagnesemia is not rare among CKD patients [50]. Urinary magnesium wasting could occur in CKD patients, especially with renal tubular injury and proteinuria, leading to low serum magnesium levels despite reduced glomerular filtration rate. The current study supposed that CKD patients with hypomagnesemia are vulnerable to phosphate toxicity. Increasing magnesium levels might restore α-Klotho expression, promote urinary phosphate excretion and eventually mitigate kidney damage. Although magnesium has long received little attention in the clinical practice of CKD, it may serve as a solution to ameliorate phosphate toxicity in CKD patients. Whether magnesium supplementation prevents CKD progression requires clinical trials.
In conclusion, this study demonstrated that a low magnesium diet aggravates high phosphate–induced kidney injuries through not only increasing intestinal phosphate absorption, but also decreasing urinary phosphate excretion. Renal α-Klotho downregulation was likely to be responsible for the low magnesium–induced suppression of urinary phosphate excretion. The mechanism by which magnesium regulates α-Klotho expression needs to be further studied. Our study proposes magnesium as a novel and feasible option to restore α-Klotho expression and prevent the toxicity of phosphate in CKD patients.
ACKNOWLEDGEMENTS
We thank Naoko Horimoto for her technical assistance. This research was supported by JSPS KAKENHI (grant number JP16K19487 to S.Y.) and a scholarship donation from MSD (to S.Y.).
AUTHORS’ CONTRIBUTIONS
Y.S. was responsible for the research idea and study design, contributed to data acquisition and analysis and wrote the draft manuscript. T.H. and I.M. were responsible for study supervision. T.H., I.M., T.O., S.Y., K.K., K.S., A.M., N.H. and Y.I. contributed for revision of the manuscript. Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved.
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments