





Abstract
Salt intake as part of a western diet currently exceeds recommended limits, and the small amount found in the natural diet enjoyed by our Paleolithic ancestors. Excess salt is associated with the development of hypertension and cardiovascular disease, but other adverse effects of excess salt intake are beginning to be recognized, including the development of autoimmune and inflammatory disease. Over the last decade there has been an increasing body of evidence demonstrating that salt affects multiple components of both the innate and adaptive immune systems. In this review we outline the recent laboratory, animal and human data, highlighting the effect of salt on immunity, with a particular focus on the relevance to inflammatory kidney disease.
INTRODUCTION
Salt [sodium chloride (NaCl)] has been a prominent feature of the human diet for a relatively short time, at least in evolutionary terms. Our ancestors survived for millions of years on a diet that contained <1 g of salt per day. Salt is required for the maintenance of extracellular and therefore plasma volume, and given the limited salt composition of this natural diet, humans evolved under an intense evolutionary pressure for the selection of salt-conserving genes. Salt was discovered as a way of preserving food ∼5000 years ago and since then salt intake has increased rapidly to current estimates of 8 g/day in the UK, with even greater intake in other parts of the world [1]. Most of this salt is found in processed foods and restaurant meals; the remaining salt is added while cooking and eating. Current salt intake as part of a western diet is therefore far in excess of the amount we are physiologically programmed to handle.
Given our ability to retain salt, in steady state the minimum amount required to replace losses is <0.5 g/day. There is good evidence from both observational and interventional studies that excess salt intake is associated with the development of hypertension [2, 3], it is generally accepted that excess salt intake is linked to the development of cardiovascular disease [4], and there is some evidence for a role in the progression of chronic kidney disease (CKD) [5]. Based on this, the World Health Organization recommends limiting salt intake to <5 g/day, which it estimates could prevent 2.5 million deaths globally each year.
There are, however, perhaps less well-appreciated adverse effects of excess salt intake that are becoming apparent. These include the development of malignancy, in particular gastric cancer [6], obesity and metabolic syndrome [7]. Excess salt is also implicated in autoimmune and inflammatory disease. Observational studies have demonstrated that increased salt intake is associated with the development of rheumatoid arthritis [8] and correlates with disease severity in multiple sclerosis [9]. Furthermore, in renal transplantation, perioperative salt loading worsens short-term outcomes [10] and adherence to a low salt diet improves renal outcomes and survival in the longer term [11]. In this review we outline the recent data from laboratory, animal and human studies that demonstrate an effect of salt on both innate and adaptive immunity and postulate how these findings may be relevant in inflammatory kidney disease.
SALT AND INNATE IMMUNITY
Initial evidence of an effect of salt on macrophages
The mechanism by which excess salt causes hypertension is unclear and cannot be fully explained by an effect on extracellular volume [12]. Over the last decade, however, two concepts have been described that not only provide novel insights into the pathogenesis of hypertension, but also provide the initial evidence of salt impacting immune cells. First, it has been shown that salt loading results in sodium storage at interstitial sites within muscle and skin [13, 14]. This results in sodium concentrations in interstitial compartments (up to 250 mM) in excess of that found in plasma, where sodium is tightly regulated due to its effect on osmolality. Second, is the increasing awareness of a role for inflammation in the development of hypertension.
A link between both these concepts was demonstrated in a seminal paper in 2009 that demonstrated a high salt diet in rats results in elevated skin sodium concentrations, which in turn activates resident macrophages, which through production of vascular endothelial growth factor C, clear this stored sodium through the lymphatic system [13]. Macrophage depletion promoted skin sodium storage and the development of hypertension. Hence salt loading leads to storage at interstitial sites, with NaCl concentrations in excess of plasma, resulting in immune cell activation and salt clearance via the lymphatic system. Either excess salt or impaired immune-mediated salt clearance can lead to hypertension.
Further studies on the effect of salt on macrophage activation and its relevance in the clearance of cutaneous infection
Following this initial work, there have been several other studies investigating how salt affects macrophage activation. Like other cells of the immune system, macrophages display a spectrum of activation states with plasticity along this spectrum. At one end are pro-inflammatory M1 macrophages, which secrete inflammatory cytokines interleukin-1 (IL-1), IL-6, IL-12 and tumour necrosis factor-α (TNF-α) and activate effector T cells (Teffs), and at the other end are reparative M2 macrophages that secrete anti-inflammatory cytokines IL-10 and transforming growth factor-β (TGF-β), with reduction in Teffs and an increase in regulatory T cell (Treg) activity [15]. M2 macrophage activity is important in wound healing, and experimentally, they have been shown to reduce the development of autoimmune disease [16, 17].
Salt has been shown to have a pro-inflammatory effect on macrophages, increasing M1 activation and reducing M2 activation (Figure 1) [18, 19]. In cell culture, the addition of 40 mM NaCl increases M1 macrophage activation and this is dependent on a signalling pathway involving p38 mitogen-activated protein kinase (MAPK) and the transcription factor nuclear factor of activated T-cells 5 (NFAT5; also known as tonicity-responsive enhancer binding protein) [18]. Sodium accumulates at sites of skin infection in mice. In humans, adding NaCl to cell cultures in vitro led to increased M1 clearance of infection, and a high salt diet in animals resulted in enhanced clearance of cutaneous leishmaniasis. In contrast, M2 activation is blunted in vitro in high salt culture conditions, and a high salt diet is associated with reduced wound healing and reduced M2 activity in a mouse model of peritonitis [19]. Unlike the salt effect on M1 macrophages, and indeed that on T helper 17 (Th17) cells, which is dependent on NFAT5, the inhibitory salt effect on M2 macrophages is dependent on a reduction in signalling through a pathway involving protein kinase B (PKB/Akt) and the mammalian target of rapamycin.
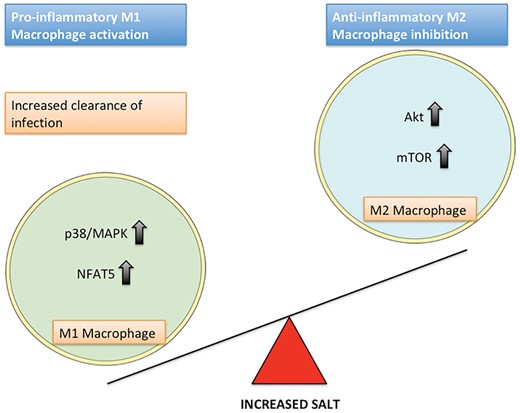
The effect of salt on M1 and M2 macrophage polarization and the intracellular pathways involved.
Taken together, these data on macrophage activation states and high interstitial NaCl concentrations provide insight into the potential evolutionary reasons why the immune system responds to salt. Infection is associated with the creation of localized hypersalinity, which allows for local activation of pro-inflammatory cells while limiting such activation in the body more generally. A similar high salt environment is created at sites of body–environmental interface (e.g. skin, kidney, genitourinary tract), providing protection from pathogens at these locations.
The effect of salt on renal mononuclear phagocytes and the development of pyelonephritis
Further evidence for this role of hypersalinity in providing protection from infection was provided in a recent study that assessed the role of interstitial sodium concentrations on tissue resident mononuclear phagocytes (MNPs) in the kidney [20]. Tissue MNPs include distinct subsets of macrophages and dendritic cells. Berry et al. [20] demonstrated that CD14+ MNPs, a subset of MNPs with enhanced phagocytic capability, were enriched in the medulla compared with the cortex of human kidneys; this positioning was dependent on NFAT5-mediated production of the chemokines chemokine ligand 2 (CCL2) and chemokine ligand 1 (CX3CL1) by renal tubular epithelial cells in response to hypersalinity. Medullary CD14+ MNPs demonstrated increased phagocytosis of uropathogenic Escherichia coli and phagocytic activity increased with increasing extracellular sodium concentrations in an NFAT5-dependent manner. The induction of nephrogenic diabetes insipidus (NDI) resulted in increased bacteraemia and death following intravesical uropathogenic E. coli challenge in mice. This was interpreted as being due to disruption of the medullary renal sodium gradient. Moreover, NDI in patients, either in the setting of tolvaptan use or as a result of sickle cell disease, is associated with increased urinary tract infection risk [20]. Taken together, these data demonstrate that high interstitial salt concentrations in the kidney, primarily required for water reabsorption and the maintenance of extracellular volume, also promote medullary localization of specific subsets of MNPs, which in turn provide protection from infection. It is tempting to speculate that autoimmune kidney disease might arise in part due to the hypertonic environment found in the kidneys, created through evolution for these volume and infectious purposes.
SALT AND ADAPTIVE IMMUNITY
CD4+ T cell polarization and the imbalance of Th17 cells and Tregs in inflammatory kidney disease
Antigen is presented to naïve CD4+ T cells on major histocompatibility complex class II (MHC II). This is done by a select group of antigen-presenting cells, including dendritic cells, macrophages and B cells. After antigen presentation, naïve CD4+ T cells become activated and polarized towards one of the number of CD4+ T cell subsets, including Th1, Th2, Th17 and Tregs, each of which has different effects on the immune response, in part through the cell-specific cytokines produced.
Th17 cells are pro-inflammatory and provide protection against extracellular bacterial and fungal infections, particularly at epithelial cell surfaces [21]. Polarization requires stimulation by IL-1β, IL-6 and TGF-β, whereas full maturation requires IL-21 and IL-23. Th17 cells secrete various cytokines, including IL-17, which acts on stromal cells and is important for neutrophil recruitment, and others such as IL-26 and IL-22, which act on non-immune cells, enhancing inflammation or serving to attenuate damage, depending on the tissue and environment. Tregs have opposing effects to Th17 cells. Naturally occurring and peripherally induced Tregs are characterized by expression of the transcription factor Forkhead Box P3 (FOXP3) and provide peripheral tolerance through inhibiting the function of effector CD4+ and CD8+ T cells, B cells and cells of the innate immune system through cell–cell contact mechanisms and release of suppressive cytokines (e.g. TGF-β, IL-10) [22].
Th17 cells and Tregs both play a major role in autoimmune kidney disease and renal transplant rejection. Th17 cells are implicated in the development of both glomerular and tubulointerstitial disease in patients and animal models, including anti-neutrophil cytoplasmic antibody–associated glomerulonephritis [23, 24], lupus nephritis [25], immunoglobulin A nephropathy [26] and primary Sjögren’s syndrome–associated tubulointerstitial nephritis [27]. Conversely, Treg activity limits inflammatory glomerular disease and autoimmune disease relapses, both clinically and in experimental models [28]. Furthermore, it is becoming increasingly clear that excessive Th17 and reduced Treg activity are important components in the development of acute and chronic allograft rejection [29]. Hence excess Th17 activation and reduced Treg activity underlie a number of inflammatory kidney diseases in both the native and transplant setting. This augmented Th17 and attenuated Treg phenomenon is the same effect that salt has on these CD4+ T cell subtypes, as we describe below.
The effect of salt on Th17 cells and the development of multiple sclerosis
Two studies published in Nature in 2013 provided the initial evidence of an effect of salt on Th17 polarization [30, 31]. Kleinewietfeld et al. [30] isolated naïve CD4+ T cells from healthy volunteers and cultured these for 7 days in optimal Th17 polarizing conditions in standard media and in media supplemented with varying concentrations of NaCl (0–80 mM). There was a dose-dependent increase in Th17 polarization with salt up to 40 mM NaCl, which provided optimal polarization and cell survival. The authors concluded, on the basis of control experiments in which media was supplemented with sodium gluconate, magnesium chloride and mannitol, that sodium, as opposed to chloride or tonicity, was mediating this pro-inflammatory effect. Further in vitro work showed that salt stimulation affected naïve rather than memory CD4+ T cells, did not influence Th1 and Th2 polarization and not only polarized Th17 cells, but also promoted a pathogenic Th17 phenotype (increased expression of IL-2, TNF-α, IL-9 and colony-stimulating factor 2/granulocyte-macrophage colony-stimulating factor). It was subsequently shown that the intracellular pathway mediating this response to hypersalinity involved upregulation of phosphorylated p38 MAPK, NFAT5 and serum- and glucocorticoid-regulated kinase 1 (SGK1) (Figure 2). Knocking down any of these molecules resulted in a sharp reduction in salt-mediated Th17 polarization. The authors then investigated the salt effect on Th17 cells in vivo in an animal model of multiple sclerosis [experimental autoimmune encephalitis (EAE)], a Th17-mediated disease. A high salt diet exacerbated disease severity, and this was associated with increased Th17 cells in the central nervous system (CNS). The use of a p38 MAPK inhibitor abrogated both salt-induced Th17 CNS infiltration and clinical markers of disease severity.
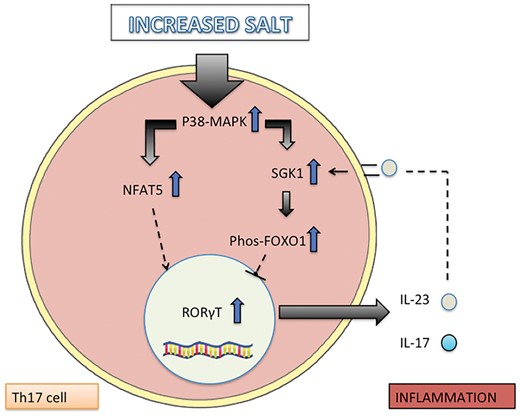
Intracellular pathways involved in salt-induced Th17 polarization.
Wu et al. [32] focused further on the role of SGK1 in salt-mediated Th17 polarization. They described the pathway downstream of SGK1, which involves phosphorylation of Forkhead box protein O1 (FOXO1) and upregulation of the main Th17 transcription factor, RAR-related orphan receptor gamma T (RORγT) (Figure 2). They also confirmed the polarizing effect of salt on Th17 cells in vitro and demonstrated that this Th17 polarization was prevented in SGK1 knockout cells. They showed that a high salt diet in mice is associated with increased gut Th17 cell frequency, confirmed that a high salt diet exacerbates EAE and showed that this was prevented in SGK1 knockout mice. Interestingly, since this initial work, the same group has demonstrated a pivotal role of SGK1 in mediating the balance between Th17 and Tregs in isotonic conditions [32]. Taken together, these initial studies demonstrate that Th17 cell polarization is enhanced by high salt conditions both in vitro and in vivo, that this is dependent on signalling through p38 MAPK, NFAT5 and SGK1 and that salt-mediated Th17 polarization exacerbates animal models of autoimmune disease.
The effect of salt on suppressor Treg function and transplant rejection
Given the interrelationship between Th17 cells and Tregs and the effect of salt on Th17 polarization, subsequent work investigated whether there was an effect of salt on Treg function, first in vitro and then in mouse models of graft-versus-host disease (GVHD), colitis and mismatched cardiac transplantation [33, 34]. Hernandez et al. [33] demonstrated that the addition of salt to co-culture of Tregs with effector T cells inhibited regulatory cell function in a dose-dependent manner over the range of 10–40 mM NaCl. Further analysis of purified Tregs cultured under high salt conditions demonstrated that the mechanism of this loss of suppressor function was a Treg shift to a Th1 phenotype with upregulation of pro-inflammatory molecules such as interferon γ (IFN-γ); knockdown of IFN-γ resulted in maintenance of the suppressor function. The authors demonstrated that the same pathway increased SGK1 activity and phosphorylation of FOXO1 and FOXO3, which is involved in Th17–Treg balance in isotonic conditions, and in the polarization of Th17 in hypertonic conditions, which also mediates the salt effect on Tregs. Inhibition or knockdown of SGK1 resulted in recovery of the suppressor function; high salt conditions increased phosphorylation of FOXO1 and FOXO3, which resulted in impaired stabilization of the FOXP3 locus. This inhibitory effect of salt on Tregs was then confirmed in vivo. A high salt diet was associated with increased IFN-γ-secreting Tregs in the spleen and mesenteric lymph nodes, and this was associated with increased SGK1 expression. Moreover, a high salt diet increased IFN-γ-secreting Tregs and worsened disease in a mouse model of GVHD, while transfer of Tregs cultured under high salt, as opposed to standard conditions, led to exacerbation of disease in an animal model of colitis [33].
Safa et al. [34] investigated the effect of high salt on the alloimmune response, using a mouse model of MHC II–mismatched cardiac transplantation, which is dependent on Treg suppressor function for graft survival. Despite having no effect on blood pressure, a high salt diet reduced allograft survival, and this was associated with a reduction in Tregs within the transplant and the spleen. This high salt effect on rejection was abolished in SGK1 knockout mice. Together, these initial Treg studies demonstrate an inhibitory effect of salt on Treg suppressor function, which is mediated through an SGK1–FOXO1/3 pathway. High salt exacerbates animal models of autoimmune disease and worsens murine transplant rejection.
The effect of salt on the development of autoimmune diseases
Subsequent studies have investigated further aspects of the effect of salt on immunity in models of multiple sclerosis (EAE), colitis and lupus nephritis and also in the setting of the progression of acute kidney injury (AKI) to CKD. The results of this work are summarized below.
Animal models of multiple sclerosis
Wilck et al. [35] proposed an alternative indirect in vivo mechanism by which high salt affects Th17 cells and the development of EAE. This was through alteration in the gut microbiome and provides a link to studies that demonstrate gut-derived Th17 cells mediate the development of extra-intestinal disease, including inflammatory kidney disease [36]. Salt loading inhibited the growth of Lactobacillus sp. in vitro in mice and in healthy human volunteers. In an EAE model, Lactobacillus administration ameliorated Th17 CNS infiltration and the development of disease. The mechanism of the effect on Th17 polarization was proposed to be due to reduced bacterial production of indole derivatives, which have previously been shown to associate with improvement in EAE. Hence multiple extracellular cues may affect Th17 polarization and several pathways may mediate the pro-inflammatory salt effect in vivo.
Jorg et al. [37] investigated whether alteration of dendritic cell function occurred in response to salt and mediated the in vitro effects of salt on Th17 and the development of EAE. They found no effect on dendritic cell function, suggesting the salt effect on Th17 is a direct effect on Th17 cells themselves. In contrast, Barbaro et al. [38] demonstrated that high salt conditions increased both macrophage and dendritic cell activation and that co-culture of salt activated dendritic cells with T cells from salt-sensitive mice led to increased pro-inflammatory cytokine production (IL-17 and IFN-γ) from CD4+ and CD8+ cells. The intracellular pathway mediating this effect involved sodium entry into dendritic cells through amiloride-sensitive sodium channels, formation of reactive oxygen species and the production of isolevuglandin protein adducts, which ultimately enhanced antigen presentation to T cells.
Animal models of inflammatory bowel disease
Several studies have investigated the role of salt on intestinal immunity and the development of colitis, perhaps unsurprising given the abundance of Th17 cells within the gut [33, 39, 40]. In these studies, salt loading of mice without disease results in increased gut Th17 cells and reduced Treg suppressor function. This is associated with increased SGK1 expression, increased gut permeability and histological features of inflammation. High salt has also been shown to worsen mouse models of inflammatory bowel disease, associated with increased gut IL-17. One study investigated the source of this IL-17 and demonstrated that salt not only affects CD4+ T cells, but also innate lymphoid cells (ILCs), in particular ILC3, the innate lymphoid equivalent of Th17 cells [40]. Indeed, recombination-activating gene knockout (i.e. lymphocyte depleted) mice still get IL-17-mediated gut inflammation in response to a high salt diet, highlighting that salt promotes IL-17 production from multiple immune cells.
Animal models of systemic lupus erythematosus
Elevated salt concentrations in vitro have been shown to increase follicular helper T cell polarization and a high salt diet worsened lupus features in an animal model [41]. A further study demonstrated that a high salt diet leads to exacerbated experimental lupus nephritis, with more significant disease on histology, increased proteinuria and worse survival [42]. This was associated with increased Th17 and Th1 cells in the spleen and fewer Tregs. The salt response of isolated CD4+ T cells from patients with systemic lupus erythematosus was then analysed. Th17 cell polarization increased with anti-CD3 and anti-CD28 stimulation in the presence of NaCl. This, as in healthy volunteers, was abrogated by the addition of an SGK1 inhibitor. Of note, high salt conditions do not promote increased polarization of naïve T cells to Th1 cells in vitro under Th1 polarizing conditions [43], but this and other studies have shown a high salt diet does promote a Th1 phenotype in vivo [42, 43]. This may reflect CD4+ T cell plasticity, with Th17 cells or Tregs developing a pro-inflammatory Th1 phenotype with expression of IFN-γ in response to high salt conditions.
Animal models of AKI
AKI, particularly if recurrent, can lead to CKD, and high salt has been shown to increase the progression of AKI to CKD in animal models [44]. The mechanism behind this phenomenon is unclear but has been proposed to be immune mediated, as immunosuppression with mycophenolate mofetil abrogates the salt effect. More recent work has demonstrated that Th17 cells mediate interstitial inflammation in AKI; salt loading worsened Th17-mediated inflammation and this resulted in more significant CKD [45]. This was prevented by angiotensin receptor blockade, which was shown in vitro to directly affect Th17 cells.
Human studies of dietary salt modification and their effect on Th17–Treg balance
Given the growing body of laboratory and animal work that have demonstrated a polarizing effect of salt on diverse immune cells (summarized in Table 1), using dietary modifications to limit salt intake in the management of patients with autoimmune disease provides an intriguing and potentially beneficial, low-cost therapeutic avenue. Initial interventional studies have investigated the effect of changing salt intake on the immune response in healthy volunteers and in patients with autoimmune disease. The results of these studies are outlined in Table 2. Together, these demonstrate dietary salt modification may be used to modulate the immune response to varying degrees, but whether this translates to changes in clinical outcomes is yet to be determined. Moreover, where salt exerts its pro-inflammatory effect on immune cells in vivo is unknown.
Summary of the known effects of salt on immune cell function, the intracellular pathways involved and the in vivo clinical consequences of the high salt effect
| Cell type | Salt effect | Intracellular signalling molecules and pathways mediating the salt effect | Clinical consequences of high salt effect in vivo |
|---|---|---|---|
| Innate immune system | |||
| M1 macrophage [18] | Activation | p38 MAPK, NFAT5 | Enhanced clearance of cutaneous infection |
| M2 macrophage [19] | Inhibition | Akt, mammalian target of rapamycin | Reduced wound healing |
| Monocytes [46] | Expansion of CD14++CD16+ (intermediate) monocytes | Formation of reactive oxygen species | Renal hypoxia and inflammation |
| Tissue resident MNPs [20] | Increased phagocytic activity of CD14+ MNPs | NFAT5 | Protection against pyelonephritis |
| Dendritic cell [38] | Activation | Nicotinamide adenine dinucleotide phosphate oxidase (reactive oxygen species formation), formation of isolevuglandin protein adducts | Hypertension |
| Neutrophils [40] | Activation as a consequence of increased IL-23 production (direct effect unknown) | Unknown | Exacerbated gut inflammation |
| ILCs [40] | Activation of ILC3 | Unknown | Exacerbated gut inflammation |
| Adaptive immune system | |||
| Th17 [30, 31, 39, 40, 42, 45] | Activation | p38 MAPK, NFAT5, SGK1, FOXO1 | Exacerbated EAE, colitis and lupus nephritis. Increased progression AKI to CKD |
| Treg [33, 34] | Inhibition | SGK1, FOXO1/3 | Exacerbated transplant rejection, EAE |
| Th1 [42] | Increased activity in vivo, possibly conversion from Th17 or Treg (no direct effect in vitro) | N/A | Exacerbated lupus nephritis |
| Th2 | No effect [30] | N/A | N/A |
| Follicular helper T cell [41] | Activation | TET2 | Exacerbated SLE |
| CD8+ T cell [38] | Increased IFN-γ and IL-17 from CD8+ cells from salt-sensitive mice when cultured with salt primed dendritic cells | Unknown | Unknown |
| B cells | Unknown | Unknown | Unknown |
| Cell type | Salt effect | Intracellular signalling molecules and pathways mediating the salt effect | Clinical consequences of high salt effect in vivo |
|---|---|---|---|
| Innate immune system | |||
| M1 macrophage [18] | Activation | p38 MAPK, NFAT5 | Enhanced clearance of cutaneous infection |
| M2 macrophage [19] | Inhibition | Akt, mammalian target of rapamycin | Reduced wound healing |
| Monocytes [46] | Expansion of CD14++CD16+ (intermediate) monocytes | Formation of reactive oxygen species | Renal hypoxia and inflammation |
| Tissue resident MNPs [20] | Increased phagocytic activity of CD14+ MNPs | NFAT5 | Protection against pyelonephritis |
| Dendritic cell [38] | Activation | Nicotinamide adenine dinucleotide phosphate oxidase (reactive oxygen species formation), formation of isolevuglandin protein adducts | Hypertension |
| Neutrophils [40] | Activation as a consequence of increased IL-23 production (direct effect unknown) | Unknown | Exacerbated gut inflammation |
| ILCs [40] | Activation of ILC3 | Unknown | Exacerbated gut inflammation |
| Adaptive immune system | |||
| Th17 [30, 31, 39, 40, 42, 45] | Activation | p38 MAPK, NFAT5, SGK1, FOXO1 | Exacerbated EAE, colitis and lupus nephritis. Increased progression AKI to CKD |
| Treg [33, 34] | Inhibition | SGK1, FOXO1/3 | Exacerbated transplant rejection, EAE |
| Th1 [42] | Increased activity in vivo, possibly conversion from Th17 or Treg (no direct effect in vitro) | N/A | Exacerbated lupus nephritis |
| Th2 | No effect [30] | N/A | N/A |
| Follicular helper T cell [41] | Activation | TET2 | Exacerbated SLE |
| CD8+ T cell [38] | Increased IFN-γ and IL-17 from CD8+ cells from salt-sensitive mice when cultured with salt primed dendritic cells | Unknown | Unknown |
| B cells | Unknown | Unknown | Unknown |
Summary of the known effects of salt on immune cell function, the intracellular pathways involved and the in vivo clinical consequences of the high salt effect
| Cell type | Salt effect | Intracellular signalling molecules and pathways mediating the salt effect | Clinical consequences of high salt effect in vivo |
|---|---|---|---|
| Innate immune system | |||
| M1 macrophage [18] | Activation | p38 MAPK, NFAT5 | Enhanced clearance of cutaneous infection |
| M2 macrophage [19] | Inhibition | Akt, mammalian target of rapamycin | Reduced wound healing |
| Monocytes [46] | Expansion of CD14++CD16+ (intermediate) monocytes | Formation of reactive oxygen species | Renal hypoxia and inflammation |
| Tissue resident MNPs [20] | Increased phagocytic activity of CD14+ MNPs | NFAT5 | Protection against pyelonephritis |
| Dendritic cell [38] | Activation | Nicotinamide adenine dinucleotide phosphate oxidase (reactive oxygen species formation), formation of isolevuglandin protein adducts | Hypertension |
| Neutrophils [40] | Activation as a consequence of increased IL-23 production (direct effect unknown) | Unknown | Exacerbated gut inflammation |
| ILCs [40] | Activation of ILC3 | Unknown | Exacerbated gut inflammation |
| Adaptive immune system | |||
| Th17 [30, 31, 39, 40, 42, 45] | Activation | p38 MAPK, NFAT5, SGK1, FOXO1 | Exacerbated EAE, colitis and lupus nephritis. Increased progression AKI to CKD |
| Treg [33, 34] | Inhibition | SGK1, FOXO1/3 | Exacerbated transplant rejection, EAE |
| Th1 [42] | Increased activity in vivo, possibly conversion from Th17 or Treg (no direct effect in vitro) | N/A | Exacerbated lupus nephritis |
| Th2 | No effect [30] | N/A | N/A |
| Follicular helper T cell [41] | Activation | TET2 | Exacerbated SLE |
| CD8+ T cell [38] | Increased IFN-γ and IL-17 from CD8+ cells from salt-sensitive mice when cultured with salt primed dendritic cells | Unknown | Unknown |
| B cells | Unknown | Unknown | Unknown |
| Cell type | Salt effect | Intracellular signalling molecules and pathways mediating the salt effect | Clinical consequences of high salt effect in vivo |
|---|---|---|---|
| Innate immune system | |||
| M1 macrophage [18] | Activation | p38 MAPK, NFAT5 | Enhanced clearance of cutaneous infection |
| M2 macrophage [19] | Inhibition | Akt, mammalian target of rapamycin | Reduced wound healing |
| Monocytes [46] | Expansion of CD14++CD16+ (intermediate) monocytes | Formation of reactive oxygen species | Renal hypoxia and inflammation |
| Tissue resident MNPs [20] | Increased phagocytic activity of CD14+ MNPs | NFAT5 | Protection against pyelonephritis |
| Dendritic cell [38] | Activation | Nicotinamide adenine dinucleotide phosphate oxidase (reactive oxygen species formation), formation of isolevuglandin protein adducts | Hypertension |
| Neutrophils [40] | Activation as a consequence of increased IL-23 production (direct effect unknown) | Unknown | Exacerbated gut inflammation |
| ILCs [40] | Activation of ILC3 | Unknown | Exacerbated gut inflammation |
| Adaptive immune system | |||
| Th17 [30, 31, 39, 40, 42, 45] | Activation | p38 MAPK, NFAT5, SGK1, FOXO1 | Exacerbated EAE, colitis and lupus nephritis. Increased progression AKI to CKD |
| Treg [33, 34] | Inhibition | SGK1, FOXO1/3 | Exacerbated transplant rejection, EAE |
| Th1 [42] | Increased activity in vivo, possibly conversion from Th17 or Treg (no direct effect in vitro) | N/A | Exacerbated lupus nephritis |
| Th2 | No effect [30] | N/A | N/A |
| Follicular helper T cell [41] | Activation | TET2 | Exacerbated SLE |
| CD8+ T cell [38] | Increased IFN-γ and IL-17 from CD8+ cells from salt-sensitive mice when cultured with salt primed dendritic cells | Unknown | Unknown |
| B cells | Unknown | Unknown | Unknown |
Summary of studies investigating the immunological effects of dietary salt modification in healthy human volunteers and in patients with autoimmune disease
| References | Subjects included | Dietary modification | Main results |
|---|---|---|---|
| Zhou et al. [46] | Healthy volunteers (n = 20) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | Increase in CD14++CD16+ (intermediate) monocytes; decrease in CD14++CD16−(classical) and CD14+CD16++ (non-classical) monocytes with high salt intake |
| Yi et al. [47] (part of Mars flight simulation study) | Healthy volunteers (n = 6) | Fixed daily salt intake of 12 g, 9 g, then 6 g for 1–2 months, then reverted back to 12 g for 1 month. Food provided to subjects | Serum cytokines analysed after each intervention period: increased pro-inflammatory cytokines (IL-6 and IL-23) and reduced anti-inflammatory cytokines (IL-10) with high salt intake |
| Luo et al. [48] | Healthy volunteers (n = 15) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | T cell subset analysis: increased Th17 and reduced Treg populations with high salt intake. Associated with increased expression of NFAT5 and SGK1 |
| Wen et al. [49] | Healthy volunteers (n = 49) | Fixed daily salt intake of 3 g (low salt) for 7 days, then 7 days high salt (18 g), then 7 days high salt (18 g) plus potassium supplementation (4.5 g KCl; 60 mmol K) | Serum IL-17 and PBMC IL-17 mRNA increased with high salt. Associated with increased SGK1. High salt effect abolished with potassium supplementation of diet |
| Scrivo et al. [50] | Patients with RA (n = 15) and SLE (n = 15) | Dietary counselling used to limit daily salt intake to 5 g for 3 weeks, then reverted back to standard diet for 2 weeks. 24-h urine Na measurement used to assess adherence to diet | RA patients: no effect in Th17 or Treg subsets. SLE patients: increase in Treg after salt restriction; no effect on Th17 cells |
| References | Subjects included | Dietary modification | Main results |
|---|---|---|---|
| Zhou et al. [46] | Healthy volunteers (n = 20) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | Increase in CD14++CD16+ (intermediate) monocytes; decrease in CD14++CD16−(classical) and CD14+CD16++ (non-classical) monocytes with high salt intake |
| Yi et al. [47] (part of Mars flight simulation study) | Healthy volunteers (n = 6) | Fixed daily salt intake of 12 g, 9 g, then 6 g for 1–2 months, then reverted back to 12 g for 1 month. Food provided to subjects | Serum cytokines analysed after each intervention period: increased pro-inflammatory cytokines (IL-6 and IL-23) and reduced anti-inflammatory cytokines (IL-10) with high salt intake |
| Luo et al. [48] | Healthy volunteers (n = 15) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | T cell subset analysis: increased Th17 and reduced Treg populations with high salt intake. Associated with increased expression of NFAT5 and SGK1 |
| Wen et al. [49] | Healthy volunteers (n = 49) | Fixed daily salt intake of 3 g (low salt) for 7 days, then 7 days high salt (18 g), then 7 days high salt (18 g) plus potassium supplementation (4.5 g KCl; 60 mmol K) | Serum IL-17 and PBMC IL-17 mRNA increased with high salt. Associated with increased SGK1. High salt effect abolished with potassium supplementation of diet |
| Scrivo et al. [50] | Patients with RA (n = 15) and SLE (n = 15) | Dietary counselling used to limit daily salt intake to 5 g for 3 weeks, then reverted back to standard diet for 2 weeks. 24-h urine Na measurement used to assess adherence to diet | RA patients: no effect in Th17 or Treg subsets. SLE patients: increase in Treg after salt restriction; no effect on Th17 cells |
KCl, potassium chloride; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Summary of studies investigating the immunological effects of dietary salt modification in healthy human volunteers and in patients with autoimmune disease
| References | Subjects included | Dietary modification | Main results |
|---|---|---|---|
| Zhou et al. [46] | Healthy volunteers (n = 20) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | Increase in CD14++CD16+ (intermediate) monocytes; decrease in CD14++CD16−(classical) and CD14+CD16++ (non-classical) monocytes with high salt intake |
| Yi et al. [47] (part of Mars flight simulation study) | Healthy volunteers (n = 6) | Fixed daily salt intake of 12 g, 9 g, then 6 g for 1–2 months, then reverted back to 12 g for 1 month. Food provided to subjects | Serum cytokines analysed after each intervention period: increased pro-inflammatory cytokines (IL-6 and IL-23) and reduced anti-inflammatory cytokines (IL-10) with high salt intake |
| Luo et al. [48] | Healthy volunteers (n = 15) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | T cell subset analysis: increased Th17 and reduced Treg populations with high salt intake. Associated with increased expression of NFAT5 and SGK1 |
| Wen et al. [49] | Healthy volunteers (n = 49) | Fixed daily salt intake of 3 g (low salt) for 7 days, then 7 days high salt (18 g), then 7 days high salt (18 g) plus potassium supplementation (4.5 g KCl; 60 mmol K) | Serum IL-17 and PBMC IL-17 mRNA increased with high salt. Associated with increased SGK1. High salt effect abolished with potassium supplementation of diet |
| Scrivo et al. [50] | Patients with RA (n = 15) and SLE (n = 15) | Dietary counselling used to limit daily salt intake to 5 g for 3 weeks, then reverted back to standard diet for 2 weeks. 24-h urine Na measurement used to assess adherence to diet | RA patients: no effect in Th17 or Treg subsets. SLE patients: increase in Treg after salt restriction; no effect on Th17 cells |
| References | Subjects included | Dietary modification | Main results |
|---|---|---|---|
| Zhou et al. [46] | Healthy volunteers (n = 20) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | Increase in CD14++CD16+ (intermediate) monocytes; decrease in CD14++CD16−(classical) and CD14+CD16++ (non-classical) monocytes with high salt intake |
| Yi et al. [47] (part of Mars flight simulation study) | Healthy volunteers (n = 6) | Fixed daily salt intake of 12 g, 9 g, then 6 g for 1–2 months, then reverted back to 12 g for 1 month. Food provided to subjects | Serum cytokines analysed after each intervention period: increased pro-inflammatory cytokines (IL-6 and IL-23) and reduced anti-inflammatory cytokines (IL-10) with high salt intake |
| Luo et al. [48] | Healthy volunteers (n = 15) | 3-day run-in on normal diet, then fixed daily salt intake of 15 g (high salt) for 7 days, then 5 g (low salt) for 7 days. Food provided to subjects | T cell subset analysis: increased Th17 and reduced Treg populations with high salt intake. Associated with increased expression of NFAT5 and SGK1 |
| Wen et al. [49] | Healthy volunteers (n = 49) | Fixed daily salt intake of 3 g (low salt) for 7 days, then 7 days high salt (18 g), then 7 days high salt (18 g) plus potassium supplementation (4.5 g KCl; 60 mmol K) | Serum IL-17 and PBMC IL-17 mRNA increased with high salt. Associated with increased SGK1. High salt effect abolished with potassium supplementation of diet |
| Scrivo et al. [50] | Patients with RA (n = 15) and SLE (n = 15) | Dietary counselling used to limit daily salt intake to 5 g for 3 weeks, then reverted back to standard diet for 2 weeks. 24-h urine Na measurement used to assess adherence to diet | RA patients: no effect in Th17 or Treg subsets. SLE patients: increase in Treg after salt restriction; no effect on Th17 cells |
KCl, potassium chloride; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
CONCLUSION
The immune system consists of opposing pro-inflammatory and anti-inflammatory cells. Salt affects the activity of multiple cells of both innate and adaptive immunity, promoting predominantly pro-inflammatory subtypes. In vitro, salt increases activation of M1 macrophages and Th17 cells, whereas it suppresses M2 macrophage and Treg function. Intracellular molecules that mediate this effect include NFAT5 and SGK1. The in vivo consequences of hypersalinity are increased clearance of infection, but also the development of autoimmune disease. Whether dietary salt manipulation can be used as an adjunctive therapy in inflammatory kidney diseases or transplantation remains to be investigated.
FUNDING
RE is a Kidney Research UK funded Clinical Training Fellow (Grant code: TF_007_20161125).
CONFLICT OF INTEREST STATEMENT
None declared.


{kind=link}
{kind=link}
Comments