





Abstract
Fibrosis, a characteristic of all chronic kidney diseases, is now recognized to be an independent predictor of disease progression. Deposition of pathological matrix in the walls of glomerular capillaries, the interstitial space and around arterioles both predicts and contributes to functional demise of the nephron and its surrounding vasculature. Recent identification of the major cell populations of fibroblast precursors in the kidney interstitium as pericytes and tissue-resident mesenchymal stem cells, and in the glomerulus as podocytes, parietal epithelial and mesangial cells, has enabled the study of the fibrogenic process in much greater depth directly in the fibroblast precursors. These cells are not only matrix-producing cells, but are also important innate immune surveillance cells that regulate the inflammatory process, exacerbate tissue damage by release of radicals and cytokines, and contribute to parenchymal and microvascular dysfunction by aberrant wound-healing responses. Innate immune signaling in fibroblasts and their precursors is intimately intertwined with the process of fibrogenesis. In addition, genomic and genetic studies also point to defective responses in loci close to genes involved in solute transport, metabolism, autophagy, protein handling and vascular homeostasis, principally in the epithelium and endothelium, as upstream drivers of the fibrotic process, indicating that cellular crosstalk is vital for development of fibrosis. As we move beyond TGFβ inhibition as a central target for fibrosis, targeting innate immune signaling and metabolic dysfunction appear increasingly tenable alternative targets for novel therapies.
INTRODUCTION
Fibrosis has been the ‘dark horse’ of kidney disease research, but recently its importance has come to the forefront, through recognition that it is a major contributor to progression of almost all forms of kidney disease [1]. While the extent of fibrosis does not correlate well with glomerular filtration rate (GFR) measurements of organ function, it predicts future progression of disease with much greater certainty [2]. No longer merely a bystander problem, in response to tubular, microvascular or podocyte disease, the cellular process of laying down matrix, known as ‘fibrogenesis’, has been recognized to amplify and sustain pathological responses in the functioning units of the kidney, and therefore to be responsible for progression of disease [3]. Moreover, as pathological matrix deposition progresses, fibrotic contraction and replacement of architecture hasten the demise of organ function. Sclerotic changes in the glomerulus likely promote loss of the glomerular filtration barrier and autoregulation of the peritubular capillaries, ultimately leading to complete loss of glomerular filtration, and loss of molecular transfer between tubule and peritubular capillary. A common finding in both diseased glomerulus and interstitium is that deposition of pathological matrix (fibrosis) and loss of capillaries are intimately associated and may be causally linked.
Although TGFβ has been regarded as the canonical ligand driving pathological fibrotic responses throughout the body, recent detailed studies of fibroblasts, their precursors, as well as findings from human clinical trials have cast doubt on the central importance of TGFβ. Rather, metabolic and innate immune responses are emerging as more tractable strategies to target the chronic disease process, including fibrosis.
The direct pathological role of fibroblasts has been demonstrated by the transplantation of fibroblasts from a diseased tissue into healthy architecture, where disease can be transferred [4]. Moreover, blockade of receptors or transcription factors restricted to the fibroblast is sufficient in animal models to preserve organ function [5–7]. Such reports indicate a pathological role for the fibroblast in disease progression and support strategies to directly target the fibroblast. Here we will initially describe the origins of the fibroblast, identifying important cell surface markers that have advanced our ability to study fibrosis. We will demonstrate key functional pathways in the kidney that may be upstream drivers of the fibrotic process, predominantly in the epithelium, and highlight current and future strategies to target fibrosis.
FIBROBLAST ORIGINS AND THE EMERGENCE OF PERICYTES AND MESENCHYMAL STEM CELLS
The origin of interstitial fibroblasts remains an area of controversy. Nevertheless, most investigations in recent years using genetic fate mapping studies in mice point to cells of mesenchymal origin, resident in the kidney from nephrogenesis, as the major population of precursor cells that become cells we recognize as fibroblasts in diseased kidney interstitium (Figure 1). These precursors have been shown to be attached predominantly to capillaries particularly at branching points of the peritubular capillary plexus, and some investigators have named them ‘pericytes’ or ‘perivascular cells’, because of their ultrastructural relationship to the vasculature, being partially or completely embedded in the capillary basement membrane (Figure 1), although other texts have identified the same cells by independent methods and named them resident fibroblasts [5, 8–12]. The terms ‘fibroblast’, ‘resident fibrocyte’ and ‘pericyte’ derive from ultrastuctural studies using electron microscopy and other forms of microscopy dating from the late 19th and early 20th centuries [13]. All these cell types are mesenchymal, lacking well-defined lysosomal structures, polarity or resting on basement membrane. Pericytes are further defined as abluminal cells possessing processes and/or cell body embedded in duplicated capillary basement membrane. Fibroblasts/fibrocytes of healthy tissue are highly similar, but are completely embedded in matrix. In adult healthy kidney of humans and rodents, only a minority of resident mesenchymal cells appear to be anatomically disconnected from a capillary and may be legitimately called a resident fibroblast (or resident fibrocyte). However, the extensive connections afforded by the long processes of these cells make discriminating a fibroblast from a pericyte purely based on anatomical grounds, challenging. Although a range of molecular markers has been proposed to selectively label pericytes and discriminate them from other cell types, these markers are not particularly robust. They appear to depend on the tissue of study, the species and the state of cell activation. For example, NG2 labels mesangial cells and vascular smooth muscle, and labels a subpopulation of pericytes in the kidney only during regeneration and development [5]. In homeostasis, most adult kidney pericytes do not express NG2. Pericytes that lack NG2 are well described in other organs such the lung and in the CNS, NG2 labels glial cells and oligodendrocyte progenitors [14–16]. The most robust marker of pericytes in healthy kidney is PDGFRβ. Nevertheless, a range of additional markers has now been reported that label resident mesenchymal cells and may discriminate between subsets (Figure 1). A 1–5% subpopulation of these pericytes and resident fibroblasts retains multi-lineage potential, is marked by expression of the transcription factor GLI1, is able to differentiate into all mesenchymal lineages (adipocyte, chondrocyte, osteoblast, fibroblast) in vitro and may be thought of as tissue resident multipotent mesenchymal stromal cell progenitor (MMSCs) (Figure 1) [5, 12]. Studies suggest that this rare subpopulation, also attached to capillaries, is able to expand to represent up to 60% of all fibroblasts in mouse models of diseased kidney, and genetic fate mapping studies of mesenchymal cells from several groups now strongly suggest that the majority of the remaining 40% come from the other pericytes and fibroblasts [17] (Figure 1). It is striking that across multiple organs in the body, including lung, liver, skin, muscle, heart and gut, the presence of tissue-resident MMSCs or pericytes has been identified and their role as precursor cells for disease-associated fibroblasts (also known as myofibroblasts) has been documented through rigorous fate mapping studies [8, 18].
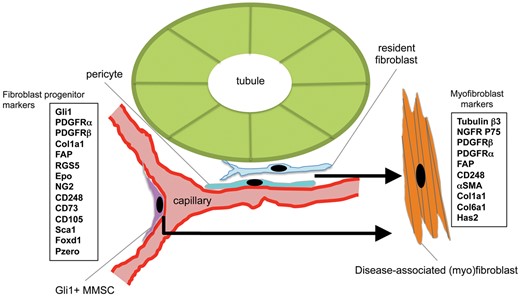
Schema showing the location of resident mesenchymal cells: resident fibroblast, pericyte and MMSCs, that are the major populations of progenitors of disease-associated myofibroblasts. Markers of these progenitor populations and the myofibroblasts are shown in boxes.
While most current research points strongly to these resident mesenchymal cells as the major source of fibroblasts, other studies have been less conclusive. For example, several groups have identified a leukocyte that can become a fibroblast, and propose that a major population of fibroblasts derives from leukocytes [19]. Other groups have proposed that endothelial cells can differentiate into pathological fibroblasts [20]. Until a few years ago, it was believed that the epithelium of the kidney was the primary source of fibroblasts, but multiple recent studies have now shown that at least in short-term animal models, diseased epithelial cells do not become fibroblasts (reviewed in [3]). The absence of epithelial to mesenchymal transition (EMT) as a source of fibroblasts has been recapitulated in multiple organs in the body by newer more rigorous studies and has led to a burgeoning area of research around epithelial to fibroblast cross-talk (see below). While the exact contribution of endothelial cells and leukocytes to fibroblast appearance remains controversial, our own studies offer some potential explanation for the discrepancies in published work. First, although all cells can acquire fibroblast characteristics on plastic culture dishes, the fate mapping studies of endothelial cells in vivo have relied on Cre/Lox systems (e.g. Tie2-Cre or Cdh5-Cre) that inadvertently also mapped the fate of leukocytes or pericytes due to expression of the transgenes in multiple cell lineages [21, 22]). Second, the markers used to detect fibroblasts are controversial; FSP1, which is better known as S100A4, is detected in rodents predominantly in activated macrophages, not fibroblasts. Vimentin also labels macrophages and injured epithelial cells as well as fibroblasts [23]. In our opinion, the contribution of endothelium to interstitial fibroblasts in vivo is unclear at this time. The contribution of circulating leukocytes (also known as ‘circulating fibrocytes’) to matrix-producing fibroblast continues to remain controversial. Some of the controversy surrounds the detection of collagen in side macrophages in the tissue, but since a major role of macrophages is matrix turnover and clearance, this finding does not confirm macrophages produce pathological matrix. Fate mapping studies either by bone marrow chimeras or by Cre/Lox systems have also provided differing results [19]. In our own studies, myeloid lineage mapped using the Lyz2-Cre; Rs26 tdTomatoR transgenic system did not identify significant numbers of fibroblasts or myofibroblasts arising from myeloid cells, but in other studies we did identify a very small population of dendritic cell-like leukocytes that activated collagen transgene [17]. Other reports using parabiotic techniques to share circulations between animals with different genetic backgrounds found no evidence of circulating cells contributing to fibroblasts at a wound site [5]. It remains possible that a minor population of fibroblasts can derive from a minor leukocyte population, but in our opinion this minor leukocyte has not been rigorously identified at this time. Finally, it is worth noting that all fate mapping studies have been performed in mice, not humans, and therefore the exact contribution of epithelial and endothelial cells and leukocytes to the fibroblast population in human kidney disease remains unclear.
The finding that most fibroblasts originate from cells attached to capillaries with MMSC-like and pericyte-like properties has led to the proposal that they are critical in tissue repair and regeneration as well as contributing to progressive chronic disease. MMSCs from bone marrow have long been recognized to have regenerative and anti-inflammatory properties, and pericytes are critical regulators of vascular growth and stability. MMSCs in bone marrow have recently been shown to be a pericyte-like population of cells in vivo [24]. Moreover, stromal cells in general have been reported to play vital trophic roles in organ patterning growth and differentiation during embryogenesis [25, 26]. Recent studies in the kidney show similar vital roles for these progenitor cells in kidney development, and also in kidney homeostasis, indicating that fibroblast precursors play crucial active roles in tissue generation and in maintenance of organ health rather than being passive bystanders [25, 27]. The breadth of cell function in homeostasis controlling epithelial nutrition, basement membrane deposition, vascular permeability and vascular tone suggest pericytes, and MMSCs are integral to normal organ function. Although an unequivocal role for MMSCs and pericytes in regeneration after acute kidney injury has not yet been demonstrated, studies of the equivalent cells in skeletal muscle, known as fibro-adipogenic precursors (FAPs), has shown their critical role in muscle regeneration after acute injury but also demonstrated their role in disease progression in chronic degenerative diseases of the muscle such as Duchenne muscular dystrophy [7, 28, 29].
One conclusion from these findings is that if we can coax pathological fibroblasts back to the precursor state, they have all the attributes to orchestrate successful resolution of tissue damage.
GENETIC PREDISPOSITION AND CELLULAR CROSS-TALK IN FIBROSIS
Genome-wide association studies from >100 000 patients with kidney disease have identified single-nucleotide polymorphisms at loci in the genomes of patients that are associated with kidney disease. Many of these polymorphisms are in non-coding regions but appear to be enriched in enhancer regions of nearby genes and likely control the expression of those nearby genes [30]. These extremely robust human studies have exposed surprising findings that are now instructing our approach to fibrosis research. Most of the genetic loci are close to genes involved in solute and toxin transport (SLC34A1, SLC22A2, SLC7A9), intracellular trafficking (DAB2), metabolism (AMPKinase, CPS1, GCKR, GATM, SOD1), autophagy (TP53INP2), DNA damage/repair (BRCA1/2), vascular integrity (VEGFA) or innate immunity (CASP9, IL1, IRAK1) [31–34]. There are no polymorphisms in loci near to genes involved in cardinal fibrotic pathways including TGFβ, connective tissue growth factor (CTGF) and platelet-derived growth factor (PDGF). The most significant locus associated with kidney disease is close to the UMOD gene and two genes associated with fatty acid metabolism, ACSM5 and ACSM2 [35]. UMOD codes for uromodulin, the most highly secreted protein by the epithelium of the kidney, and polymorphisms in the UMOD locus are associated with increased uromodulin expression, which may of itself be pathological (Figure 2). Families with mutations in the UMOD gene develop early onset progressive fibrotic kidney disease, hypertension and hyperuricemia, Mutations in the UMOD gene lead to abnormal protein, folding, preventing endoplasmic reticulum (ER) trafficking and activation of cell stress pathways, particularly ER stress [36, 37]. Such observations suggest that non-coding variations close to the UMOD gene may affect production of this protein, also contributing to dysregulated protein production, and therefore cell stress (Figure 2). Strikingly, in other inherited fibrotic diseases, misfolded proteins have been shown to be causative, including idiopathic pulmonary fibrosis (IPF) (surfactant protein-C), cardiac failure (transthyretin), muscular dystrophy (dystrophin), cystic fibrosis (CFTR) and neurodegeneration (α-Synuclein, SOD1), suggesting that cell stress responses to these mutated proteins may lie upstream of the fibrotic/degenerative process in some patients [38].

Schema showing the impact of genetic predisposition and environmental factors on injury responses in the epithelium and cross-talk to the fibroblast. Examples of genetic/genomic susceptibility loci are shown in green boxes under functional headings. In combination with environmental insults the injured epithelium has aberrant responses that exacerbate one or many of the pathways shown in white boxes. Activation of these pathways leads to activation of immune responses and consequent release of factors that activate neighboring fibroblast precursors.
Alternatively, polymorphisms at the UMOD locus may be detrimental to fatty acid oxidation (FAO) since the ACSM2 and ACSM5 genes that are involved in fatty acid metabolism reside closeby in the genome [35]. FAO is vital in the nephron for energy production and has recently been shown to be important in maintaining epithelial cell function in response to cell stress [39, 40]. In other studies, FAO has been shown to be play a key role in maintaining leukocytes in an inactivated state [41], and studies are now pointing to metabolism as an important regulator of fibroblast activation (Figure 3). In addition, gene variants in loci thought to be regulating mitochondrial biogenesis (AMPK) and other aspects of mitochondrial function including anti-oxidant pathways (SOD1, MPV17L) have been demonstrated (Figure 2) [42]. Although future experiments need to verify the role of these non-coding polymorphisms in loci of metabolic genes, it appears that control of metabolic responses in settings of tissue injury are important determinants of long-term organ outcome. In support of the role of polymorphisms affecting metabolic gene function in the pathogenesis of kidney fibrosis, families with mutations in nuclear genes that encode mitochondrial proteins, as well as mitochondrial DNA mutations, develop kidney diseases ranging from Fanconi syndrome, through interstitial nephritis to focal segmental glomerulosclerosis (FSGS) [43]. Collectively, such findings point to mitochondrial metabolic dysregulation as an important problem in many chronic kidney diseases. Consistent with these genetic and genomic findings, transcriptional analyses of human fibrotic kidney disease point strongly to downregulation of mitochondrial functions and other metabolic functions that require mitochondria (Figure 3). Rather than such a loss of mitochondrial function representing a secondary response to disease, these changes to mitochondrial disease or dysfunction may be a primary driver of kidney disease progression in certain patients. Recent studies suggest that strategies to enhance FAO, enhance mitochondrial biogenesis, or improve mitochondrial function are beneficial to kidney disease [39, 40, 44, 45]. Moreover, studies in which single genes have been targeted in the kidney epithelium that disrupt mitochondrial homeostasis have been shown to be sufficient to drive loss of organ function and kidney fibrosis [46, 47]. In particular, AMP kinase activators, peroxisome proliferator-activated receptor α (PPARα) activators and activators of nuclear factor-like 2 (NRF2) all enhance mitochondrial function and have all shown promise in improving renal outcomes in preclinical as well as clinical studies [44, 48, 49].

Highly regulated pathways in human CKD. Enrichment analysis of biological process ontology in human biopsies from CKD patients compared with healthy controls.
Among the non-coding variants that predispose to kidney disease progression are a number of non-coding RNA sequences. These microRNA and long-non-coding RNAs are increasingly being identified as important regulators of gene expression that control tissue injury responses in the kidney [50, 51]. Although beyond the scope of the current review, many of these non-coding RNAs regulate cellular responses in innate immunity and metabolism, and may be future therapeutic targets for kidney disease (see Section: Role of innate immunity in fibrosis).
These genetic and genomic findings collectively indicate that epithelial to fibroblast crosstalk is a fundamental process in driving kidney disease progression whereby injured or distressed epithelium (or endothelium) signals to interstitial cells to trigger and maintain their activation (Figure 2). In turn, the activated fibroblast provides signals and an environment that are detrimental to the health of the epithelium. This crosstalk sets up a hostile environment in the interstitium that can drive disease perpetuation.
As studies have emerged that show epithelium does not directly differentiate into fibroblasts, genetic and genomic studies strongly implicate the epithelium in driving kidney disease progression. Rather than cellular trans-differentiation, studies now propose that injured epithelium signals to the interstitial space to drive fibrosis (Figure 2). These signals are wide and varied and include ligands such as PDGF, αvβ6-integrin-activated TGFβ, CTGF, inflammatory cytokines and chemokines such as oncostatin M (OSM), TNF-like weak inducer of apoptosis (TWEAK), monocyte chemoattractant protein (MCP1) and interleukin (IL)18, potentially toxic radicals as well as danger-associated molecular patterns (DAMPs) such as HMGB1 [52, 53]. Epithelial injury per se is sufficient to cause fibrogenesis [54, 55]. The pathological epithelial responses now well documented there may be driving interstitial disease include impaired autophagy, mitochondrial dysfunction, ER stress, DNA damage and the cell senescence response, as well as a ‘partial EMT’ phenotype (Figure 2) [56–59].
ROLE OF INNATE IMMUNITY IN FIBROSIS
Although we think of fibroblasts as essentially matrix-producing cells that deposit a pathological matrix, recent insights, enabled by the identification of the progenitor cells that give rise to fibroblasts, and by genetic methods to isolate, purify and target these cells, have demonstrated that fibroblasts produce a wide array of pro-inflammatory cytokines and chemokines, as well as producing oxygen radicals (Figure 4). As such they are a type of inflammatory cell, but not one derived from circulating leukocytes. Several studies now point to the fibroblast as the dominant cell to produce cytokines and chemokines in chronically diseased tissue [40, 60–65]. In the absence of pericytes and resident fibroblasts, leukocytes fail to be recruited, and in states of genetically induced over-activation of fibroblasts, intense recruitment of leukocytes can be triggered [27, 66]. It is likely therefore that a very important but overlooked role for the fibroblast is in controlling inflammatory responses. In studies from our laboratory, fibroblasts are more sensitive to sterile injury molecules released from damaged cells (DAMPs) than macrophages, and produce higher levels of pro-inflammatory factors than other cells in comparable studies [61, 62, 67].
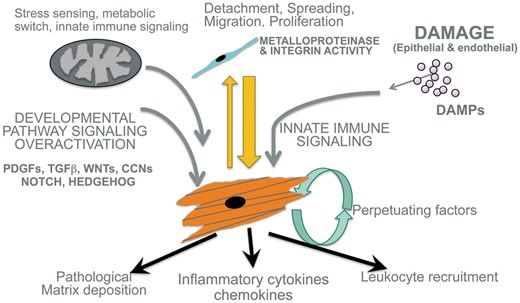
Schema showing major mechanisms that activate fibroblast precursor cells to become and remain as pathological myofibroblasts. Note that in addition to the canonical pathways including TGFβ, CTGF and WNT, additional pathways including innate immune signaling and mitochondrial metabolic changes are critical for pathological responses. Note in addition that myofibroblasts secrete large amounts of cytokines and inflammatory factors in addition to depositing pathological matrix.
What has transpired from these and other studies is that external signals of the innate immune system including DAMPs acting through Toll-like receptors (TLRs), chemokines such as CXCL-11 and -12, tumor necrosis factor (TNF) superfamily members, and IL family members, including IL1β, IL6 and IL11 acting through cognate receptors, are all capable of promoting a more pro-fibrotic phenotype in the fibroblast in vitro and in vivo [60, 61, 65, 68]. Moreover, new studies suggest synergy between cardinal pro-fibrotic signals such as TGFβ or wingless/INT (WNT) may co-exist with innate immune signals [61, 67], potentially explaining why injured tissues develop fibrosis, whereas embryonic tissues with migratory stromal cells that have active TGFβ and WNT signaling do not develop fibrosis (Figure 4). What is more, in terms of therapeutic strategies, genetic studies also point to inherited predisposition to fibrotic kidney disease through dysregulation of innate immunity. For example, polymorphisms in the IL1β and IL1 receptor-associated kinase (IRAK1) loci and coding polymorphisms in TLR4 are associated with chronic kidney disease (CKD) [32–34, 69, 70]. These findings may suggest that inhibition of innate immune signaling could dampen pro-fibrotic responses and inflammation, without the potential safety hazards of blocking other signaling pathways such as NOTCH, WNT, TGFβ or HEDGEHOG that have homeostatic functions.
Several studies have recently pointed to successful anti-fibrotic effects from blocking innate immune pathways. The TNF superfamily receptors such as FN14 exert strong pro-fibrotic responses in fibroblasts and blockade of FN14 signaling with antibodies or soluble receptors has proven to be effective. Inhibition of TLR and IL1β/IL18 downstream signaling by blockade of intracellular kinases such as IRAK1 and IRAK4 has also been shown to highly effective in blocking progression to fibrosis after injury [60, 67, 71–73]. In addition, Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling that is downstream of multiple inflammatory receptors for IL6, IL11, Oncostatin-M and type-I interferons has recently been highlighted as an activated pathway in early diabetic kidney disease (DKD) and to drive fibrogenesis [73, 74]. Clinical studies to block this pathway have shown promise in early trials (see section on Clinical Development).
MECHANICAL FACTORS CONTRIBUTING TO PERSISTENCE OF PATHOLOGICAL FIBROBLASTS
An additional mechanism contributing to persistence of pathological fibroblasts has gained increasing acceptance. With aging, and repeated cycles of injury and repair, we deposit extracellular matrix that is not fully resorbed during repair and regeneration. This matrix becomes stiff with increasing cross-linking. Many studies now show that an important regulator of the pathological fibroblast phenotype is the sensing of the stiffness of the environment. Although there are no obvious genetic or genomic leads that corroborate the importance of these findings, recent experiments have identified the transcription factors , Yes-activated protein (YAP), transcriptional coactivator with PDZ binding motif (TAZ) and myocardium-related transcription factor (MRTF), as central regulators of the sensing of tissue tension that may maintain fibroblasts in a pathological state [75–81]. YAP and TAZ are part of the Hippo signaling pathway that can be activated by multiple cell surface receptor complexes including WNT, TGFβ and the αv-integrins. Although no therapeutic interventions regulating the Hippo pathway are available, several approaches to limit matrix cross-linking such as blockade of the lysyl oxidases and transglutaminases have been developed [82]. Blockade of lysyl oxidase 2 (LOXL2) with antibodies that inhibit the catalytic subunit have appeared highly effective at reducing tissue tension and fibrosis in animal models of lung fibrosis, although these have not translated to therapeutic benefit in human lung fibrosis, and clinical responses in patients with liver cirrhosis are still awaited. In the kidney, four isoforms of the LOXL family are expressed in disease. Potentially developing inhibitors of more than one crosslinking enzyme will be required to effect the tissue tension that evolves in chronic human disease.
CLINICAL DEVELOPMENT OF THERAPIES TO TREAT KIDNEY FIBROSIS
Recent human clinical trials to retard CKD progression by blocking the profibrotic cytokine TGFβ in the kidney have fallen short. Anti-TGFβ1 antibody, that blocks all three forms of TGFβ showed efficacy in preclinical models of kidney disease, but in a phase III study in patients with DKD the humanized form of this monocloncal antibody, LY2382770, failed to show efficacy. Similarly, Genzyme’s pan anti-TGFβ antibody fresolimumab was not taken forward in patients with steroid-resistant FSGS following phase II studies. Although there are numerous caveats to explain why there has been a lack of success, these studies mark the end of a long experience with attempts to safely block the TGFβ pathway to treat fibrosis by different strategies in human disease. Our own experience of studying human CKD biopsy material has been the lack of strong engagement of the TGFβ signaling pathway apparent on transcriptional analysis, as compared with other pathways (Figure 3). Such a finding may suggest that the animal models upon which the molecular hypothesis is based are not faithful representations of human disease. One major challenge with identifying good therapeutic targets for fibrosis is the problem of ‘time’. Animal models are studied over days and weeks, whereas human disease occurs over months and years. Many of the pathways important in the early activation steps of the fibrotic response are likely not so important later in disease perpetuation. Moreover, with the recent identification of fibroblasts and their precursors with greater certainty, it has become apparent that many cell signaling pathways convert fibroblast precursors into pathological cells in assays performed ex vivo, indicating that the myofibroblast phenotype is a wounding response rather than a TGFβ response. If we look more deeply into key pathways, there is evidence for activation of multiple signaling pathways that can drive fibroblast persistence including WNT, Notch and Hedgehog, but most striking is the enrichment for innate immune signaling, including IL6, Interferon, IL18, IL1β and pro-inflammatory chemokine production (Figure 3). We predict that blockade of innate immune signaling with factors that inhibit parts of the NFκB pathway, the JAK–STAT pathway and parts of the complement cascade may inhibit multiple signals that can activate and perpetuate the pathological fibroblast. In keeping with such findings, studies using a small molecule inhibitor of JAK1/2, Baricitinib, in DKD have demonstrated a reduction in proteinuria over a 6-month period. Such an inhibitor would be expected to inhibit signals from multiple innate immune signaling receptors, and therefore may represent a node for targeting in more advanced trials.
Recently, there have been successes demonstrated by U.S. Food & Drug Administration (FDA) approval of two anti-fibrotic therapies for the treatment of Idiopathic Pulmonary Fibrosis (IPF) [83, 84]. This disease shares many similarities with CKD. Genetic studies point to initiation of the disease in the distal bronichial or alveolar epithelium, particularly Type-II epithelial cells. Cell stress pathways, responses to misfolded proteins and DNA damage responses are implicated as upstream drivers of disease. The stressed/injured epithelium in turn activates underlying resident pericytes and fibroblasts to become pathological, and therefore shows striking similarities to pathological mechanisms in kidney disease. Pathological fibroblasts in lung are able to sustain the disease and drive organ failure [85]. The new drugs shown to delay progression of lung fibrosis are pirfenidone and nintedanib. The latter is a multikinase inhibitor, which potently blocks both PDGFRα and PDGFRβ [86]. It is striking that pathological fibroblasts express these receptors, whereas other cell types, including lung epithelium, do not. A wealth of literature points to the benefits of blocking PDGFRα or PDGFRβ in fibrotic disease, but previous attempts to block these receptors with another kinase inhibitor, imatanib, have provided mixed results in lung and systemic fibrosis [87–89]. One potential explanation is that imatanib is rather weak inhibitor of PDGFRα, but showing higher potency for PDGFRβ as well as Abelson (ABL). In the kidney, the pathological (myo)fibroblasts express both of these receptors and have been shown to be viable targets for reducing tissue injury and inflammation as well as fibrosis. It is likely that nintedanib could be considered for progressive kidney disease, although its side-effect profile may result in its use only in subpopulations of patients that are rapidly progressing. Blockade of these receptors, particularly PDGFRβ, in kidney fibroblasts potently blocks proliferation and migration as well as deposition of fibrotic matrix. Other potent PDGFRβ kinase inhibitors are available including crenolanib, and should be considered for potential studies in CKD [90].
Although pirfenidone has gained approval for IPF and is fairly well tolerated, its mechanism of action is poorly understood. At efficacious concentrations in humans it does not appear to inhibit known profibrotic pathways. It may reduce TNF release from monocytes (and potentially other cells) and therefore may be acting through TNF-related signaling pathways [91]. Pirfenidone was given to patients with DKD in a phase II clinical study with mixed results [92]. At lower doses, small but significant effects were observed. At higher doses, no benefits were observed and the drug was poorly tolerated. Although pirfenidone is unlikely to gain wide acceptance for DKD, it may be informing us about the potential importance of TNF signaling in the kidney parenchyma, since a TNF-signaling pathway signature is reproducibly apparent in many forms of kidney disease.
As stated above, targeting mitochondrial dysfunction is an attractive approach that encapsulates genetic predisposition seen in many patients with activated cell stress pathways in the epithelium as well as the fibroblast. It is notable that activators of PPARα, another important transcription factor in mitochondrial biogenesis and FAO, have been used to treat hyperlipidemia in patients. Although weak activators, fibrates have been given to patients with kidney disease in a lipid-lowering trial (FIELD study) and post hoc analysis has shown they retard progression of CKD [49]. This powerful study of 10 000 patients may be pointing to PPARα activation as an important therapeutic strategy. To that end, recent studies of non-coding RNA, known as microRNA, that are over expressed in kidney disease identified miRNA21 as an important amplifier of kidney disease [40]. miRNA21 specifically silences several genes that have been identified as potential drivers of kidney disease in genome-wide association study studies [51]. A major target for miRNA21 is PPARα and multiple genes involved in FAO, including ACAT and ASCM5. In addition, miRNA21 silences several important transporters important in epithelial function including organic anion transporter 1 (OAT1) and the sodium phosphate transporter SLC34A1. In direct studies, miR21 activation inhibits mitochondrial function and biogenesis, and promotes mitochondrial ROS generation, diminishing epithelial functions [40, 93]. It also remarkably contributes to sustaining activation of pathological fibroblasts, presumably by switching them from mitochondrial respiration to glycolysis (Figure 4). A novel the anti-microRNA21 drug has been developed that has excellent penetration of the kidney. Silencing of miRNA21 promotes mitochondrial function and enhances FAO and the anti-oxidant state of the kidney. Anti-miRNA21, optimized for use in humans (RG-012/SAR-339375) is now in phase II clinical trials for Alport nephropathy, but may have broader applications in other forms of CKD.
Another approach to improving the metabolic state of the kidney has been to promote activation of the anti-oxidant transcription factor NRF2. NRF2 is an important regulator of many anti-oxidant genes including the glutathione system and other anti-oxidants including superoxide dismutase I. Moreover, NRF2 may play additional roles directly in mitochondrial turnover. NRF2 is retained in the cytoplasm in an inactivated state by a chaperone known as Kelch-like ECH-associated protein 1 (KEAP1). KEAP1 inhibition releases NRF2 where it can translocate and act in the nucleus. Recently, a KEAP1 inhibitor, bardoxolone, which activates NRF2, was tested in DKD and showed strikingly positive results in phase II studies [48]. The compound was advanced to phase III studies, which were subsequently abandoned due to increased incidence of heart failure in some patients [94]. Although bardoxolone does activate NRF2, it is a covalent irreversible modifier that binds many targets irreversibly, and is therefore prone to off-target side-effects. The target profile of NRF2 remains very attractive to improve redox state and mitochondrial metabolism in the kidney, and therefore the development of KEAP1 inhibitors for kidney disease remains attractive. It is notable that dimethylfumarate, marketed as tecfidera, is a short-acting NRF2 activator and is approved by the FDA for the treatment of chronic multiple sclerosis. Dimethylfumarate has shown efficacy in short-term animal models of kidney fibrosis [95].
FUTURE DIRECTIONS
Although fibrotic kidney diseases lack modern therapies, the wealth of new data on cell lineage, and on genetic predisposition to kidney diseases, is helping guide the development of smarter therapies to target validated signaling nodes which control disease amplification, support disease progression or prevent normal resolution. As we dissect human fibrotic kidney disease more closely, defining new patient subpopulations that have definitive activation, or lack of activation, of key pathways we wish to target therapeutically, may enable us to design smarter and shorter trials to validate new therapies.
The overlap of mechanisms driving chronic fibrotic disease of the kidney with mechanisms driving degenerative and fibrotic diseases elsewhere in the body should enable us to capitalize on therapies being developed for one indication being evaluated for indications in other organs such as the kidney.
ACKNOWLEDGEMENTS
The Duffield laboratory is funded by Biogen.
CONFLICT OF INTEREST
I.A.L. and J.S.D. have Biogen stock, and have a patent for the inhibition of IRAK4 to treat kidney disease. J.S.D. has a patent for the use of anti-miRNA21 to treat Alport syndrome and other kidney diseases, and has stock in Regulus Therapeutics. J.S.D. has patents for the use of WNT pathway regulation to treat kidney disease. J.S.D. has stock options in Promedior Inc., which is developing anti-fibrotic therapies.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments