





Abstract
Biomarker development in lupus nephritis (LN) has traditionally relied on comparing the characteristics of candidate markers to clinical findings in patients and controls from cross-sectional cohorts. In this work, two additional strategies for LN biomarker development that are gaining ground will be discussed. One approach compares analytes directly to kidney histology. The second strategy utilizes longitudinal measurements of biomarker levels at regular intervals as patients move from disease quiescence to disease flare. These approaches have begun to empower biomarkers as diagnostic and prognostic tools in LN and have revealed novel and sometimes unexpected roles for these biomarkers in the pathogenesis and prediction of LN disease activity.
INTRODUCTION
To successfully manage lupus nephritis (LN) it is important to be able to accurately distinguish active nephritis from chronic kidney damage. Currently disease activity and organ damage are assessed by renal histology after kidney biopsy and by clinical measurements of kidney function and proteinuria. Importantly, pathologic studies provide limited information because patients are not biopsied frequently and clinical measures provide limited information since they do not reflect intrarenal injury very well. Therefore identification of novel biomarkers of LN activity represents an important unmet need in LN management.
A search of PubMed using the index terms human, lupus nephritis, biomarkers and disease activity returned >240 papers published in the last decade. More than 10% of these articles are reviews. These publications describe >45 different cytokines, growth factors, autoantibodies, hormones, adhesion molecules, serum components and cell types that reflect active LN. Yet exactly zero new biomarkers of LN disease activity have been incorporated into routine clinical use. This disconnect between the level of effort being put into biomarker research and the lack of translation into usable clinical tests is concerning. Coupled with a so far unsuccessful but significant effort to develop new LN therapies, optimizing disease management may appear to be an impossible problem to solve.
We suggest this task is difficult, but not impossible. Assessing the efficacy of new therapies will in fact be facilitated by developing biomarkers of LN. Several promising LN biomarker candidates have been identified and with a bit more work may meet the requirements for use in clinical practice. Here we will consider some of these candidates, along with data from our own biomarker development program, to illustrate a process for successful biomarker development in LN and the roadblocks that have been encountered in this area of investigation.
The term LN biomarker is vague, and without proper clinical context almost meaningless. We envision three broad categories of biomarkers under the general category of managing disease activity. These include biomarkers of renal histology, biomarkers to guide the choice of treatment and biomarkers of renal flare. Histology biomarkers would be useful to noninvasively determine how well a therapy is doing to resolve inflammatory renal injury and to monitor the development of chronic disease. Treatment-specific biomarkers would help match patients to a treatment regimen most relevant to their LN. Biomarkers of flare would clarify the pathways critical to the development of LN flare. This could alter treatment paradigms for LN flare by adding to the list of treatment-specific biomarkers, by identifying new therapeutic targets and by providing tools to forecast impending LN flares so they could be preemptively treated, potentially shortening exposure to toxic drugs and limiting permanent kidney damage.
BIOMARKERS OF KIDNEY HISTOLOGY IN LN
Kidney biopsies are critical in the initial assessment of LN. They not only confirm the diagnosis, but provide information on disease class [based on International Society of Nephrology/Renal Pathology Society (ISN/RPS) criteria] and the extent of tissue damage [based on the National Institutes of Health (NIH) activity and chronicity indices]. Histologic data are also valuable in assessing response to therapy. However, frequently repeating biopsies is an unrealistic management tool to monitor response. Clinical measures of kidney function and proteinuria have traditionally served as surrogates of histologic activity despite considerable evidence demonstrating discordance between kidney pathology and clinical markers in LN [1]. The need for real-time evaluation of renal histology during the treatment of LN has organized our current and ongoing efforts to develop accurate surrogate markers of kidney pathology.
To illustrate important considerations in the process of identifying, characterizing and validating histologic biomarkers, previously unpublished data on biomarkers of renal interstitial injury from our laboratory will be discussed. We sought biomarkers that differentiated LN patients who had interstitial inflammation or interstitial fibrosis involving ≤25% (none–mild) or >25% (moderate–severe) of the renal cortex. The rationale for these cutoffs came from the observation that LN patients who showed >25% interstitial inflammation or fibrosis on their diagnostic biopsy were more likely to develop kidney failure than those with less interstitial pathology [2]. The biomarker identification process was based on the hypotheses that the urine of patients with LN would be a good source of surrogate biomarkers and that no single biomarker will be sufficient to represent a specific histologic lesion.
To develop surrogate biomarkers of interstitial injury we used established clinical endpoints, plausible biomarkers based on LN pathogenesis and candidates generated de novo using urine proteomics. Proteinuria and serum creatinine concentration (SCr) were included because any novel biomarker must provide added value to clinical endpoints that can be measured easily and inexpensively. Urine analytes initially considered for our biomarker panels were monocyte chemoattractant protein-1 (uMCP-1), osteopontin (uOPN), hemopexin (uHPX) and endothelial protein C receptor (uEPCR). uMCP-1 was included as a plausible candidate biomarker because of its proinflammatory and profibrotic effects and because it is significantly upregulated during active LN [3]. uOPN, uHPX and uEPCR were included because, in a discovery cohort (n = 15) using liquid chromatography–tandem mass spectrometry (LC-MS/MS) to evaluate the urine proteome in an unbiased fashion [4], they were found to be differentially expressed in the urine of LN patients whose kidney biopsies showed bland versus actively inflamed versus sclerotic interstitial compartments. Although other proteins were also differentially expressed in this discovery urine set by LC-MS/MS analysis, the selection of osteopontin, EPCR and hemopexin was based on previous reports of their associations with LN [5–9].
Biomarker models were built using a training set of 81 LN patients. Importantly, each of these analytes was measured in urine or serum collected at the time of diagnostic kidney biopsy. Biopsies were segregated by the degree of interstitial inflammation and interstitial fibrosis as described on the biopsy report (≤25%, >25%). This semiquantitative assessment was made by a clinical nephropathologist and based on the degree of cortical involvement by active inflammation or interstitial fibrosis. Linear discriminant analysis (LDA) was then used to derive composite biomarker panels for interstitial inflammation and interstitial fibrosis, as we previously described [9]. Initially, all six analytes were used to model interstitial pathology. The iterative process of LDA sequentially eliminated noncontributory analytes, finally yielding optimally weighted equations for discriminating between patients with none-mild or moderate-severe interstitial inflammation or fibrosis.
The results of this analysis are shown in Figure 1. The equations presented were derived by taking the ratios of likelihoods of the multivariate, normally distributed data from the none–mild and moderate–severe groups. Logarithmic transformation ensured a normal fit to each of the biomarker variables in the equations. Intercepts were chosen to maximize the sum of sensitivity and specificity. SCr was retained in the LDA statistical model and contributed to each biomarker equation, but proteinuria did not (Figure. 1). Model predictive power was increased with the addition of uHPX and uEPCR to the fibrosis equation and uHPX and uMCP-1 to the inflammation equation.
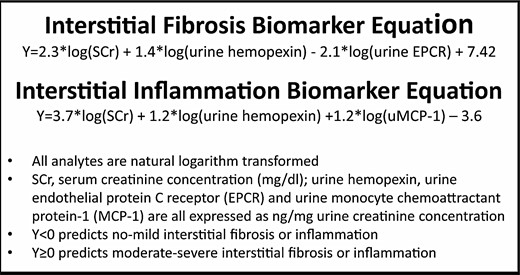
Biomarkers of interstitial injury in LN. These composite biomarkers of interstitial injury were derived using linear discriminant analysis to combine urine and serum analytes into equations that gave the highest sum of sensitivity and specificity for distinguishing between none–mild and moderate–severe interstitial fibrosis and interstitial inflammation.
The sensitivity and specificity of these composite biomarkers to discriminate between biopsies having none–mild and moderate–severe interstitial injury were calculated (Table 1). From the sensitivities and specificities, receiver operating characteristic (ROC) curves were constructed. The areas under the ROC curves were 0.85 and 0.89 for fibrosis and inflammation, values generally associated with good diagnostic tests. However, the interstitial inflammation and fibrosis biomarker equations are plastic in that additional analytes can be incorporated into the equations to improve sensitivity and specificity. A reasonable approach would be to test the contributions of additional candidates identified in the literature that have good diagnostic characteristics. For example, pentraxin 3 belongs to a family of acute phase reactants that may play a role in innate immunity, modulating complement activation and clearing apoptotic cells. In a discovery cohort of 213 patients and a validation cohort of 75 patients, urine pentraxin 3 levels were found to correlate best with interstitial inflammation (r = 0.35, P < 0.03) [10]. Although this correlation is modest for a stand-alone biomarker, pentraxin 3 could conceivably add to and improve the interstitial inflammation biomarker panel shown in Figure 1. Similarly, urine kidney injury molecule-1 (uKIM-1) was found to associate with tubulointerstitial inflammation in LN, having a sensitivity of 88% and a specificity of 61% to discriminate between mild and moderate involvement [11].
Diagnostic characteristics of interstitial pathology biomarkers to differentiate between mild and moderate–severe injury
| Parameter | Interstitial fibrosis biomarker (%) | Interstitial inflammation biomarker (%) |
|---|---|---|
| Training set | ||
| Sensitivity | 70 | 72 |
| Specificity | 81 | 79 |
| Misclassification rate | 22.7 | 22.5 |
| Validation Set | ||
| Sensitivity | 44 | 80 |
| Specificity | 91 | 92 |
| Misclassification rate | 17.6 | 9.4 |
| Parameter | Interstitial fibrosis biomarker (%) | Interstitial inflammation biomarker (%) |
|---|---|---|
| Training set | ||
| Sensitivity | 70 | 72 |
| Specificity | 81 | 79 |
| Misclassification rate | 22.7 | 22.5 |
| Validation Set | ||
| Sensitivity | 44 | 80 |
| Specificity | 91 | 92 |
| Misclassification rate | 17.6 | 9.4 |
Diagnostic characteristics of interstitial pathology biomarkers to differentiate between mild and moderate–severe injury
| Parameter | Interstitial fibrosis biomarker (%) | Interstitial inflammation biomarker (%) |
|---|---|---|
| Training set | ||
| Sensitivity | 70 | 72 |
| Specificity | 81 | 79 |
| Misclassification rate | 22.7 | 22.5 |
| Validation Set | ||
| Sensitivity | 44 | 80 |
| Specificity | 91 | 92 |
| Misclassification rate | 17.6 | 9.4 |
| Parameter | Interstitial fibrosis biomarker (%) | Interstitial inflammation biomarker (%) |
|---|---|---|
| Training set | ||
| Sensitivity | 70 | 72 |
| Specificity | 81 | 79 |
| Misclassification rate | 22.7 | 22.5 |
| Validation Set | ||
| Sensitivity | 44 | 80 |
| Specificity | 91 | 92 |
| Misclassification rate | 17.6 | 9.4 |
Because these composite biomarkers are meant to be surrogates for repeating the kidney biopsy, the accuracy of the biomarkers compared with the kidney biopsy was calculated as the percent of patients misclassified by the biomarker equations (Table 1). Patients were considered misclassified if the biomarker equation suggested mild interstitial disease but the kidney biopsy showed moderate–severe disease or if the biomarker equation suggested moderate–severe disease but the biopsy showed mild disease. The fibrosis biomarker tended to misclassify equally in both directions, while most of the misclassifications for the inflammation equation were due to identifying moderate–severe disease when the biopsy showed mild or no interstitial inflammation (false positives).
Ideally the accuracy of histology biomarkers should be close to the accuracy expected for kidney biopsies. A small number of studies have examined clinical biopsy accuracy. When needle biopsies were compared with autopsy evaluation of the kidneys, there was 96% agreement for glomerular lesions and 31–88% agreement for interstitial lesions [12]. Agreement was better in diffuse disease than focal disease, but most of the patients in this series did not have glomerulonephritis. Another investigation of glomerular disease calculated theoretical misclassification rates of 25 and 15% if 10 or 20 glomeruli were present in the biopsy specimen [13], but the interstitium was not modeled. The misclassification rate of chronic allograft nephropathy, a tubulointerstitial lesion of the transplanted kidney, was found to be ∼12% in one investigation [14]. Thus a misclassification rate of 10–20% may be acceptable for biomarkers of kidney histology.
These biomarker findings were supported by validation studies conducted in an independent set of 53 LN biopsies that were performed and interpreted at a second center. Contemporaneous urine and serum samples collected at the time of biopsy were used for analyte measurement. The interstitial fibrosis biomarker equation performed similarly in the validation set as in the training set, but the interstitial inflammation biomarker equation did better in the validation set (Table 1). Interestingly, misclassification rates were lower for both biomarkers in the validation cohort. A limitation in interpreting these findings is the modest size of the discovery and validation cohorts, a universal challenge in studies of rare diseases like LN.
The specificity of these interstitial biomarkers for LN was tested by applying the equations to 41 biopsies from patients with nonlupus glomerular diseases such as IgA nephropathy, membranous nephropathy, focal segmental glomerulosclerosis and diabetic nephropathy. Each equation misclassified 29% of biopsies, suggesting somewhat higher specificity of these biomarkers for LN. Interestingly, when considering just IgA nephropathy, another immune complex disease, the misclassification rate for each biomarker was 14%, but only seven IgA patients were tested. Furthermore, the interstitial injury equations were not good biomarkers for glomerular lesions. The misclassification rates for glomerulosclerosis, glomerular crescents and glomerular necrosis were 37–40%, suggesting biomarker specificity for the tubulointerstitial compartment.
Biomarker panels with specificity for glomerular lesions can be developed in the same way as for interstitial pathology. To date there have been few individual markers correlated to specific glomerular lesions. Associations between glomerular histology and several urine biomarker candidates were examined in adult and pediatric LN patients, but many of the urine samples were collected up to 2 months after kidney biopsy [15]. Considering urine samples collected at the time of biopsy, urine neutrophil gelatinase-associated lipocalin (uNGAL) was significantly lower in the presence of cellular crescents than their absence (P < 0.004). Interestingly, both urine pentraxin 3 and uKIM-1 correlated, albeit modestly, with cellular crescents in addition to interstitial inflammation in LN [10, 11]. Urine pentraxin 3 had a correlation coefficient of 0.33 for crescents and uKIM-1 differentiated between patients who had and did not have crescents with a sensitivity of 100% but a specificity of only 63%. The fact that the same analytes may be biomarkers of both glomerular and interstitial inflammatory lesions is not surprising. It is likely, however, that in a composite biomarker the weighting of common analytes will be different for glomerular and interstitial pathology.
Several other promising candidate markers of kidney pathology have been proposed. For example, urine white blood cells from patients (n = 19) with active proliferative LN were enumerated and compared with SLE patients (n = 79) with inactive LN [16]. Not surprisingly, patients with active LN shed significantly more macrophages, T cells and B cells into their urine than controls. ROC analyses were done to determine if specific urine leukocyte subsets could differentiate active from inactive LN. Urine CD8+ T cells had an area under the ROC curve (AUC) of 1.0 and were 100% sensitive and specific for differentiating active from inactive LN, suggesting outstanding diagnostic discrimination. Urine T cells were also diagnostically superior to proteinuria (AUC = 0.92, sensitivity = 94%, specificity = 84%) and SCr (AUC = 0.60, sensitivity = 47%, specificity = 79%). This work will need external validation in an independent cohort. Additionally, only 74% of these patients had a kidney biopsy to verify histologic activity at the time urine was collected for biomarker analysis. It is critical that biomarkers of pathology be measured against kidney biopsy findings, the gold standard of renal pathology in LN.
In another approach to develop a noninvasive measure of renal histologic activity, biomarkers were sought that reflected the degree of inflammatory kidney damage (glomerular crescents, necrosis, proliferation and interstitial inflammation) as measured by the NIH activity index. In this study, putative LN biomarkers described in the literature (n = 16) plus the clinical biomarkers of complement, proteinuria and eGFR were tested to identify a diagnostic panel that could differentiate between pediatric LN patients with an NIH activity index >10 and ≤10 [17]. Urine for biomarker analysis was obtained at the time of diagnostic kidney biopsy. Using stepwise logistic modeling, the optimal biomarker panel included uMCP-1, uKIM-1 and uNGAL, but no clinical variables. The diagnostic metrics of this panel were excellent, with an AUC of 0.92, sensitivity of 90%, specificity of 86%, positive likelihood ratio of 6.3 and a false positive rate of 14%. This biomarker panel needs to be tested in independent LN cohorts, including adults, before it can be applied clinically. A potential limitation of the panel is the cutoff level of 10 for the activity index. This may not be as useful as being able to detect lower levels of histologic renal activity, because it has been shown that a residual activity index as low as 3 after induction therapy portends a poor overall prognosis for long-term kidney function [18]. Several other individual urine analytes have been correlated with the activity index of LN biopsies. These include VCAM-1 (r = 0.42, P = 0.05), colony stimulating factor-1 (CSF-1; r = 0.47, P < 0.005) and the M2 macrophage cell surface marker CD163 (r = 0.64, P < 0.01) [19–21].
A remaining concern with the putative biomarkers that track histologic activity is that none have been evaluated prospectively to determine if they truly reflect the dynamic nature of kidney injury in LN and change in parallel with changes in activity as LN is treated or as it flares. This type of evaluation is necessary if these biomarkers are to be used to assess the histologic response to treatment and will require validation in a cohort of patients who have undergone serial kidney biopsies.
BIOMARKERS TO GUIDE THE CHOICE OF
THERAPY IN LN
The impetus to find biomarkers that predict a favorable response to therapy has increased as novel and expensive drugs that act on specific immune pathways are brought to clinical trial. Such companion diagnostics are often the targets of these biologic and small molecule drugs. Perhaps the best example of this type of biomarker comes from a study of anifrolumab, a monoclonal antibody against interferon-α receptor 1, in nonrenal SLE [22]. This study showed anifrolumab was significantly more effective than placebo in patients with a high interferon gene signature but not in patients with a low signature. The interferon gene signature is measured in peripheral blood leukocytes as the intensity of expression of a subset of transcripts known to be induced by type 1 interferons. Importantly, in a recently launched clinical trial of anifrolumab in LN (ClinicalTrials.gov identifier: NCT02547922) patients will be stratified into high and low interferon-α gene signature groups during the screening period.
Another potential source of biomarkers to guide therapy is the kidney itself. We recently measured intrarenal gene expression in the diagnostic biopsies of patients with LN flares [23]. After standard-of-care induction therapy with corticosteroids plus MMF or cyclophosphamide, patients were evaluated to determine who had achieved an early complete clinical renal remission and who had not yet responded to treatment. Principal component analysis of renal gene expression in the diagnostic kidney biopsy (the biopsy just before the start of induction therapy) segregated patients into responders and nonresponders, suggesting that molecular pathways active in the kidney at the time of LN flare determine whether a patient will be an early treatment responder or a nonresponder. For example, the proinflammatory nuclear factorκB pathway appeared to be more activate in complete responders than nonresponders. Identification of differentially activated molecular pathways can potentially facilitate the search for urine or serum biomarkers of treatment response by focusing investigation on components of those pathways. This process could be applied to patients treated with novel LN therapies to identify treatment response biomarkers. Measuring such biomarkers before initiating treatment would help in determining the best approach for the individual patient.
IDENTIFYING TRUE BIOMARKERS OF LN
FLARE: LESSONS LEARNED FROM THE
OHIO SLE STUDY
Although several candidate biomarkers for LN flare have been identified, almost all studies have used cross-sectional data collection. Few putative flare biomarkers have been tested serially during periods leading up to flare. Only through such longitudinal testing can biomarkers of LN flare be properly classified as markers of flare (changing at the time of flare) or as forecasters, or possibly triggers, of flare (changing prior to flare onset).
To address this issue, our laboratory initiated the Ohio SLE Study (OSS; described in Zhang et al. [9] and Ross et al. [30]) in 2001. This study assessed SLE patients every 2 months, regardless of disease activity, effectively removing ascertainment bias as a confounding variable in data analysis. At each bimonthly visit a detailed medical evaluation was done, medications were recorded and blood and urine samples were collected. During the ∼60 days between each visit, each patient recorded variables such as sun exposure, body temperature, perceived stress levels and illness on a daily basis. Study investigators reviewed the medical charts to adjudicate flares, based on prespecified criteria for both nonrenal and renal flares. At the end of follow-up in 2008 the OSS consisted of 116 lupus patients (74 with LN) followed for a median of 45 months, during which time 114 nonrenal flares and 88 renal flares were identified in >2200 follow-up visits. The OSS has provided a platform to classify previously identified LN biomarkers as markers or forecasters of flare and also facilitated the identification of new LN flare biomarkers. The power of this longitudinal design is perhaps best exemplified by OSS biomarker studies focused on the complement system.
Complement and LN
The complement system clearly plays a role in the pathogenesis of lupus, especially LN, but the specific nature of that role is complex and often confounding. Part of this confusion is due to multiple activation pathways, multiple regulators and points of regulation in the activation pathways, many complement receptors that mediate diverse functions and genetic variations in complement activation proteins and regulators. Added to this is the apparent contrary roles observed for complement in lupus pathogenesis, where complement appears to protect against lupus onset and disease activity (via the classical pathway), but also directly participates in the tissue damage associated with lupus, especially in the kidney.
Circulating levels of C3 and C4. A decrease in circulating levels of C3 and C4 indicative of complement activation has long been viewed as an indicator of increased LN disease activity, and low C3 and C4 levels have been recently added to the revised criteria for diagnosing lupus [24]. Nevertheless, the utility of following C3 and C4 levels for managing LN patients remains unclear and controversial [25, 26]. The reason for this, at least in part, is that few studies have measured C3 and C4 levels at regular intervals leading up to flare. To directly address the potential for C3 and C4 to serve as biomarkers of flare, C3 and C4 levels were measured bimonthly in 71 OSS LN patients who were followed for an average of 35 months, during which time 70 renal flares were identified [27]. Comparing complement levels 2 months before and at flare with mean baseline complement levels by univariate analysis showed that C3 and C4 decreased at flare, but not 2 months before flare. Sensitivity and specificity were modest at ≤75%. However, when a multivariate regression analysis was done that included >1000 bimonthly measurements of C3 and C4 levels and other clinical values [C-reactive protein (CRP), erythrocyte sedimentation rate (ESR)], demographics and genetic variants that affect C3 activation, a clearer picture emerged. First, a significant decrease in C4 levels (along with younger age and higher ESR levels), but not C3 levels, occurred 2 months prior to the onset of renal flare. Second, a significant decrease in C3 levels (again along with younger age and higher ESR levels), but not C4 levels, occurred at the time of renal flare. Importantly, the risk for renal flare occurring as C3 decreased due to activation was much higher in individuals who were homozygous for a dysfunctional genetic variant of factor H (FH 402HH; Figure 2A), a regulator of C3 activation.
![Studies of the complement system in the OSS. All figures reproduced in modified form from previous reports [27, 32, 36], with permission. (A) Risk curves, according to factor H 402Y->H genotype, for renal flare as plasma C3 levels decrease. The vertical arrows show the median plasma C3 levels at the ‘no-flare’ and ‘at-flare’ follow-up visits. (B) Mean E-CR1 levels in nonrenal and LN patients at baseline, 2 months before (−2), at the time of (0) and 2 months after (+2) a nonrenal or renal flare. **P < 0.001 for a decrease in detectable E-CR1 level at nonrenal flare compared with nonrenal flare baseline in LN. (C) Anti-C1q IgG levels at 8, 6, 4 and 2 months before (−8, −6, −4, −2) and at (0) renal flare in patients who were anti-C3b negative or anti-C3b-positive. *P = 0.02 for an increase at renal flare for anti-C3b positive patients.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/32/suppl_1/10.1093_ndt_gfw300/5/m_gfw30002.jpeg?Expires=1750239015&Signature=qK7US8OH2XO7HnXx8CWO4BZrqJbwO6OhXIc17KsnbLh2lxFZLqybToSD2nROcvAHiuoeO8y6nUFOt3Uf3h9dOxWVBa~sBDf1ymiBascZtmEBh4awMbg6k1wpcXeGfaQX~srmStV43ktaG9xrWv9tBY7Lzxk9EKg6SRl4ukrgFVpbOY~PiTFzahtGn7yf4~gpHoDTkzh34b~c5AvhwThKZPB6iV82mZn0CQ9opHNEtlop8Fb9qgivQFLkFfJcUmPyFQyAtvOJsLXcZHkGM7xFB9wgf8zqW-KcXDQAdG2hkwDIpXOPnE4gtlXpHBQ47GXJVYjjDweL7rlvb5sa9IbpCQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Studies of the complement system in the OSS. All figures reproduced in modified form from previous reports [27, 32, 36], with permission. (A) Risk curves, according to factor H 402Y->H genotype, for renal flare as plasma C3 levels decrease. The vertical arrows show the median plasma C3 levels at the ‘no-flare’ and ‘at-flare’ follow-up visits. (B) Mean E-CR1 levels in nonrenal and LN patients at baseline, 2 months before (−2), at the time of (0) and 2 months after (+2) a nonrenal or renal flare. **P < 0.001 for a decrease in detectable E-CR1 level at nonrenal flare compared with nonrenal flare baseline in LN. (C) Anti-C1q IgG levels at 8, 6, 4 and 2 months before (−8, −6, −4, −2) and at (0) renal flare in patients who were anti-C3b negative or anti-C3b-positive. *P = 0.02 for an increase at renal flare for anti-C3b positive patients.
These findings reveal some interesting relationships among C3, C4 and renal flare. The unique roles of decreased C4 as a significant forecaster of LN flare and C3 as a significant marker of LN flare imply that classical pathway activation can trigger a flare but the alternative pathway is what drives complement-mediated kidney damage reflected by the clinical manifestations of a renal flare (abnormal urine sediment, proteinuria, lower GFR). This is supported by the observation that genetic variation of factor H is also implicated as a marker of flare, but not as a forecaster of flare. The specific nature of that variation is also revealing in that patients who are homozygous for the dysfunctional form of factor H are less efficient at regulating C3 activation, leading to an increased likelihood of driving complement-mediated tissue damage. Conversely, patients who have the fully functional form of factor H are more capable of controlling C3 activation, and thus the relationship is weak between decreased C3 levels and renal flare. These insights were only possible through rigorous longitudinal testing.
Erythrocyte type 1 complement receptor (E-CR1). The type 1 complement receptor (CR1) is found on many circulating cells, where it binds the activated complement proteins C3b, C4b, iC3b as well as C1q. Humans uniquely express CR1 on erythrocytes (E-CR1), while nonhuman primates express a closely related CR1-like protein [28]. Nonprimate species express neither. These receptors in humans and nonhuman primates allow erythrocytes in the circulation to bind complement-opsonized substrates such as immune complexes (ICs), a phenomenon known as immune adherence. This process promotes safe, regulated removal of ICs from the circulation. Interruption of this process leads to tissue trapping of ICs in vulnerable organs like the kidneys. Low levels of circulating E-CR1 occur in lupus and correlate with high disease activity [29, 30]. This loss is acquired, likely the consequence of E-CR1 interacting with complement-opsonized ICs and possibly mediated by erythrocyte-fixed C3b [31]. Thus E-CR1 has been viewed both as a protective factor against LN flare and potentially a biomarker of LN activity.
The OSS provided an opportunity to test if decreased E-CR1 levels could serve as a biomarker of LN flare [32]. Mean E-CR1 levels were measured in fresh blood by radioimmunoassay bimonthly in 43 OSS patients for an average of 22 months, during which time 66 flares were identified. From these measurements two key observations were made. First, E-CR1 levels fluctuated greatly from one bimonthly visit to the next, making any single pre-flare measurement useless as a baseline value. Second, when compared with E-CR1 values averaged from values ≥4 months from a flare as a baseline, a significant decrease occurred at the time of nonrenal flare in LN patients, followed by a rebound in levels, while no significant change was observed at the time of renal flare (Figure 2B). Levels of soluble plasma CR1 did not change during any of these periods.
We interpret these unexpected findings as revealing three important aspects about E-CR1 changes during active LN and LN flare. First, acquired decreases in E-CR1 levels that have been reported to be associated with active LN from numerous cross-sectional studies often are not true decreases in receptor levels, at least acutely. Rather they reflect a decrease in the ability to detect E-CR1 due to altered epitopes recognized by anti-CR1 antibodies used in the detection assays as a result of interactions with complement-opsonized ICs in the circulation. This is in agreement with earlier studies in nonhuman primates exposed to daily IC injections to induce glomerulonephritis [33, 34]. Second, fluctuations in detectable levels indicate that E-CR1 is performing its function of interacting with circulating ICs, and protecting the kidney against IC deposition. Third, when this function is lost, perhaps due to eventual damage of the CR1 binding site from excessive IC interactions, E-CR1 epitopes remain unaltered and maximally exposed to detecting antibodies, thus creating the appearance of high E-CR1 levels, despite loss of function. Loss of function means loss of protection from IC deposition in the kidney, resulting in renal involvement during flare.
Autoantibodies to C1q and C3b. Autoantibodies to complement proteins such as C1q and C3b occur in lupus patients and likely reflect an autoimmune response to products of complement activation. Anti-C1q antibodies have been extensively studied in SLE and have been considered to be specific biomarkers for LN compared with nonrenal lupus, and for active LN compared with inactive LN [35]. The relationship with active LN has led to conjecture that anti-C1q could serve as a biomarker of renal flare. This was tested in the OSS along with anti-C3b autoantibodies [36]. In this study, anti-C1q was first compared with anti-C3b in a cross-sectional analysis by testing serum samples from the OSS patients collected at OSS entry. This analysis showed that anti-C3b was less sensitive than anti-C1q (36 versus 63%) but more specific than anti-C1q (98 versus 71%) for LN compared with nonrenal lupus. Importantly, only anti-C3b was associated with OSS LN patients with documented renal flares (with 51% sensitivity and 81% specificity). For the longitudinal analysis to assess these antibodies as biomarkers of renal flare, 24 renal flares from LN patients who were anti-C1q positive were tested bimonthly for circulating anti-C1q levels from 8 months before flare to the time of flare (defined as a flare interval). Anti-C3b was present in 16 of these flare intervals, and the levels of anti-C3b antibody were also measured in these intervals. Multivariate regression analysis revealed that, when considering all 24 flare intervals, mean anti-C1q levels did not change during the flare interval. Thus anti-C1q did not function as an independent biomarker of renal flare. However, when considering just the 16 flare intervals in which anti-C3b was present, anti-C1q levels increased significantly at flare (Figure 2C) and thus did function as a biomarker of flare. There was also a trend for anti-C3b antibodies to increase at flare, though this change fell short of statistical significance (P = 0.07). Interestingly, in this same study cohort, a decreased serum C3 level was not a biomarker of renal flare in anti-C3b-negative samples, but was in anti-C3b-positive samples.
These results provide further insight into the relationship of complement activation with LN and renal flare. Anti-C1q antibody is a surrogate for classical pathway activation, and the inability of this antibody alone to serve as a biomarker of renal flare suggests that this pathway by itself is not sufficient to drive kidney tissue damage, in agreement with the OSS study on circulating C3 and C4 described above [27]. Anti-C3b antibody is a surrogate for C3 cleavage, a step that is amplified by the alternative pathway. The observations that, compared with anti-C1q, anti-C3b is more specific for LN, is uniquely associated with LN patients prone to renal flare and identifies patients in whom anti-C1q and other markers of complement activation can serve as biomarkers of renal flare suggests that this autoantibody reflects a level of complement activation sufficient to drive kidney damage. Accordingly, the presence of anti-C3b antibody likely identifies LN patients in whom therapies targeting the complement system may be particularly effective.
This study may also explain some of the discrepancies in the few previous studies that have assessed serial anti-C1q measures in an LN cohort. For instance, a longitudinal evaluation of 23 LN patients concluded that the detection of anti-C1q antibodies alone was not helpful in the management of LN [37]. However, the appearance or increase in anti-C1q plus anti-dsDNA antibodies was associated with a future (2–4 months later) LN flare with positive and negative predictive values of ∼70%, suggesting a high risk of impending flare in patients having both antibodies. It is intriguing to speculate that the dual presence of anti-dsDNA and anti-C1q antibodies may coincide with, and possibly drive, a level of complement activation that leads to the onset of anti-C3b antibodies.
Other studies of LN biomarkers in longitudinal cohorts
Although cross-sectional studies dominate the biomarker literature, recently several longitudinal cohorts have been used to evaluate LN biomarkers. For example, in a study of 64 pediatric LN patients, uMCP-1 and uNGAL were measured at three serial visits, each roughly 3 months apart [38]. This investigation found that uMCP-1 could predict improvement in disease activity (AUC in ROC analysis 0.81) and uNGAL could predict disease worsening (AUC = 0.76) when followed over time. Interestingly, in another longitudinal cohort, changes in NGAL levels after LN flare and during treatment were predictive of complete remission [39]. In this investigation, 13 of 45 patients experienced an LN flare. Patients who achieved a complete response to treatment showed a significant increase in the ratio of the fractional excretion of NGAL to the fractional excretion of protein at least 3 months before remission. The negative predictive value of this ratio was excellent at 96%, but the positive predictive value was only 16%. Serum CSF-1 was tested in two small longitudinal cohorts (14 and 17 patients, respectively) as a biomarker for new-onset LN and LN flare [20]. Increasing serum CSF-1 predicted new-onset LN and impending LN flare with a positive predictive value of 83% and a negative predictive value of 63%. Importantly, CSF-1 performed better than conventional clinical markers of lupus activity, including anti-dsDNA antibody titers, complement C3 levels and nonspecific markers of inflammation (ESR, CRP). These data suggest CSF-1 should be examined in a larger LN cohort and a more diverse lupus population, as almost all of these patients were white.
SUMMARY AND FUTURE DIRECTIONS
A large number of putative LN biomarkers have been identified, but none have been incorporated into clinical practice. Several factors contribute to this roadblock between discovery and clinical application. Biomarkers have generally been identified by comparing novel candidates to clinical indicators of disease activity. By this approach, a biomarker can never be more informative than the clinical data. Such biomarkers, while contributing to the overall understanding of LN pathogenesis, are not likely to become clinical tools. Conversely, if a biomarker represents diagnostic or prognostic information that cannot be accurately or readily ascertained on clinical grounds alone, it conceivably will add value to the clinical assessment and could become a novel management tool.
Additionally, LN biomarker studies have often been done in cross-sectional cohorts. Because of the dynamic nature of LN, it is necessary to understand the longitudinal behavior of a biomarker in active disease, during treatment and during remission. The lack of well-phenotyped LN cohorts with annotated biospecimens and long-term follow-up contributes to this roadblock, and also limits the availability of validation cohorts.
Finally, most biomarkers tested have been single analytes. It seems far more likely that in order to assess a complex process like LN, biomarker panels will need to be developed that are composed of several analytes, each providing independent information.
Figure 3 outlines a strategy for biomarker development in LN. Although biomarker discovery will be an ongoing process that is likely to improve as disease pathogenesis becomes increasingly clear, and as more sophisticated discovery techniques are developed, there are currently several candidate biomarkers, some reviewed here, that have been evaluated through the verification stage. A focus on validation efforts for these promising candidates now seems warranted.
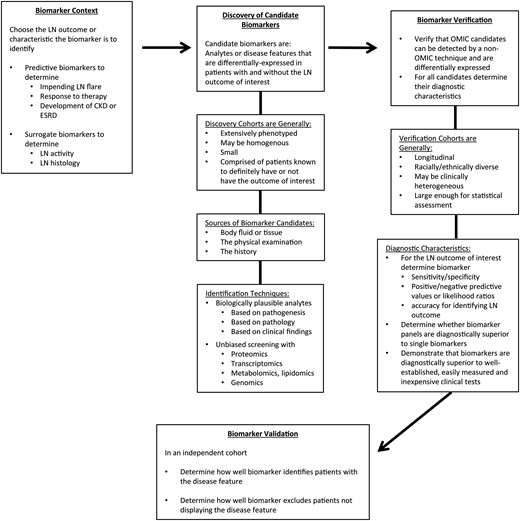
A strategy for biomarker development in LN: from context to validation. The phrase ‘LN biomarker’ is vague. The purpose of a biomarker in managing LN should be precisely defined. Some important unmet needs in the care of patients with LN include how to predict who will flare before they do so and who is likely to respond to therapy or not respond and develop chronic kidney disease (CKD) or end-stage renal disease (ESRD). Disease activity is generally assessed clinically, but clinical impressions do not necessarily reflect histologic activity. Understanding intrarenal histologic activity in a noninvasive way would improve LN management. Biomarkers that predict these outcomes or are surrogates of a kidney biopsy will be of high clinical value. Candidate LN biomarkers are identified in discovery cohorts of LN patients. Such cohorts are generally small but well-characterized clinically. LN outcomes are known for each patient. For example, to identify biomarkers of histologic activity the comparator for potential candidates should be the kidney biopsy. Biomarker candidates can be identified by a variety of techniques. Screening with omic techniques is time efficient. Candidates identified in the discovery phase of biomarker development need to be verified before proceeding with validation. This is especially important for candidates derived using omic techniques. Depending on how large omic data sets are analyzed, candidates found to be significant may not be differentially expressed when measured by an alternate technique specific for that analyte. Alternatively, differentially expressed transcripts may not translate to changes in protein levels, and would not be further developed. It is also important to understand how well the biomarker candidates reflect the clinical outcomes of interest in a cohort that is larger and possibly more diverse than the discovery cohort. By studying a larger cohort, the diagnostic metrics of a biomarker candidate can be determined. This will be important in deciding whether a candidate is superior to existing clinical diagnostics, and thus should be taken forward, and whether a candidate can stand on its own, or would be more robust if part of a composite biomarker (a biomarker panel). Finally, biomarkers need to be validated using independent cohorts by other investigators.
ACKNOWLEDGEMENTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) P01 DK55546 (D.J.B., B.H.R.), NIDDK U01 DK085673 (B.H.R., J.B.K.), NIDDK R01 DK093574 (S.S.W.) and National Institutes of Health UL1 RR025755 (to Ohio State University). We would like to thank Dr Xiaolan Zhang and Huijuan Song (Ohio State University Wexner Medical Center, Nephrology Division) for urine analyte measurements. All kidney biopsies were read during the course of clinical evaluations by the Nephropathology Divisions at the Ohio State University Wexner Medical Center and Brigham and Women's Hospital.
CONFLICT OF INTEREST STATEMENT
The results presented in this paper have not been published previously in whole or part, except in abstract format.
REFERENCES
Author notes
D.J.B. and M.M. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
Comments