





Glomerular injury often incites a progression to chronic kidney disease, which affects millions of patients worldwide. Despite our current understanding of this disease's pathogenesis, there is still a lack of therapy available to curtail its progression. However, exciting new data strongly suggest the podocyte—an actin-rich, terminally differentiated epithelial cell that lines the outside of the glomerular filtration barrier—as a therapeutic target. The importance of podocytes in the pathogenesis of human nephrotic syndrome is best characterized by identification of genetic mutations, many of which regulate the actin cytoskeleton. The intricate regulation of the podocyte actin cytoskeleton is fundamental in preserving an intact glomerular filtration barrier, and this knowledge has inspired new research targeting actin-regulating proteins in these cells. This review will shed light on recent findings, which have furthered our understanding of the molecular mechanisms regulating podocyte actin dynamics, as well as discoveries that have therapeutic implications in the treatment of proteinuric kidney disease.
INTRODUCTION
The kidney glomerulus is a highly specialized structure that allows selective ultrafiltration of plasma so that essential proteins are retained in circulation while by-products of cellular metabolism are eliminated into the urine. The glomerular filtration barrier is composed of a fenestrated endothelium, the glomerular basement membrane (GBM) and podocytes [1]. Structural defects at any level of this barrier can result in loss of essential proteins such as albumin and immunoglobulins. Proteinuric kidney diseases caused by a dysfunctional glomerulus account for ∼80% of all cases that progress to end-stage kidney disease, ultimately requiring renal replacement therapy. Thus, an understanding of the fundamental biology is critical for the development of precise therapies for proteinuric kidney diseases. Podocytes lie within the glomerular apparatus. They are highly specialized, terminally differentiated cells with a large cell body that extends the thick primary processes. The primary processes branch further into fine secondary and tertiary processes (foot processes) that are supported by microtubular and actin cytoskeletons, respectively. Functional podocytes rely on these cytoskeletons to stabilize the glomerular capillaries and participate in the barrier function of the glomerular filter via interactions with cell–cell junction and cell–matrix proteins. Utilizing a two-photon microsocopy on live translucent zebrafish expressing enhanced green fluorescent protein, it has been demonstrated that podocytes are relatively stationary during normal conditions [2]. Yet, following podocyte injury, such as in the unilateral ureteral ligation-induced and adriamycin-induced injury models, there are dynamic podocyte movements observed using serial multiphoton microscopy in vivo [3]. These observations suggest that signals that induce lamellipodial extensions and focal adhesion turnover ultimately lead to retraction and movement of the foot processes and may contribute to the pathogenesis of podocyte foot process effacement (FPE), a term used to describe the loss of the intricate interdigitations of the foot processes. Podocyte FPE can occur following glomerular injury due to the alteration of the podocyte actin-related proteins with the GBM. Although the pathological relevance of FPE is currently unsettled, an excellent detailed review by Kriz et al. [4] suggests that FPE is a method for podocytes to prevent detachment, and the severity may not be uniform in all proteinuric kidney disease processes. Further supporting the pivotal role of the podocyte actin cytoskeleton in the pathogenesis of nephrotic syndrome is the discovery of human genetic mutations in actin-related genes, such as ACTN4 (gene encoding actinin-4 protein), MYO1E (gene encoding myosin IE protein), INF2 (gene encoding inverted formin-2 protein), ITGA3 (gene encoding alpha 3 integrin protein), ARHGAP24 (gene encoding Rho GTPase activating protein 24 protein), ARHGDIA (gene encoding Rho GDP dissociation inhibitor alpha protein), ANLN (gene encoding anillin protein) and CD2AP (gene name encoding CD2-associated protein) [5–8]. Mutations in the genes above have been demonstrated to cause profound changes in the podocyte actin cytoskeleton. Therefore, it has been speculated that interventions targeting pathways that regulate the actin cytoskeleton can play an important role in the treatment of proteinuric kidney diseases. Our goal in this review is to highlight the regulation of molecules governing the podocyte actin cytoskeleton, as well as potential therapeutic applications that target them.
ACTIN CYTOSKELETON AND FUNCTION IN PODOCYTES
The actin cytoskeleton provides architectural and functional support that produces the framework for the cell and its connection to the extracellular matrix [9]. Actin cytoskeletal remodeling orchestrates many features of cellular physiology, including cell shape, adhesion and motility [10]. Podocytes rely on an intact actin cytoskeleton due to the cell's complex cell architecture—cellular foot processes extend from the cell body, which encircles the adjacent capillaries. The cell–matrix adhesion and cell–cell junction proteins that are linked directly or indirectly to the podocyte actin cytoskeleton must withstand the external forces elicited from daily filtration [11]. Previous studies have indicated that when podocytes are injured, cytoskeletal reorganization is the common final pathway resulting in FPE. The highly ordered parallel contractile actin filament bundles in the foot processes become disordered, short and branched under pathological conditions, thus resulting in proteinuria [12, 13]. Although the dysregulation of the actin cytoskeleton following podocyte injury underlies FPE and subsequent proteinuria, which may induce the progression of renal injury in proteinuric kidney disease, it appears that podocytes also display a remarkable capacity to recover when the insult is removed. For example, in minimal change disease, the foot processes can recover from the effaced phenotype to its normal upright position, as determined by ultrastructural observations made from transmission electron microscope images. This phenomenon is also observed in some transient proteinuric models in rodents, such as the protamine sulfate infusion and lipopolysaccharide (LPS) injection, where there is reversible FPE and proteinuria [14–16]. The exact mechanism inducing podocytes to undergo FPE is being actively investigated, as mechanistic insight may unearth new potential therapies. In the following section, we will discuss the proteins that regulate the actin cytoskeleton, while also focusing on the current therapeutic targets that have been identified in mouse models of proteinuria and in human nephrotic syndrome.
PODOCYTE RHO GTPASES: A DYNAMIC CHANGE IN THE ACTIN CYTOSKELETON
The Rho family of small GTPases controls signal transduction pathways that influence many aspects of cell behavior, including cytoskeletal dynamics [17, 18]; the family includes RhoA, Rac1 and Cdc42, which are highly expressed in podocytes. Each GTPase acts as a molecular switch, cycling between an active GTP-bound state regulated by guanine nucleotide exchange factor (GEF) and an inactive GDP-bound state. Cycling occurs through the hydrolysis of GTP to GDP by GTPase-activating proteins (GAP). In the GTP-bound state, Rho family member RhoA acquires an active conformation which has been implicated in stress fiber formation by promoting actin:myosin contraction at the rear of the cell. Conversely, the subclasses Rac1 and Cdc42 promote cell motility through the formation of lamellipodia and filopodia, respectively. In migrating cells, Cdc42 regulates the direction of migration at the leading edge [19–21].
In podocytes, the role of RhoA was initially investigated through the generation of a podocyte-specific constitutively active (CA) RhoA mouse that led to albuminuria and FPE due to the disorganization of the actin cytoskeleton. Podocytes expressing CA-RhoA had increased stress fiber formation and reduced motility [13]. On the other hand, the podocyte-specific ablation of RhoA displayed normal glomerular morphology and intact foot processes, suggesting that RhoA is dispensable during the development and maintenance of podocytes [13]. More recently, human mutations in the kidney ankyrin repeat containing protein (KANK1,2,4) have been shown to cause steroid-resistant nephrotic syndrome. In podocytes, the loss of KANK2 resulted in an increase in RhoA activity and reduced cell migration, similar to what had been described in the podocyte-specific CA-RhoA mouse model [22]. These results indicate that increased RhoA activity can lead to FPE and clinical manifestation of proteinuria [13]. Further suggesting that RhoA activation may be detrimental to podocyte health, mice treated with inhibitors for RhoA-dependent kinase (ROCK) attenuated proteinuria and prevented kidney failure in a various mouse models of nephropathy [23–26]. Moreover, the acid sphingomyelinase inhibitor dramatically attenuated proteinuria and glomerulosclerosis in the db/db mice model for diabetic kidney disease (DKD) by inhibiting the acid sphingomyelinase-like phosphodiesterase 3b (SMPDL3b)-induced RhoA activation [27]. What will require further clarification is whether reduced podocyte motility observed in cell culture following RhoA activation correlates to the observed phenotype of proteinuria and FPE in vivo. Utilizing state-of-the art technology by two-photon or intravital in vivo imaging in mice with fluorescently tagged podocyte Rho GTPases may shed further light on this issue.
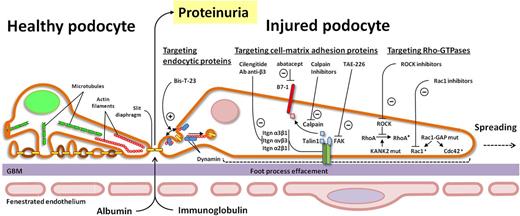
Cartoon schematic depicting potential therapeutic targets on small Rho GTPase, cell matrix adhesion proteins and endocytic proteins. ROCK, Rho-associated protein kinase; CDC42, cell division control protein 42 homolog; FAK, focal adhesion kinase; KANK2, KN motif and ankyrin repeat domains 2.>
Unlike Rac1 and RhoA, mice with podocyte-specific deletion of Cdc42 display severe proteinuria and podocyte FPE with disorganized stress fibers and glomerulosclerosis. It has also been shown that Cdc42 in podocytes is required for actin nucleation at sites of the slit diaphragm protein, nephrin [22]. Further supporting the importance of the Rho GTPase pathway, loss of podocyte Rhophilin-1 in mice resulted in abnormal actinomyosin regulation and RhoA signaling events [33].
The ability of these small Rho GTPase proteins to profoundly modulate the podocyte actin cytoskeleton suggests their critical role not only in the pathogenesis of nephrotic syndrome, but also as potential therapeutic targets.
PODOCYTE ACTIN CYTOSKELETON–GBM INTERACTIONS
The actin cytoskeleton in podocyte foot processes is also linked with the GBM by a variety of cell–matrix adhesion receptors, including integrins and focal adhesion proteins. Integrins are αβ heterodimeric cell adhesion receptors, while focal adhesions are cytoplasmic proteins that anchor the cell to the extracellular matrix. The combination of these structures provides physical reinforcement for the podocytes to withstand mechanical stress while transmitting signals within the cell and regulating actin polymerization, cell shape and cell motility [34, 35]. The importance of integrins and focal adhesion proteins has been demonstrated in both genetic mouse models of diseases and in human genetic mutations, that result in nephrotic syndrome. For example, α3β1, the major integrin heterodimer in the podocyte, when both ablated individually in mouse podocytes resulted in severe proteinuria and FPE [36, 37]. Human mutations in ITGA3 have also been identified as causal for nephrotic syndrome [38, 39]. Focal adhesion proteins such as talin1 and integrin-linked kinase (ILK) have also been implicated in maintaining a normal filtration barrier in mice, while ACTN4 mutations have been discovered to cause FSGS [5, 40]. Recently, investigators have placed significant emphasis on manipulating integrin and focal adhesion signaling for therapeutic purposes. It has been shown that inhibiting focal adhesion kinase (FAK), a non-receptor tyrosine kinase, with TAE-226 in mouse models of glomerular injury leads to FPE, proteinuria and podocyte migration [15]. Furthermore, podocyte-specific ablation of Crk, a downstream interactor of FAK, results in protection from protamine sulfate-induced podocyte FPE [14]. Focal adhesion turnover is also regulated by glycogen synthase kinase 3β (GSK3β), where activation has been demonstrated to induce podocyte injury [41, 42]. In the adriamycin-induced nephropathy model, treatment with GSK3β inhibitor, lithium, significantly ameliorated the albuminuria and FPE, while also reducing podocyte migration in vitro [42]. Another critical integrin expressed in podocytes is αvβ3 integrin, which can be activated by uroplasminogen type I activator receptor (uPAR) (in podocytes) [43], or its soluble form, suPAR (from the circulation) [44]. The activation of αvβ3 integrin induced FPE through actin cytoskeletal rearrangement [45]. Blocking αvβ3 with an anti-β3 antibody or the small-molecule inhibitor, cilengitide, alleviated proteinuria induced by uPAR [43, 44]. Emerging evidence also suggests that the use of abatacept in patients with focal segemental glomerulosclerosis inhibits the binding of talin1, an integrin-activating protein, to podocyte-associated B7-1, a costimulatory protein best characterized in antigen-presenting cells. Blocking B7-1, which is expressed in injured podocytes, allowed for normalization of integrin signaling [46] (Table 1). It will be important in the coming years to further validate these findings as podocyte B7-1 induction in proteinuric diseases, its detection in kidney biopsy specimens and the therapeutic efficacy of CTLA4-Ig in proteinuric patients have not all been in accord [51–53]. Further highlighting the importance of talin1 function is its proteolysis secondary to calpain activation following injury [54]. Calpain is a calcium-dependent cysteine protease that has enzymatic activity to cleave diverse substrates. Treatment with calpain inhibitor abrogates talin1 cleavage and reduced kidney injury. Recent evidence also suggests the importance of targeting α2 integrin in podocytes, as podocyte-specific deletion of ITGA2 results in significantly less proteinuria and glomerulosclerosis following adriamycin-induced injury when compared with wild-type mice. Selective pharmacological inhibition of integrin α2β1 in wild-type mice also reduced proteinuria [48] (Figure 1). More research must be done in order to confirm the mechanisms of how these exciting new inhibitors affect the podocyte integrin pathways [42].
Key points focusing on the podocyte actin cytoskeleton as a therapeutic target
| 1. The inhibition of small Rho GTPases RhoA and Rac1 could potentially attenuate proteinuria, and improve kidney function and histological damage [23–25, 47] |
| 2. Inhibition of key focal adhesion proteins such as FAK and Crk1/2 reduced podocyte FPE and proteinuria by decreased podocyte migration in mouse models of glomerular injury [14, 15] |
| 3. Modulating activation of integrin β1 through abatacept (CTLA-4-Ig) or integrin αv inhibitor, cilengitide or integrin α2β1 has potential therapeutic application in proteinuric disease [43, 44, 46, 48] |
| 4. TRPC5 inhibitor ML204 protected LPS-induced proteinuria and FPE induced by LPS and protamine sulfate [49] |
| 5. BisT23 induced dynamin oligomerization and actin polymerization may have implications in the treatment of various causes of nephrotic syndrome [50] |
| 1. The inhibition of small Rho GTPases RhoA and Rac1 could potentially attenuate proteinuria, and improve kidney function and histological damage [23–25, 47] |
| 2. Inhibition of key focal adhesion proteins such as FAK and Crk1/2 reduced podocyte FPE and proteinuria by decreased podocyte migration in mouse models of glomerular injury [14, 15] |
| 3. Modulating activation of integrin β1 through abatacept (CTLA-4-Ig) or integrin αv inhibitor, cilengitide or integrin α2β1 has potential therapeutic application in proteinuric disease [43, 44, 46, 48] |
| 4. TRPC5 inhibitor ML204 protected LPS-induced proteinuria and FPE induced by LPS and protamine sulfate [49] |
| 5. BisT23 induced dynamin oligomerization and actin polymerization may have implications in the treatment of various causes of nephrotic syndrome [50] |
LPS, lipopolysaccharides.
Key points focusing on the podocyte actin cytoskeleton as a therapeutic target
| 1. The inhibition of small Rho GTPases RhoA and Rac1 could potentially attenuate proteinuria, and improve kidney function and histological damage [23–25, 47] |
| 2. Inhibition of key focal adhesion proteins such as FAK and Crk1/2 reduced podocyte FPE and proteinuria by decreased podocyte migration in mouse models of glomerular injury [14, 15] |
| 3. Modulating activation of integrin β1 through abatacept (CTLA-4-Ig) or integrin αv inhibitor, cilengitide or integrin α2β1 has potential therapeutic application in proteinuric disease [43, 44, 46, 48] |
| 4. TRPC5 inhibitor ML204 protected LPS-induced proteinuria and FPE induced by LPS and protamine sulfate [49] |
| 5. BisT23 induced dynamin oligomerization and actin polymerization may have implications in the treatment of various causes of nephrotic syndrome [50] |
| 1. The inhibition of small Rho GTPases RhoA and Rac1 could potentially attenuate proteinuria, and improve kidney function and histological damage [23–25, 47] |
| 2. Inhibition of key focal adhesion proteins such as FAK and Crk1/2 reduced podocyte FPE and proteinuria by decreased podocyte migration in mouse models of glomerular injury [14, 15] |
| 3. Modulating activation of integrin β1 through abatacept (CTLA-4-Ig) or integrin αv inhibitor, cilengitide or integrin α2β1 has potential therapeutic application in proteinuric disease [43, 44, 46, 48] |
| 4. TRPC5 inhibitor ML204 protected LPS-induced proteinuria and FPE induced by LPS and protamine sulfate [49] |
| 5. BisT23 induced dynamin oligomerization and actin polymerization may have implications in the treatment of various causes of nephrotic syndrome [50] |
LPS, lipopolysaccharides.
TRANSIENT RECEPTOR POTENTIAL CATION CHANNEL AND THE ACTIN CYTOSKELETON IN PODOCYTES
Transient receptor potential cation channels (TRPCs) are non-selective cationic channels with a preference for calcium ions, which play a major role in pathogenesis of renal and cardiovascular disease [55]. In podocytes, many of these TRPCs have been shown to be expressed, including TRPC1, TRPC3, TRPC4, TRPC5 and TRPC6 [56–60]. The discovery that gain-of-function TRPC6 mutations lead to familial FSGS [60, 61] and that TRPCs are activated in various acquired glomerulopathies highlights the importance of these channels [63].
TRPCs allow the influx of Ca2+ into the podocytes and can regulate multiple transcriptional pathways to exert physiological effects. Pharmacological inhibitor studies have demonstrated that TRPC3, TRPC5 and TRPC6 channels can play a vital role in calcium entry in the podocytes [56, 64–68]. For example, TRPC5 was identified as a mediator of cytoskeletal changes in podocytes because it induces Rac1 activation and synaptopodin degradation [49, 65]. TRPC5 and TRPC6 appear to have opposite effects on podocyte actin dynamics and cell motility through the regulation of RhoA and Rac1, respectively. The loss of TRPC6 in podocytes results in loss of stress fibers and synaptopodin, deactivation of Rac1 and increased motility. This cellular phenotype is rescued by CA-RhoA. On the contrary, loss of TRPC5 results in enhanced stress fiber formation, deactivation of RhoA and decreased motility. This cellular phenotype is rescued by CA-Rac1 [65]. In mice, loss of TRPC5 or pharmacological inhibition of TRPC5 in podocytes protects podocytes from cytoskeleton remodeling and thus prevents LPS-induced FPE and albuminuria [49]. These data suggest that Ca2+ signaling and cytoskeletal changes in podocytes are orchestrated by balancing TRPC5 and TRPC6 activity. Furthermore, recent evidence suggests that down-regulation of the microRNA-30 family induces an increase in calcium signaling, resulting in podocyte injury [69], thus motivating further research evaluating the influence of TRPC channel-mediated Ca2+ influx on the actin cytoskeleton in podocytes.
SYNAPTOPODIN AND THE ACTIN CYTOSKELETON IN PODOCYTES
Synaptopodin is a unique class of proline-rich actin-associated protein, which is highly expressed in podocytes and telancephalic dendrites [70]. Synaptopodin interacts with α-actinin and regulates the actin-bundling activity of α-actinin [71]. Synaptopodin also interacts directly with CD2AP and MAGI-1 (membrane-associated guanylate kinase, WW and PDZ domain containing 1), two adaptor proteins that link cell surface receptors to the actin cytoskeleton at the slit-diaphragm and the basement membrane, respectively [72, 73]. Besides providing a physical linkage to the actin cytoskeleton, synaptopodin has been shown to be required for stress fiber formation in podocytes [19, 71]. Synaptopodin can induce stress fiber formation by inhibiting Smurf1-mediated ubiquitination of RhoA, thereby preventing the targeting of RhoA for proteasomal degradation [19]. Calcineurin inhibitors, such as cyclosporine A (CsA) and FK506, have been used to treat human proteinuric kidney diseases. Their efficacy was originally thought to be due to their immunosuppressive effects on T cells. However, it has been shown that calcineurin mediates the degradation of synaptopodin by inducing protease cathepsin L. CsA protects synaptopodin from cathepsin L-mediated degradation, thus stabilizing the podocyte actin cytoskeleton [74]. Future studies evaluating the inhibition of cathepsin L and the stabilization of synaptopodin are required to ascertain whether these proteins will eventually lead to new therapeutic targets.
ROLE OF ENDOCYTIC PROTEINS IN REGULATING THE ACTIN CYTOSKELETON
In podocytes, recent evidence suggests that there is a close relationship between clathrin-mediated endocytic proteins and the actin cytoskeleton. For example, loss of synaptojanin1, a 5′ inositol phosphatase and the GTPase dynamin, which is best known for regulating the fission reaction of clathrin-mediated endocytosis, results in aberrant actin dynamics. The loss of dynamin 1 and dynamin 2 in mouse podocytes resulted in severe proteinuria, renal failure and FPE, partly due to the robust accumulation of actin nucleating protein Arp2/3 at sites of clathrin-coated pits [75]. Moreover, it has also been suggested that dynamin oligomerization has been implicated in regulating the actin cytoskeleton both by initiating actin polymerization through the removal of the capping protein gelsolin from the barbed ends of actin filaments and by cross-linking actin filaments [76]. Exciting novel discoveries have exploited dynamin's link with the actin cytoskeleton, as the use of the small molecule Bis-T-23 promotes actin-dependent dynamin oligomerization. Schiffer et al. [50] have demonstrated that the use of Bis-T-23 can ameliorate proteinuria and improve glomerular morphology in zebrafish and a variety of rodent models of podocyte injury (Figure 1). These models include toxin-mediated (LPS and puromycin aminonucleoside-induced proteinuria) genetic mouse models of disease such as ACTN4, PKCε and CD2AP knockout, and in DKD induced by streptozotocin. Furthermore, the administration of Bis-T-23 to cultured mouse podocytes stabilized stress fibers, which are usually lost upon LPS administration. To further investigate whether dynamin oligomerization protects against podocyte injury, the investigators generated mice expressing an inducible dynamin transgene with an enhanced propensity to oligomerize. These mutant mice were protected from LPS-induced proteinuria. Indeed, these protective effects highlight the possibility of targeting podocyte actin polymerization in a precise manner [50].
CONCLUSION
Previous therapies for various forms of nephritic and nephrotic syndromes have concentrated on modulating the immune system, with the use of glucocorticoids and cytotoxic agents being the mainstay. Yet in glomerular diseases such as membranous nephropathy and anti-neutrophil cytoplasmic antibodies-related vasculitis, the underlying pathobiology of each disease is vastly different. The recent concerted efforts to curtail glomerular diseases have led to exciting advances in our understanding of podocyte actin cytoskeleton regulation and its role in the maintenance of glomerular filtration barrier integrity. These novel discoveries will hopefully lead to a new era in the treatment of proteinuric kidney diseases, potentially targeting specific actin cytoskeletal pathways in the podocyte. Identifying early molecular ‘footprints’ in the podocytes through the use of human biopsies and mouse models of disease will be essential. Final validation with randomized controlled trials in humans will be required.
CONFLICT OF INTEREST STATEMENT
None declared.
ACKNOWLEDGEMENTS
We would like to personally thank Arnaud Marlier for his assistance with the graphical art. This study was supported by U.S. National Institute of Health grants DK083294 and DK093629. We apologize to investigators whose work could not be cited due to word limitations.


{kind=link}
Comments