





Summary of the key findings
Membranoproliferative glomerulonephritis type II (MPGN II), also termed dense deposit disease (DDD), is associated with dysregulation of the alternative pathway of complement in the circulation and on biosurfaces that leads to the local deposition of complement activation products along the glomerular basement membrane (GBM). As both Factor I and Factor H control alternative complement activation in a solution and as deficiency of Factor H leads to MPGN, Matthew Pickering and colleagues explored the possibility that the deficiency of Factor I might lead to a somewhat similar renal phenotype. They generated Factor I-deficient mice and compared the renal disease in these animals to that observed in Factor H-deficient mice and animals with combined Factor H and Factor I deficiency [ 1 ].
Renal disease in Factor H-deficient mice is characterized by C3 deposition on glomerular capillary walls, mesangial hypercellularity, peripheral capillary loop thickening and double contouring of the GBM, entirely consistent with the diagnosis of MPGN II [ 2 ]. In Factor I-deficient mice, although glomerular changes included hypercellularity, mesangial expansion and capillary wall thickening, light microscopic features of MPGN II, including capillary wall double contours, were absent and glomerular C3 staining was only mesangial in distribution, in striking contrast to the linear capillary wall staining pattern in the Factor H-deficient animals. But what was most surprising was that MPGN II did not develop in mice deficient in both Factor H and Factor I and that glomerular C3 staining was identical to that seen in mice deficient in only Factor I. This finding shows that Factor H protects the GBM from C3 deposition. The implication of this finding is that Factor I activity is an absolute requirement for the development of MPGN II, and this discovery has several important clinical implications.
Complement dysregulation—a cause for MPGN
MPGN II is a rare and severe kidney disease characterized by an amorphous electron-dense material that accumulates along the GBM. End-stage renal failure is the ultimate outcome in about half of affected patients who have had the disease for at least 10 years, and renal transplantation is associated with the histological recurrence of the dense deposits in nearly all cases and eventual graft loss in nearly half [ 3,4 ].
Uncontrolled activation of the alternative pathway of complement in the circulation in these patients can be confirmed by low C3 and Factor B plasma levels and by the accumulation of C3 degradation products like C3d and C3dg in plasma. Immunohistochemical analyses of the GBM reflect the deposition of C3, C5 and C9 but are notable for the absence of immunoglobulins. The alternative pathway is a constitutively active immune surveillance system—low levels of active C3 are spontaneously and continuously generated in the circulation and can translocate onto any biological surface, cell or biomembrane [ 4,5 ]. At these sites, surface-attached inhibitors of complement activation are present and control the fate of newly generated transient C3b (formed by cleaving C3 to C3a and C3b). If surface-deposited C3b is regulated, further complement activation is inhibited; if this regulation does not occur, complement activation continues, enhances C3 deposition and leads eventually to terminal pathway activation and the formation of MAC (membrane attack complex); see Figure 1 .
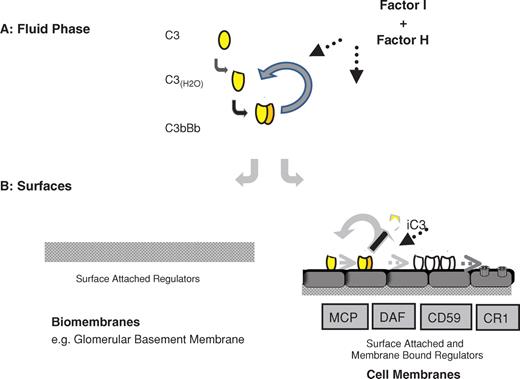
Alternative complement pathway activation occurs in the fluid phase and is controlled by Factor I and Factor H. In the absence of either Factor I or Factor H regulators, activation is uncontrolled and proceeds continuously resulting in the consumption of C3 and Factor B. In this situation, little active C3 is present that can deposit on biological surfaces. However, at biological surface either membrane-bound or surface-attached regulators like Factor H exist to control the further progression of complement activation. In the absence of Factor H or Factor H deficiency even the low-level C3 deposition is uncontrolled and complement activation occurs e.g. at the glomerular basement membrane, and results in the deposition of complement activation products. If Factor I is absent complement is uncontrolled in the fluid phase and consumed, however, the presence of Factor H restricts further low-level complement activation at the surface of the glomerular basement membrane.
Factor H mutations have been found in some patients with MPGN II that result in intracellular accumulation of the Factor H protein. Because secretion is blocked, levels of Factor H in the circulation are low or absent [ 6 ]. In other patients, Factor H is present in the circulation but mutations prevent its complement regulatory activity and binding with C3b [ 7 ]. In still other patients, autoantibodies that bind to and inactivate the complement regulatory region of Factor H lead to the disease [ 8 ]. In fact, the majority of patients have an IgG autoantibody called C3 nephritic factor (C3NeF) that binds to and stabilizes C3 convertase, a molecular complex of C3b and Bb [ 3 , 9 , 10 ]. Binding of C3NeF to C3 convertase protects the convertase from the action of inhibitors thereby preventing inactivation and degradation.
Recent evidence reveals that MPGN II and the renal disease atypical haemolytic uraemic syndrome (aHUS) are both caused by defective complement regulation. Apparently, the same and related genes are affected, indicating related or similar pathological principles. In MPGN II, the effect seems to be more severe as the reported Factor H mutations affect both alleles with homozygous as well as compound heterozygous mutations. In aHUS the majority of mutations occur mostly in heterozygous settings [ 11 ]. Based on these and additional findings, it was recently proposed that both MPGN II and aHUS are related diseases that represent different outcomes of defective complement control [ 12,13 ]. Knowledge about the underlying molecular defects is important to initiate appropriate therapy.
The study by Pickering and colleagues has added an invaluable piece to the puzzle of this disease. Because defective complement activation in the fluid phase and inappropriate surface inhibition are an absolute requirement for the development of MPGN II, we can now focus on complement activation and complement activation products to study their role in disease pathogenesis. This finding is of tremendous diagnostic, prognostic and therapeutic importance.
What is in it for the practising nephrologist
The diagnosis of MPGN II is not straightforward. The characteristic histopathological and electron microscopic findings must be present on biopsy, and evidence of alternative pathway activity must be present in the circulation by following the complement activation profile (CH50 and APH 50) and by measuring plasma levels of complement activation products (C3, C3d, C3dg, Factor B) as well as by following autoantibody levels (to Factor H and C3 nephritic factor). But the results of Pickering and colleagues suggest that complement activation at surfaces as well as the ratio of C3 and C3b to iC3b, C3c and C3dg should be measured. This ratio could represent a valuable primary efficacy endpoint to follow in the treatment of MPGN II. However, following this ratio requires the unambiguous identification of these proteins and that is not straightforward. Nevertheless, focused efforts in this area could reap large dividends, as this ratio could also carry prognostic significance. Levels of iC3b, C3c and C3dg that remain high relative to levels of C3 and C3b in spite of treatment could imply disease progression and be a harbinger of end-stage renal failure, be it in the native or transplanted kidney.
The work by Pickering and colleagues also holds the promise of MPGN II-specific therapies. Whatever the mechanism may be that leads to uncontrolled complement activation at biosurfaces in patients with MPGN II, establishing appropriate local control and preventing the formation of iC3b, C3c and C3dg may be curative. Therapeutic options that should be considered should focus on preventing degradation of C3b or mopping up the C3b degradation products that do form in the circulation. Agents for the former might include antibodies to specific regions of C3 to prevent Factor I-mediated activity; agents for the latter might include the natural ligands of iC3b, C3c and/or C3dg.
Certainly, the future for patients with MPGN II is looking brighter and there is a cautious hope that an effective treatment will be available in the near future. New reagents that target and block complement are emerging and being translated into the clinic. A new phase in the therapeutic handling of these types of complement-mediated diseases has started.
Take-home message
MPGN II is caused by dysregulation of the alternative pathway of the complement cascade both in the fluid phase and on surfaces, and consequently results in the accumulation of C3 degradation products in the GBM. Treatment for this disease should focus on the prevention of C3 degradation or the removal of these degradation products from the circulation.
Work of the authors is supported by the Deutsche Forschungsgemeinschaft and the National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases, R01 DK074409).
Conflict of interest statement . None declared.
References
Author notes
Comment on Rose KL, Paixao-Cavalcante D, Fish J et al . Factor I is required for the development of membranoproliferative glomerulonephritis in Factor H-deficient mice. J Clin Invest 2008; 118: 608ff.


{kind=link}
Comments