





Abstract
Developing efficient gene regulation tools is essential for optimizing microbial cell factories, but most existing tools only modulate gene expression at the transcriptional level. Regulation at the translational level provides a faster dynamic response, whereas developing a programmable, efficient and multiplexed translational regulation tool remains a challenge. Here, we have developed CRISPRi and CRISPRa systems based on hfCas13X that can regulate gene translation in Bacillus subtilis. First, we constructed a CRISPRi system to regulate gene translation based on catalytically deactivated hfCas13X (dhfCas13X). Second, we designed unique mRNA–crRNA pairs to construct DiCRISPRa (degradation-inhibited CRISPRa) and TsCRISPRa (translation-started CRISPRa) systems, which can activate downstream gene translation by enhancing mRNA stability or initiating mRNA translation. In addition, we found that fusing dhfCas13X with the RNA-binding chaperone BHfq significantly improved the activation efficiency of the DiCRISPRa and TsCRISPRa systems (43.2-fold). Finally, we demonstrated that the constructed CRISPR systems could be used to optimize the metabolic networks of two biotechnologically relevant compounds, riboflavin and 2′-fucosyllactose, increasing their titers by 3- and 1.2-fold, respectively. The CRISPRa and CRISPRi systems developed here provide new tools for the regulation of gene expression at the translation level and offer new ideas for the construction of CRISPRa systems.
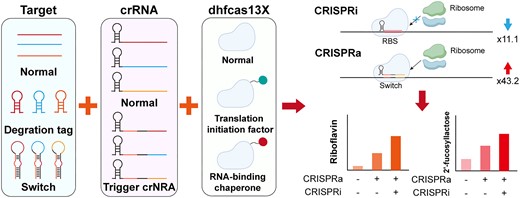
Using synthetic biology to construct microbial cell factories that convert renewable raw materials into high-value chemicals such as functional nutrients, pharmaceuticals and fuels is a crucial approach for environmental protection and circular economy (1–3). However, the metabolic networks within microbial cell factories are inherently complex and often prioritize the allocation of building blocks towards cell growth, thereby reducing the titer of target products. Therefore, balancing the trade-off between product synthesis and cell growth is crucial for enhancing the synthesis efficiency of microbial cell factories. Recently, various dynamic regulation tools have been developed for programmable regulation of gene expression and synergistic control of metabolic fluxes in microbial cell factories, thereby enhancing the production efficiency of desired products, such as inducible promoter and genetic circuits (4–6). However, most of these regulatory tools can only modulate gene expression at the transcriptional level (7–9). Compared to transcriptional regulation strategies, regulation at the translational level offers a faster dynamic response and allows for the independent regulation of each gene within the polycistronic operons of bacteria (10,11).
Among the various metabolic regulation tools, CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated protein) system is the most widely used due to its ability to simultaneously activate and repress genes with high precision, versatility and programmability, enabling rapid and scalable genetic modifications (12). To date, the majority of CRISPR regulatory tools have been developed based on dCas9 or dCas12a (13–15). However, they can only regulate gene expression at the transcriptional level. Compared to dCas9 or dCas12a, Cas13 is a unique CRISPR nuclease that targets RNA (16). Cas13 can regulate gene expression at the translation level rather than the transcription level, providing a tool for multidimensional control of gene expression. Identified Cas13 types currently include Cas13a, Cas13b, Cas13d and Cas13X (17). In mammals, Cas13 is capable of cleaving specific mRNA, thereby reducing the translation of the target gene (18). In bacteria, only a few Cas13 regulatory tools have been developed, including Cas13a and Cas13d (19,20). Due to the high collateral degradation activity of Cas13, researchers typically use deactivated Cas13 (dCas13) to inhibit gene translation (21). However, research on Cas13-based CRISPR activation (CRISPRa) systems is scarce (22). Moreover, existing CRISPRa systems mostly focus on modifying Cas proteins, with relatively less emphasis on the design and modification of the other essential component, the crRNA.
Bacillus subtilis is a commonly used industrial model microorganism and Generally Recognized as Safe (GRAS), widely employed in the production of industrial enzymes, functional nutrients and pharmaceuticals (23,24). Based on dCas9 or dCas12a, a series of transcription-regulated CRISPR interference (CRISPRi) and CRISPRa systems have been constructed in B. subtilis (6,25). However, the effectiveness of the CRISPRa system has proven to be unsatisfactory. In this study, a translation-regulated CRISPRi/a system was developed in B. subtilis based on hfCas13X (High Fidelity Cas13X) (Figure 1A and B). First, the functionality of hfCas13X in B. subtilis was tested, and a translation-regulated CRISPRi system was constructed based on catalytically deactivated hfCas13X (dhfCas13X). By adjusting the binding sites of the crRNA and the expression level of dhfCas13X, the inhibitory effect of the translation-regulated CRISPRi system was enhanced. Subsequently, dhfCas13X was fused with translation initiation factors (IFs) or the RNA-binding protein Hfq to create the DiCRISPRa (degradation-inhibited CRISPRa) system, in which crRNA targeted the recognition sites of nucleases, preventing the degradation of the mRNA. To further increase the activation effect, a TsCRISPRa (translation-started CRISPRa) system was designed and constructed. Under the guide of Trigger crRNA, dhfCas13X-hfq could turn on the Switch structure in the 5′-UTR of target mRNA, activating mRNA translation. Finally, the constructed CRISPRi/a systems were applied to bidirectional regulation of human milk oligosaccharide 2′-fucosyllactose (2′-FL) and riboflavin metabolic networks, increasing their titers by 1.2- and 3.0-fold, respectively (Figure 1C). This study presents the first CRISPR system for regulating translation in Gram-positive bacteria, and its translation activation effect is the highest reported to date, providing a new tool for metabolic engineering of the strain.

Design of CRISPRi and CRISPRa systems for regulation of gene translation. (A) The dhfCas13X is fused with a functional domain. Guided by specific crRNAs, the fused protein targets the 5′-UTR of mRNA, blocking the initiation or elongation of ribosome translation, thus repressing gene expression. The blue arrow indicates translation repression. (B) This fused protein also has the capability to activate mRNA translation. Whether it activates or inhibits translation depends on the 5′-UTR targeted by the crRNA. The red arrow indicates translation activation. (C) By using different combinations of crRNAs, the CRISPRi and CRISPRa systems can simultaneously activate and repress gene expression in cell factories, enhancing the synthesis of desired products.
Materials and methods
Strains and cultivation conditions
The strains constructed in this study are listed in Supplementary Table S1. Escherichia coli was used to clone and construct plasmids. Both E. coli and B. subtilis were cultured in LB medium at 37°C and 220 rpm. Appropriate antibiotics (ampicillin 100 μg/ml, chloramphenicol 5 μg/ml, tetracycline 25 μg/ml, spectinomycin 50 μg/ml, zeocin 30 μg/ml) were supplemented when required. The fermentation medium for 2′-FL and riboflavin consisted of 30 g/L glucose, 2.5 g/L KH2PO4, 12.5 g/L K2HPO4·3H2O, 12 g/L yeast extract, 12 g/L tryptone and 10 ml/L trace metal solution. An additional 10 g/L of lactose was added for the fermentation production of 2′-FL. The trace metal solution was composed of 4 g/L CaCl2, 4 g/L FeSO4·7H2O, 1 g/L MnSO4·5H2O, 0.2 g/L NaMoO4·2H2O, 0.4 g/L CoCl2·6H2O, 0.2 g/L ZnSO4·7H2O, 0.1 g/L CuCl2·H2O, 0.1 g/L AlCl3·6H2O and 0.05 g/L H3BO4. The standards for 2′-FL and riboflavin were purchased from Sigma-Aldrich (St. Louis, MO, USA), while other chemical reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Restriction endonucleases and T4 ligase were purchased from New England Biolabs (Beijing, China). PrimeSTAR® Max DNA Polymerase was purchased from Takara (Tokyo, Japan).
Plasmid construction
The plasmids constructed in this study, as well as the primers used, are listed in Supplementary Tables S2 and S3. The construction process of the Donor Plasmid are as follows. The hfCas13X gene (26) was synthesized and codon-optimized by Genewiz (Suzhou, China). Subsequently, the hfCas13X gene was inserted into the plasmid pSTOP using the Gibson Assembly® Cloning Kit (NEB) to generate the plasmid pSTOP-PxylR-hfCas13X. Reverse PCR (Polymerase Chain Reaction) was employed to deactivate the nucleolytic activity of hfCas13X, resulting in the plasmid pSTOP-PxylR-dhfCas13X. The mutation sites of dhfCas13X are as follows: R84A, H89A, R739A, R740A, H744A and H745A. The P43 promoter was amplified from the plasmid pHT01-P43-GFP and replaced the PxylR promoter in the plasmids pSTOP-PxylR-hfCas13X and pSTOP-PxylR-dhfCas13X using Gibson Assembly, yielding the plasmids pSTOP-P43-hfCas13X and pSTOP-P43-dhfCas13X, respectively. Translation initiation factors, IF1, IF2, IF3, and RNA chaperone protein Bhfq were amplified from the B. subtilis 168 genome and inserted into the plasmid pSTOP-P43-dhfCas13X using Gibson Assembly, resulting in the recombinant plasmids pSTOP-P43-dhfCas13X-IF1, pSTOP-P43-dhfCas13X-IF2, pSTOP-P43-dhfCas13X-IF3 and pSTOP-P43-dhfCas13X-Bhfq, respectively. The RNA chaperone protein Ehfq was amplified from the E. coli K12 MG1655 genome and inserted into the plasmid pSTOP-P43-dhfCas13X to obtain the recombinant plasmid pSTOP-P43-dhfCas13X-Ehfq.
The construction process of the Reporter Plasmid are as follows. The DR and Spacer sequence of hfCas13X crRNA were replaced with the Cas12a crRNA array in the plasmid pcrF19NM2 using reverse PCR, resulting in the recombinant plasmid pcrFCas13. Reverse PCR was used to mutate the Eco31I restriction site in the plasmid pHT01-P43-GFP, obtaining the recombinant plasmid pHT02-P43-GFP. Subsequently, the Pveg-crRNA-PSPL-17-mcherry cassette was cloned from pcrFCas13 and replaced the P43-gfp cassette in the plasmid pHT02-P43-GFP, yielding the recombinant plasmid pHT02-crRNA-mcherry. The Pveg-crRNA-PSPL-17-mcherry cassette was inserted into the plasmid pHT02-P43-GFP, resulting in the recombinant plasmid pHT02-crRNA-mcherry-GFP. The construction of crRNAs was performed as previously described. To enhance the expression level of crRNAs, the Pveg-crRNA-PSPL-17-mcherry cassette was inserted into the plasmids pSTOP or pSTOP-P43-dhfCas13X, generating the plasmids pSTOP-crRNA and pSTOP-crRNA-P43-dhfCas13X, respectively. Degradation-tuning RNAs dR81, dR82 and RNaseY recognition sequence thrs were inserted into the 5′-UTR of mcherry using reverse PCR, resulting in the plasmids pHT02-crRNA-dR81-mcherry, pHT02-crRNA-dR82-mcherry and pHT02-crRNA-dRthrs-mcherry. The sequence of dRthrs, a 47-bp segment, was located 24 bp upstream of the thrs gene in the B. subtilis genome.
Switch design and construction methods
Switch–Trigger pairs were designed using the online tool NUPACK (27) (https://www.nupack.org/). Using the NUPACK toolkit, we obtained 247 Switch–Trigger pairs, and their binding energies in different states were also calculated by NUPACK. Next, the translation initiation rates of the Switches were calculated using the RBS calculator (28) (https://salislab.net/software/predict_rbs_calculator). The translation initiation rates of Switch–Trigger pairs were calculated by deleting the first 30 bp of the Switch sequence (opening the stem-loop structure). Subsequently, the top 20 predicted Switches were inserted into the 5′-UTR of mcherry in the plasmid pHT02-crRNA-mcherry using reverse PCR, obtaining the plasmid pHT02-crRNA-Switch-mcherry.
Strain construction
All molecular biology manipulations of B. subtilis were performed as previously described (29). To regulate the riboflavin metabolic network in B. subtilis, the native promoter of the rib operon ribDEAHT was replaced by PSPL-17-Switch102, PSPL-17-Switch126 and PSPL-17-Switch130, resulting in strains FS102, FS126 and FS130, respectively. Next, the TetR-Ptet cassette was cloned from plasmid pHT01-Ptet-GFP and fused with crRNA102, crRNA126 and crRNA130 through fusion PCR, yielding the cassettes Ptet-crRNA102,Ptet-crRNA126andPtet-crRNA130. These cassettes were then integrated into the genomes of strains FS102, FS126 and FS130, resulting in strains FSC102, FSC126 and FSC130. The plasmid pHT01-P43-dhfCas13X-Bhfq was constructed by replacing the gfp gene in plasmid pHT01-P43-GFP with dhfCas13X-Bhfq. Finally, pHT01-P43-dhfCas13X-Bhfq was transformed into strains FSC102, FSC126 and FSC130, obtaining strains FST102, FST126 and FST130. To repress the expression of the pfkA gene, the native promoter of pfkA in strain FS130 was replaced by the PSPL-17 promoter, yielding strain FSP130. Then, the cassettes Ptet-crRNARBS1, crRNARBS2 and crRNARBS3 were constructed and integrated into the genome of strain FSP130, resulting in strains FSP130-1, FSP130-2 and FSP130-3. Finally, pHT01-P43-dhfCas13X-Bhfq was transformed into strains FSP130-1, FSP130-2 and FSP130-3, resulting in strains FST130-1, FST130-2 and FST130-3.
The construction process of the 2′-FL production strain are as follows. First, the genes manB, manC, gmd and wcaG were cloned from the E. coli K12 MG1655 genome and integrated into the B. subtilis 168 genome, obtaining strain BH. The gene futC from Helicobacter pylori was synthesized, codon-optimized by Genewiz and integrated into the BH genome, resulting in strain HS. The P43 promoter of the futC gene in strain BH was replaced by PSPL-17-Switch102, PSPL-17-Switch126 and PSPL-17-Switch130, resulting in strains HS102, HS126 and HS130, respectively. Subsequently, the cassettes Ptet-crRNA102, Ptet-crRNA126andPtet-crRNA130 were integrated into the genomes of strains HS102, HS126 and HS130, resulting in strains HSC102, HSC126 and HSC130. The plasmid pHT01-P43-dhfCas13X-Bhfq was transformed into strains HSC102, HSC126 and HSC130, obtaining strains HST102, HST126 and HST130. To repress the expression of the pfkA gene, the native promoter of pfkA in strain HSC130 was replaced by the PSPL-17 promoter, yielding strain HSP130. Then, the cassettes Ptet-crRNARBS1, crRNARBS1 and crRNARBS1 were constructed and integrated into the genome of strain HSP130, resulting in strains HSP130-1, HSP130-2 and HSP130-3. Finally, pHT01-P43-dhfCas13X-Bhfq was transformed into strains HSP130-1, HSP130-2 and HSP130-3, resulting in strains HST130-1, HST130-2 and HST130-3.
Fluorescence detection
To detect the fluorescence intensity of different strains, the strains were first streaked onto LB solid medium containing 5 μg/ml chloramphenicol and 25 μg/ml tetracycline and incubated at 37°C for 12 h. Next, single colonies were picked and inoculated into 15 ml culture tubes containing 2 ml LB medium with 5 μg/ml chloramphenicol and 25 μg/ml tetracycline, and cultured at 37°C and 220 rpm for 12 h. Subsequently, 3 μl of the culture was transferred into a 96-well plate containing 150 μl of LB medium with 5 μg/ml chloramphenicol and 25 μg/ml tetracycline, and incubated at 37°C and 220 rpm for 24 h. Additionally, different concentrations of xylose were added to the culture medium as required. The Cytation microplate reader (BioTek, USA) was used to detect mCherry (excitation, 580 nm; emission, 610 nm), eGFP (excitation, 488 nm; emission, 520 nm) fluorescence and OD600.
Detection of expression levels of protein ManB and PfkA
To detect the expression levels of the ManB and PfkA proteins in different strains, 10 ml of fermentation broth cultured for 24 h was collected and ultrasonically disrupted. The supernatant was obtained by centrifugation at 6000 × g for 5 min. The Microorganism 6-PFK1 ELISA Kit (BAILILAI, China) and Microorganism MPM ELISA Kit (BAILILAI, China) were used to measure the expression levels of the ManB and PfkA proteins, respectively. The experimental procedures were carried out according to the ELISA kit protocols.
Shake flask fermentation and product detection
First, the strains were streaked onto LB solid medium containing 5 μg/ml chloramphenicol. After incubating at 37°C for 12 h, single colonies were picked and inoculated into 50 ml of culture tubes containing 5 ml of LB medium, and cultured at 37°C and 220 rpm for 12 h. Then, 1 ml of the culture was inoculated into 250 ml shake flasks containing 15 ml of fermentation medium with 5 μg/ml chloramphenicol, and incubated at 37°C and 220 rpm. After 6 h of fermentation, 5 μM anhydrotetracycline (aTC) was added to induce the CRISPRa/i system.
The glucose concentration was detected using a glucose-lactate analyzer (M100, Shenzhen Sieman Technology Co., Ltd., China). To detect the production of 2′-FL, 1 ml of the fermentation sample was centrifuged at 12 000 × g for 15 min. The supernatant was filtered through a 0.22-μm membrane and the concentration of 2′-FL was detected using high-performance liquid chromatography (HPLC, Agilent 1260). An Aminex HPX-87H column (Bio-Rad) and refractive index detector were used to measure 2′-FL production. The mobile phase was 5 mM sulfuric acid with a flow rate of 0.6 ml/min at 55°C.
The method for detecting riboflavin production was based on previous study (30). 200 μl of the fermentation broth was mixed with 800 μl of 0.05 M NaOH and centrifuged at 10 000 × g for 2 min. The supernatant was collected and diluted with 0.1 M acetate-sodium acetate buffer. Finally, the absorbance of the sample was measured at 444 nm.
Data analysis
Three independent replicates were performed for all experiments. Data analysis was performed using t-tests in SPSS 25.0 software. Statistical significance is indicated as * for P < 0.05 and ** for P < 0.01.
Results
Construction and optimization of the hfCas13X-based CRISPRi system
To develop a CRISPRi tool that inhibits gene translation, we utilized a high-fidelity Cas13X variant, hfCas13X (26,31). This Cas13 ortholog is notable for its minimal size, efficient on-target activity, low collateral degradation activity and lack of protospacer flanking sequence preference. Given hfCas13X’s low collateral degradation activity, we initially retained its nuclease activity to construct the CRISPRi tool (Figure 2A). In the reporter plasmid pHT01, crRNA and the target gene mCherry were controlled by constitutive promoters Pveg and PspL-17, respectively. In the donor plasmid pStop, hfCas13X was regulated by the xylose-inducible promoter Pxyl. Both the reporter and donor plasmids were co-transformed into B. subtilis 168. We designed and constructed crRNA1, crRNA2 and crRNA3 to target the upstream, middle and downstream regions of the mcherry mRNA, respectively.
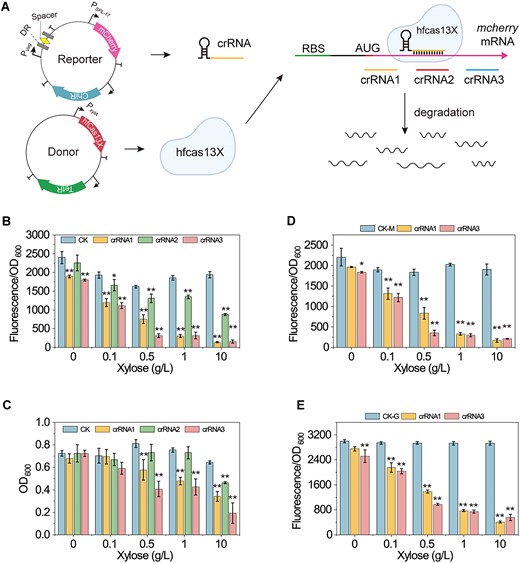
Evaluation of hfCas13X performance in B. subtilis. (A) Illustration of the hfCas13X-based CRISPRi system. A reporter plasmid is used to express the reporter gene mcherry and crRNA. The expression of mcherry and crRNA is controlled by the promoters Pspl-17 and Pveg, respectively. A donor plasmid is used to express hfCas13X. The expression of hfCas13X is controlled by the promoter PxylA. Guided by crRNA, hfCas13X targets and degrades the mRNA of mcherry, thereby inhibiting mcherry expression. mcherry, red fluorescent protein coding gene; Pspl-17 and Pveg, constitutive promoters; PxylA, xylose-inducible promoter; ChiR, chloramphenicol resistance protein; TetR, tetracycline resistance protein; DR, directed repeat sequence of the crRNA array; Spacer, 30 nt spacer sequence of the crRNA array. (B) Effect of different combinations of crRNA and hfCas13X on mcherry expression. Different concentrations of xylose were added to induce the expression of hfCas13X. CK is a non-targeting crRNA. crRNA1, crRNA2 and 3 target the upstream, middle and downstream of mcherry mRNA, respectively. (C) Effect of different combinations of crRNA and hfCas13X on cell growth. (D) Effect of different combinations of mcherry-targeting crRNA and hfCas13X on mcherry expression after a green fluorescent protein (GFP) expression cassette is inserted into the reporter plasmid. CK-M, relative fluorescence intensity of mCherry. (E) Effect of different combinations of mcherry-targeting crRNA and hfCas13X on GFP expression after a GFP expression cassette is inserted into the reporter plasmid. CK-G, relative fluorescence intensity of GFP. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. * and ** indicate P < 0.05 and P < 0.01, respectively.
As shown in Figure 2B, when co-expressed with hfCas13X, crRNA1, crRNA2 and crRNA3 significantly inhibited mcherry mRNA expression. Additionally, the inhibitory effect increased with higher concentrations of xylose, which induced the expression of hfCas13X. When the induction concentration of xylose reached 10 g/L, the inhibition folds of crRNA1, crRNA2 or crRNA3 on mCheery mRNA expression reached 12.7-, 1.2- and 11.4-fold, respectively. However, as xylose concentration increased, cell growth was gradually inhibited (Figure 2C). Strains showing more significant mCherry expression inhibition also experienced greater growth inhibition. Furthermore, when using the strong constitutive promoter P43 to express crRNA, strains containing crRNA1/crRNA3 and hfCas13X die during cultivation, and the strain with crRNA2 showed significant growth inhibition (Supplementary Figure S1). These results suggest that hfCas13X retains collateral degradation activity in B. subtilis. After cleaving mcherry mRNA, hfCas13X indiscriminately cleaves intracellular mRNA, inhibiting cell growth. To confirm the collateral degradation activity of hfCas13X in B. subtilis, we additionally expressed the reporter gene gfp under the control of promoter P43 in the reporter plasmid. As shown in Figure 2D and E, mcherry-targeted crRNA1 and crRNA3 inhibited both mCherry and GFP expression. Additionally, cell growth was also inhibited (Supplementary Figure S2). These results further confirm the collateral degradation activity of hfCas13X in B. subtilis.
To eliminate hfCas13X-induced collateral effects, we inactivated its nuclease activity, resulting in dhfCas13X. We then tested whether dhfCas13X could inhibit mcherry mRNA expression and its impact on cell growth. The results showed that when co-expressed with dhfCas13X, crRNA1, crRNA2 and crRNA3 no longer inhibited mcherry mRNA expression and cell growth (Supplementary Figure S3). These results indicate that targeting the CDS region of the target gene mRNA with dhfCas13X does not inhibit gene expression. This contrasts with CRISPRi systems based on Cas9 or Cas12a, which can target the DNA CDS region to block RNA polymerase extension and inhibit transcription (32). This result indicates that dhfCas13X’s binding affinity to mRNA is insufficient to block ribosome extension and thus inhibit mRNA translation.
Therefore, we designed and constructed crRNARBS1, crRNARBS2 and crRNARBS3 targeting the ribosome binding site (RBS) of the mRNA, aiming to block ribosome binding and inhibit mRNA translation (Figure 3A). As shown in Figure 3B, when co-expressed with dhfCas13X, crRNARBS1, crRNARBS2 and crRNARBS3 inhibited mcherry mRNA expression. When the induction concentration of xylose reached 10 g/L, the inhibition folds of crRNA1, crRNA2 or crRNA3 on mCheery mRNA expression reached 1.1-, 0.9- and 0.3-fold, respectively. Importantly, crRNARBS1, crRNARBS2 and crRNARBS3 did not inhibit bacterial growth (Figure 3C). However, the regulatory effect of dhfCas13X was significantly lower than that of hfCas13X. This may be because hfCas13X can directly cleave mRNA, allowing one molecule of hfCas13X and crRNA to cleave multiple mRNA molecules. Conversely, one molecule of dhfCas13X and crRNA can only inhibit the translation of one mRNA molecule. This suggests that increasing the expression levels of dhfCas13X and crRNA may enhance their inhibitory effect. Therefore, we placed the crRNA expression cassette on the high-copy donor plasmid pStop and replaced the promoter of dhfCas13X with the constitutive promoter P43. The results showed that the inhibition effects of crRNARBS1, crRNARBS2 and crRNARBS3 increased significantly and reached 11.1-, 6.1- and 1.9-fold, respectively (Figure 3D), and cell growth was not inhibited (Figure 3E).
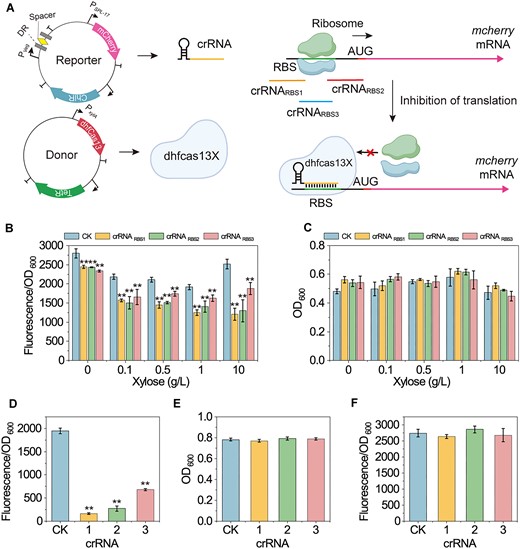
Construction and characterization of a dhfCas13X-based CRISPRi System (A) Illustration of the dhfCas13X-based CRISPRi system. Guided by crRNA, dhfCas13X targets the RBS sequence of mcherry mRNA, preventing ribosome binding to the RBS, thereby inhibiting mcherry expression. RBS, ribosome binding site; mcherry, red fluorescent protein coding gene; Pspl-17 and Pveg, constitutive promoters; PxylA, xylose-inducible promoter; ChiR, chloramphenicol resistance protein; TetR, tetracycline resistance protein; DR, directed repeat sequence of the crRNA array; Spacer, 30 nt spacer sequence of the crRNA array. (B) Effect of different combinations of crRNA and dhfCas13X on mcherry expression. Different concentrations of xylose were added to induce the expression of dhfCas13X. CK is a non-targeting crRNA. (C) Effect of different combinations of crRNA and dhfCas13X on cell growth. (D) Effect of different crRNAs on mcherry expression after expressing dhfCas13X under the strong constitutive promoter P43. (E) Effect of different crRNAs on cell growth after expressing dhfCas13X under the strong constitutive promoter P43. (F) Effect of different combinations of mcherry-targeting crRNA and dhfCas13X on GFP expression after a GFP expression cassette is inserted into the reporter plasmid. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. ** indicate P < 0.01.
We then used gfp as a reporter gene to further test the collateral degradation activity of dhfCas13X. The results showed that crRNARBS1, crRNARBS2 and crRNARBS3 did not inhibit gfp expression or cell growth (Figure 3F and Supplementary Figure S4). This result further confirms that dhfCas13X does not induce collateral effects. In summary, by inactivating the nuclease activity of hfCas13X, we eliminated hfCas13X-induced collateral effects and enhanced the inhibition efficiency of the CRISPRi system by increasing the expression levels of hfCas13X and crRNA. Importantly, the inhibition efficiency of the CRISPRi system can be modulated by adjusting the distance between crRNA and RBS.
Lastly, in mammals, there is an unknown regulatory mechanism that can process pre-crRNA into crRNA that is required since Cas13X lacks pre-crRNA processing capability (33). Therefore, we tested whether dhfCas13X could process pre-crRNA in B. subtilis. In the donor plasmid, we designed three crRNA arrays (crRNAmCherry-gfp1, crRNAmCherry-gfp2 and crRNAmCherry-gfp3) targeting both mcherry and gfp mRNAs. The results showed that when co-expressed with dhfCas13X, crRNAmCherry-gfp1, crRNAmCherry-gfp2 and crRNAmCherry-gfp3 could not inhibit the expression of mCherry and GFP (Supplementary Figure S5). This suggests that B. subtilis lacks the enzymes needed to assist in pre-crRNA processing. In the next experiments, independent mature crRNAs will need to be expressed separately for multi-site regulation.
Design, construction and optimization of the DiCRISPRa system
Given that no CRISPRa system targeting RNA has been constructed in Gram-positive bacteria, we decided to develop the first CRISPRa system based on dhfCas13X in B. subtilis. Previous studies have shown that fusing the translation initiation factor IF3 to the C-terminus of dCasRx can enhance the stability of the target mRNA in E. coli, thereby activating gene translation (22). Building on this, we designed and constructed a degradation-Inhibition CRISPRa (DiCRISPRa) system (Figure 4A). First, we inserted an RNA degradation tag into the 5′-UTR of the mcherry gene in the reporter plasmid to promote mcherry mRNA degradation and reduce its expression. By targeting the RNA degradation tag with crRNA, dhfCas13X can shield the tag, preventing mRNA degradation and activating gene translation. Additionally, we fused the C-terminus of dhfCas13X with translation factors (IF1, IF2, IF3) or RNA chaperone protein Hfq (34) (either from B. subtilis or E. coli) using a flexible 3x(GGGGS) peptide linker. This fusion may enhance downstream gene translation or strengthen the interaction between the RNA degradation tag, crRNA and dhfCas13X.
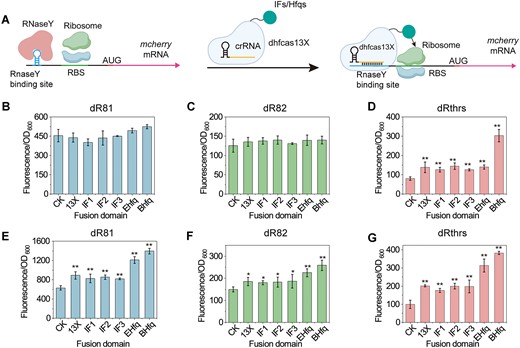
Construction and characterization of a dhfCas13X-based DiCRISPRa System. (A) Illustration of the dhfCas13X-based DiCRISPRa system. An RNaseY binding site was inserted into the 5′-UTR of mcherry mRNA. In the absence of dhfCas13X and crRNA, RNaseY recognizes and binds to the RNaseY binding site, leading to the degradation of mcherry mRNA. When dhfCas13X and crRNA are present, dhfCas13X masks the RNaseY binding site, preventing RNaseY binding to mcherry mRNA, thereby activating mcherry mRNA expression. Additionally, dhfCas13X can be fused with translation IFs or RNA chaperone proteins to promote the translation initiation of mcherry mRNA or the interaction of mRNA–crRNA–dhfCas13X. AUG, start codon. (B) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA targets the RNaseY binding site dR81. CK, no expression of dhfCas13X and crRNA; 13X, dhfCas13X; IF1, dhfCas13X-IF1; IF2, dhfCas13X-IF2; IF3, dhfCas13X-IF3; Ehfq, dhfCas13X-Ehfq; Bhfq, dhfCas13X-Bhfq. IF, the translation IF fused to the C-terminus of dhfCas13X; EHfq, E. coli-derived Hfq fused to the C-terminus of dhfCas13X; BHfq, B. subtilis-derived Hfq fused to the C-terminus of dhfCas13X. (C) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA targets the RNaseY binding site dR82. (D) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA targets the RNaseY binding site dRthrs. (E) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA is overexpressed and targets the RNaseY binding site dR81. (F) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA is overexpressed and targets the RNaseY binding site dR82. (G) Effect of dhfCas13X or dhfCas13X fusion proteins on mcherry expression when crRNA is overexpressed and targets the RNaseY binding site dRthrs. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. * and ** indicate P < 0.05 and P < 0.01, respectively.
As shown in Supplementary Figure S6, inserting an RNA degradation tag into the 5′-UTR of the mcherry gene significantly reduced its expression. Among them, dR81 and dR82 are synthetic degradation-tuning RNAs designed and constructed by Zhang et al. (35). dRthrs is part of the thrs riboswitch in B. subtilis, which contains the RNaseY recognition site. The dRthrs tag showed the highest degradation efficiency, followed by dR82, and dR81 had the lowest efficiency. These degradation tags reduced mCherry expression by 2.7-, 11.5- and 20.3-fold, respectively.
Next, we tested whether individual crRNAs (crRNAdR81, crRNAdR82 and crRNAdRthrs) could relieve the inhibition imposed by the RNA degradation tags. Results showed that individual crRNAs could not activate mCherry expression (Supplementary Figure S6), indicating that crRNAs alone do not stabilize mcherry mRNA. We then introduced donor plasmids containing P43-dhfCas13X, P43-dhfCas13X-IF or P43-dhfCas13X-Hfq expression cassettes into the strains. Results showed that expressing dhfCas13X, dhfCas13X-IF or dhfCas13X-Hfq without crRNA did not change mCherry expression (Supplementary Figure S7), indicating that these constructs alone cannot activate gene expression. When crRNAs targeted dR81 (crRNAdR81) or dR82 (crRNAdR82), none of the constructs activated mCherry expression (Figure 4B and C). When targeting dRthrs (crRNAdRthrs), the activation effects of dhfCas13X reached 0.6-fold. However, the activation effects of dhfCas13X-IF1, dhfCas13X-IF2, dhfCas13X-IF3 and dhfCas13X-EHfq were similar to dhfCas13X. When dhfCas13X was fused with BHfq, the activation effect further increased to 2.79-fold (Figure 4D). These results indicate that we successfully constructed the DiCRISPRa system, where crRNAs targeting RNA degradation tags enable dhfCas13X, dhfCas13X-IF or dhfCas13X-Hfq to activate downstream gene expression. Adding degradation tags result in a weaker gene expression, which enhances the regulatory effect of the DiCRISPRa system. When the degradation tag activity is low, and gene expression is strong, the system’s regulatory effect is minimal. Additionally, due to the 3′-UTR of mRNA also serving as recognition sites for RNases, we targeted the 3′-UTR of mcherry with dhfCas13X. However, targeting the 3′-UTR of mcherry (crRNA3UTR) did not promote gene expression when co-expressed with dhfCas13X and crRNA3UTR (Supplementary Figure S8).
The activation effect of the DiCRISPRa system was still suboptimal, likely due to insufficient crRNA expression. Therefore, the crRNA expression cassette was transferred to the donor plasmid, which significantly enhanced the regulatory effect of the DiCRISPRa system. When targeting dR81 (crRNAdR81), dR82 (crRNAdR82) and dRthrs (crRNAdRthrs), dhfCas13X was able to activate the expression of mcherry, with activation levels reaching 40%, 24% and 100%, respectively. Even with low degradation tag activity and strong background gene expression (dR81), the DiCRISPRa system showed significant activation, confirming that enhancing crRNA expression improves the system’s activation effect. However, fusing IF to the C-terminus of dhfCas13X did not further promote mcherry expression. This might suggest that the fusion of IFs does not enhance the translation initiation of mcherry. When dhfCas13X was fused with BHfq, the activation effect further increased to 1.2-, 0.7- and 2.8-fold, respectively (Figure 4E–G). In addition, dhfCas13X-EHfq also enhanced the activation effect of dhfCas13X, but it was less effective compared to dhfCas13X-BHfq. This may be due to Hfq enhancing the interaction between crRNA and target mRNA, thereby improving the regulatory efficiency of the system.
However, the maximum activation of the DiCRISPRa system was limited to 2.8-fold, and the highest gene expression post-activation was lower than the initial expression driven by the strong constitutive promoter PSPL-17. These limitations may restrict the system's applicability, especially in scenarios requiring strong promoters to enhance the expression of rate-limiting steps.
Design, construction and optimization of the TsCRISPRa system
The results above indicate that the dhfCas13X system can activate target gene expression. Moreover, they show that the interaction between crRNA and mRNA may affect the regulatory efficiency of the dhfCas13X-based CRISPRa system. Therefore, we aimed to design more stringent crRNA–mRNA pairs to enhance the activation effect of the CRISPRa system. The toehold switch is a synthetic RNA-based hairpin loop structure located in the 5′-UTR of a gene that can control gene expression with high specificity and versatility (36). The trigger RNA, which is complementary to the toehold region, can open the toehold switch’s hairpin loop structure, thereby activating downstream gene expression. Building on this concept, we designed and constructed the translation-started based CRISPRa (TsCRISPRa) system (Figure 5A). First, a toehold switch was inserted into the 5′-UTR of the mcherry gene, hiding its RBS within the toehold switch’s hairpin loop structure, preventing mcherry mRNA translation. Then, the crRNA spacer was designed to target the toehold switch and induce its conformational change, thereby activating downstream mcherry translation. Additionally, dhfCas13X, dhfCas13X-IF or dhfCas13X-Hfq can maintain mRNA stability, promote gene initiation or facilitate the crRNA–Switch interaction, further activating mCherry expression.
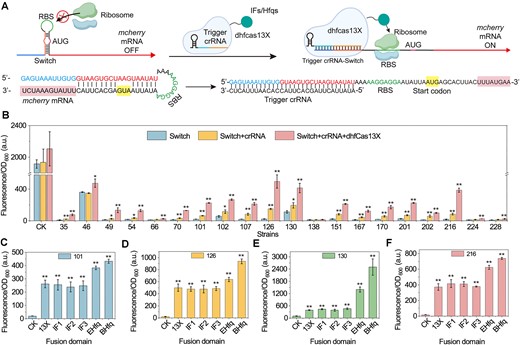
Construction and characterization of a dhfCas13X-based TsCRISPRa system. (A) Illustration of the dhfCas13X-based TsCRISPRa system. A Switch structure was inserted into the 5′-UTR of mcherry mRNA. In the absence of dhfCas13X and crRNA, the hairpin structure within the Switch masks the RBS, thereby inhibiting mcherry mRNA translation. When dhfCas13X and Trigger crRNA are present, the Trigger crRNA and dhfCas13X open the hairpin structure of the Switch, exposing the RBS region and activating mcherry mRNA expression. Additionally, dhfCas13X can be fused with translation IFs or RNA chaperone proteins to promote the initiation of mcherry mRNA translation or the interaction of Switch–Trigger crRNA-dhfCas13X. AUG, start codon. (B) Effect of different Switch, Switch–Trigger crRNA complexes and Switch–Trigger crRNA-dhfCas13X complexes on mcherry expression. CK, no Switch inserted into the 5′-UTR of mcherry mRNA. (C) Effect of different Switch101-Trigger crRNA101-dhfCas13X fusion protein complexes on mcherry expression. CK, no expression of dhfCas13X and crRNA; 13X, dhfCas13X; IF1, dhfCas13X-IF1; IF2, dhfCas13X-IF2; IF3, dhfCas13X-IF3; Ehfq, dhfCas13X-Ehfq; Bhfq, dhfCas13X-Bhfq. IF, the translation IF fused to the C-terminus of dhfCas13X; EHfq, E. coli-derived Hfq fused to the C-terminus of dhfCas13X; BHfq, B. subtilis-derived Hfq fused to the C-terminus of dhfCas13X. (D) Effect of different Switch126-Trigger crRNA126-dhfCas13X fusion protein complexes on mcherry expression. (E) Effect of different Switch130-Trigger crRNA130-dhfCas13X fusion protein complexes on mcherry expression. (F) Effect of different Switch216-Trigger crRNA216-dhfCas13X fusion protein complexes on mcherry expression. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. * and ** indicate P < 0.05 and P < 0.01, respectively.
To construct diverse Switch–Trigger pairs in B. subtilis, we designed 247 orthogonal Switch–Trigger pairs using the NUPACK (27) tool (Supplementary Table S4). To ensure the RBS within the Switch structure could initiate translation in B. subtilis, we designed an 8 bp conserved RBS sequence (AAGGAGGA) within the Switch’s loop structure. Furthermore, to ensure the Trigger can open the Switch’s hairpin loop, we set the interaction free energy between the Switch and Trigger to be sufficiently (> −40 kcal/mol). The RBS Calculator (28) was then used to predict changes in translation initiation rates of downstream genes upon Switch–Trigger interaction. Finally, we selected 20 Switch–Trigger pairs with the largest predicted changes in translation efficiency (Supplementary Table S4).
Next, we replaced the 5′-UTR of the mcherry gene in the reporter plasmid with the Switch structure, which significantly reduced mCherry expression, confirming that the designed Switch structure could control downstream gene expression (Figure 5B). Subsequently, crRNA with a space sequence of specific Trigger RNA was expressed on the Donor plasmid. Unlike the DiCRISPRa system, a single TRIGGER crRNA is now capable of activating the expression of the mcherry gene containing the corresponding Switch structure, including Switch35, Switch49, Switch54, Switch66, Switch70, Switch101, Switch102, Switch107, Switch126, Switch130, Switch151, Switch167, Switch170, Switch201, Switch202, Switch216, Switch224 and Switch228 (Figure 5B). Their activation effects reached 1.2-, 0.9-, 1.0-, 0.4-, 1.3-, 2.3-, 1.0-, 3.0-, 5.7-, 0.7-, 2.6-, 2.4-, 2.0-, 1.8-, 3.6-, 3.5-, 0.4- and 0.5-fold, respectively. This result proves that these Trigger crRNA could open the harpin structure of Switch in B. subtilis. We then co-expressed dhfCas13X to test whether it could enhance the regulatory effect of the Switch–Trigger crRNA pairs. The results showed that co-expression of crRNA and dhfCas13X further activated mCherry expression. Their activation effects increased to 6.8-, 8.9-, 6.3-, 1.0-. 8.3-, 11.4-, 3.7-, 9.3-, 21.2-, 2.7-, 8.3-, 8.3-, 8.2-, 7.7-, 6.8-, 20.6-, 1.2- and 1.3-fold, respectively (Figure 5B). These results confirm that dhfCas13X can enhance the regulatory effect of the Switch–Trigger crRNA pairs.
Subsequently, we fused IFs or Hfqs to the C-terminus of dhfCas13X and tested their effect on activating Switch101, Switch126, Switch130 and Switch216. Switch101, Switch126 and Switch216 were chosen because their significant activation by crRNA and dhfCas13X, while Switch130 was selected due to its weaker inhibition effect on mCherry expression, potentially making it more responsive to the TsCRISPRa system. Results showed that when targeting Switch101, Switch126, Switch130 and Switch216, dhfCas13X-IF1, dhfCas13X-IF2 and dhfCas13X-IF3 did not further enhance the TsCRISPRa system’s regulatory effect (Figure 5C). In contrast, dhfCas13X-EHfq and dhfCas13X-BHfq significantly improved the system’s activation effect. In addition, the activation effect of dhfCas13X-Bhfq remains higher than that of dhfCas13X-EHfq. For Switch101, Switch126, Switch130 and Switch216, the activation effects of dhfCas13X-Bhfq reached 21.2-, 43.2-, 13.4- and 41.4-fold, respectively. Notably, the combination of Switch130, Trigger crRNA130 and dhfCas13X-Bhfq not only demonstrated a high activation fold but also achieved higher gene expression levels than the initial strong constitutive promoter PSPL-17. These results further confirm that fusing dhfCas13X with Hfqs may enhance the interaction between crRNA, mRNA and dhfCas13X, thereby strengthening the activation effect of the CRISPRa system.
Finally, given the significant activation effect of the dhfCas13X-Bhfq fusion protein in both DiCRISPRa and TsCRISPRa systems, we tested its ability to inhibit gene expression. Results showed that co-expressing dhfCas13X-Bhfq with crRNARBS1, crRNARBS2 and crRNARBS3 could inhibit mCherry mRNA expression by 16.1-, 6.5- and 2.3-fold, respectively (Supplementary Figure S9). This result demonstrates that dhfCas13X-Bhfq can also be used as a CRISPRi system, facilitating practical applications.
Multiple fine-tuning of riboflavin and 2′-FL metabolic networks using CRISPRi/a systems
To further demonstrate that dhfCas13X-Bhfq can effectively regulate gene expression, we used the metabolic regulation of riboflavin as a model system. Riboflavin, also known as vitamin B2, is an essential vitamin required for cellular growth and development (37). It is a precursor for the synthesis of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) in cells, which serve as cofactors providing reducing power for cellular metabolism. In B. subtilis, riboflavin synthesis uses ribulose 5-phosphate (Ru5P) and guanosine 5′-triphosphate (GTP) as precursors. Ru5P and GTP are further catalyzed through multiple steps by the rib operon ribDEAHT to produce riboflavin (Figure 6A). The expression of the rib operon ribDEAHT is controlled by an FMN riboswitch located in the 5′-UTR of the operon (38). When intracellular FMN levels are high, the FMN riboswitch feedback inhibits the expression of the rib operon ribDEAHT. To relieve this feedback regulation, we replaced the 5′-UTR of the rib operon ribDEAHT in B. subtilis 168 with Switch102, Switch126 and Switch130, resulting in strains FS102, FS126 and FS130, respectively. Next, we integrated the expression cassettes Ptet-crRNA102, Ptet-crRNA126 and Ptet-crRNA130 into the genomes of strains FS102, FS126 and FS130, obtaining strains FSC102, FSC126 and FSC130. Here we used Ptet, a promoter induced by aTC. Subsequently, the plasmid pHT01-P43-Bhfq was transformed into the strains FSC102, FSC126 and FSC130, yielding strains FST102, FST126 and FST130. Finally, a TsCRISPRa system was established to relieve the feedback regulation by the FMN riboswitch and overproduce riboflavin.
![Regulation of riboflavin and 2′-FL biosynthesis using dhfCas13X-Bhfq. (A) Illustration of the riboflavin metabolic network. Key metabolites and enzymes involved in riboflavin biosynthesis include: Glu, Glucose; G6P, Glucose-6-phosphate; F6P, Fructose-6-phosphate; Gln6P, Gluconate-6-phosphate; Ru5P, Ribulose-5-phosphate; GMP, Guanosine monophosphate; GTP, Guanosine-5′-triphosphate; DARPP, N-(2,5-Diamino-6-oxo-1,6-dihydro-4-pyrimidinyl)-5-O-phosphono-β-D-ribofuranosylamine; ARPP, N-(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl)-5-O-phosphono-β-D-ribofuranosylamine; ArPP, 1-[(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl) amino]-1-deoxy-5-O-phosphono-D-ribitol; ArP, 1-[(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl)amino]-1-deoxy-D-ribitol; DRL, 1-Deoxy-1-(6,7-dimethyl-2,4-dioxo-3,4-dihydro-8(2H)-pteridinyl)-D-ribitol; Rib, Riboflavin; zwf, Encodes glucose-6-phosphate dehydrogenase; pfkA, Encodes phosphofructokinase; ribA, Encodes GTP cyclohydrolase; ribD, Encodes Diaminohydroxy-phosphoribosylaminopyrimidine deaminase/5-amino-6-(5-phosphoribosylamino) uracil reductase; ribT, Encodes 6,7-dimethyl-8-ribityllumazine synthase; ribH, Encodes 3,4-dihydroxy-2-butanone-4-phosphate synthase; ribE, Encodes riboflavin synthase. (B) Riboflavin production in strains FST102, FST126, and FST130. 168, B. subtilis 168; FST102, FST126, FST130: Strains using TsCRISPRa to regulate rib operon expression; aTC, Anhydrotetracycline. (C) Image of strains 168, FST102, FST126 and FST130 under UV irradiation. Riboflavin fluoresces under ultraviolet light. (D) Riboflavin production in strains FST130-1, FST130-2 and FST130-3. FST130-1, FST130-2, FST130-3: Strains using CRISPRi to regulate pfkA expression (E) Image of strains FST130-1, FST130-2 and FST130-3 under UV irradiation. (F) Illustration of the 2′-Fl metabolic network. Key metabolites and enzymes involved in 2′-FL biosynthesis include: Man6P, Mannose-6-phosphate; M1P, Mannose-1-phosphate; GDP-Man, GDP-mannose; GDP-deo-Man, GDP-4-oxo-6-deoxy-D-mannose; GDP-L-Fuc, GDP-L-fucose; Lac, Lactose; manA, Encodes mannose 6-phosphate isomerase; manB, Encodes phosphomannomutase; manC, Encodes α-D-mannose 1-phosphate guanylyltransferase; gmd, Encodes GDP-mannose 6-dehydrogenase; wcaG, Encodes GDP-L-fucose synthase; futC, Encodes α1,2-fucosyltransferase. (G) 2′-FL production in Strains HST102, HST126, and HST130. CK, Control strain HS; HST102, HST126, HST130: Strains using TsCRISPRa to regulate manB expression. (H) 2′-FL production in Strains HST130-1, HST130-2, and HST130-3. HST130-1, HST130-2, HST130-3: Strains using CRISPRi to regulate pfkA expression. (I) 2′-FL production in Strains BS18 and BS130. BS18: 2′-FL overproducer; BS130: Using Switch130 to regulate manB expression in BS130. (J) 2′-FL production in Strains BS130-1, BS130-2, and BS130-3. BS130-1, BS130-2 and BS130-3: Using CRISPRi to regulate pfkA expression in BS130. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. * and ** indicate P < 0.05 and P < 0.01, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/53/1/10.1093_nar_gkae1293/3/m_gkae1293fig6.jpeg?Expires=1750193861&Signature=jBhEtSOZPy2dxY2uwxv4aNlhbzswYE8R4Ax63ySef-wCDkylljWZVQcFQKmVAX2iuenTKGkO2qIqZWRIqUmoTCUdwdYqUFVHk30Bv4Lnafl~IZtsk~Bh4pazf9DXzQ0MH69KCSaajajPpbkBVmBgkTn59HCq1NyOkllBYehMYvB8bHN3ze68a5r8rD-VYsu4usCcjOrUSGjXsvISR-vOE1yW4~nX7-dT-1YxmKHrkUYVhJhlJnOZ-jF~E3GWVfThr5nupHny7pBnyd64~gokO3lY88FxNl7WRpq7KlEf3hZvvl4mIHl6mB7TXEpnpsEMNYf2FIGJH88aMHP2s6FsEg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Regulation of riboflavin and 2′-FL biosynthesis using dhfCas13X-Bhfq. (A) Illustration of the riboflavin metabolic network. Key metabolites and enzymes involved in riboflavin biosynthesis include: Glu, Glucose; G6P, Glucose-6-phosphate; F6P, Fructose-6-phosphate; Gln6P, Gluconate-6-phosphate; Ru5P, Ribulose-5-phosphate; GMP, Guanosine monophosphate; GTP, Guanosine-5′-triphosphate; DARPP, N-(2,5-Diamino-6-oxo-1,6-dihydro-4-pyrimidinyl)-5-O-phosphono-β-D-ribofuranosylamine; ARPP, N-(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl)-5-O-phosphono-β-D-ribofuranosylamine; ArPP, 1-[(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl) amino]-1-deoxy-5-O-phosphono-D-ribitol; ArP, 1-[(5-Amino-2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl)amino]-1-deoxy-D-ribitol; DRL, 1-Deoxy-1-(6,7-dimethyl-2,4-dioxo-3,4-dihydro-8(2H)-pteridinyl)-D-ribitol; Rib, Riboflavin; zwf, Encodes glucose-6-phosphate dehydrogenase; pfkA, Encodes phosphofructokinase; ribA, Encodes GTP cyclohydrolase; ribD, Encodes Diaminohydroxy-phosphoribosylaminopyrimidine deaminase/5-amino-6-(5-phosphoribosylamino) uracil reductase; ribT, Encodes 6,7-dimethyl-8-ribityllumazine synthase; ribH, Encodes 3,4-dihydroxy-2-butanone-4-phosphate synthase; ribE, Encodes riboflavin synthase. (B) Riboflavin production in strains FST102, FST126, and FST130. 168, B. subtilis 168; FST102, FST126, FST130: Strains using TsCRISPRa to regulate rib operon expression; aTC, Anhydrotetracycline. (C) Image of strains 168, FST102, FST126 and FST130 under UV irradiation. Riboflavin fluoresces under ultraviolet light. (D) Riboflavin production in strains FST130-1, FST130-2 and FST130-3. FST130-1, FST130-2, FST130-3: Strains using CRISPRi to regulate pfkA expression (E) Image of strains FST130-1, FST130-2 and FST130-3 under UV irradiation. (F) Illustration of the 2′-Fl metabolic network. Key metabolites and enzymes involved in 2′-FL biosynthesis include: Man6P, Mannose-6-phosphate; M1P, Mannose-1-phosphate; GDP-Man, GDP-mannose; GDP-deo-Man, GDP-4-oxo-6-deoxy-D-mannose; GDP-L-Fuc, GDP-L-fucose; Lac, Lactose; manA, Encodes mannose 6-phosphate isomerase; manB, Encodes phosphomannomutase; manC, Encodes α-D-mannose 1-phosphate guanylyltransferase; gmd, Encodes GDP-mannose 6-dehydrogenase; wcaG, Encodes GDP-L-fucose synthase; futC, Encodes α1,2-fucosyltransferase. (G) 2′-FL production in Strains HST102, HST126, and HST130. CK, Control strain HS; HST102, HST126, HST130: Strains using TsCRISPRa to regulate manB expression. (H) 2′-FL production in Strains HST130-1, HST130-2, and HST130-3. HST130-1, HST130-2, HST130-3: Strains using CRISPRi to regulate pfkA expression. (I) 2′-FL production in Strains BS18 and BS130. BS18: 2′-FL overproducer; BS130: Using Switch130 to regulate manB expression in BS130. (J) 2′-FL production in Strains BS130-1, BS130-2, and BS130-3. BS130-1, BS130-2 and BS130-3: Using CRISPRi to regulate pfkA expression in BS130. Data displayed as mean ± s.d. (n = 3). The statistical analysis is based on Student’s t-test. All data were the average of three independent experiments with standard deviations. * and ** indicate P < 0.05 and P < 0.01, respectively.
As shown in Figure 6B and C, when the inducer aTC was not added, the riboflavin production of strains FST102 and FST126 was similar to that of wild-type B. subtilis 168. The riboflavin titer of strain FST130 improved by 24.4% compared to B. subtilis 168, reaching 5.1 mg/L. This improvement may be due to the leaky expression of Switch130 compared to Switch102 and Switch126. In addition, when the inducer aTC was added, the titer of riboflavin in strains FST102, FST126 and FST130 increased by 50%, 80% and 190% compared to the wild-type B. subtilis 168, reaching 6.6, 7.7 and 12.6 mg/L, respectively (Figure 6B). Additionally, the growth of strains FST102, FST126 and FST130 was not significantly different from B. subtilis 168 (Supplementary Figure S10A). These results indicate that the constructed TsCRISPRa system can be used to activate gene expression. In addition, we constructed a strain, FS1, where the ribD gene expression was controlled by the constitutive strong promoter P43. As shown in Supplementary Figure S11, the riboflavin titer of strain FS1 was lower than that of strain FST130, in which ribD expression was regulated by the switch130. This result demonstrates that the activation effect of the switch130-crRNA130-dhfCas13X was superior to that of the constitutive strong promoter P43.
To confirm that dhfCas13X-Bhfq can also inhibit gene expression, we constructed a CRISPRi system to repress the expression of the competing pathway gene pfkA (encoding 6-phosphofructokinase). First, the native promoter of the pfkA gene in strain FSC130 was replaced with PSPL-17. Subsequently, we expressed crRNAs targeting the RBS of PSPL-17 (crRNARBS1, crRNARBS2 and crRNARBS3) and dhfCas13X-Bhfq in the genome and plasmid, respectively, obtaining strains FST130-1, FST130-2 and FST130-3. Fermentation results showed that without the inducer aTC, the riboflavin production and cell growth in strains FST130-1, FST130-2 and FST130-3 were not significantly different from wild-type B. subtilis 168 (Figure 6D and E and Supplementary Figure S10B). When aTC was added, the titer of riboflavin in strains FST130-2 was increased by 34.4%, compared to FST130, reaching 17.2 mg/L. The titer of riboflavin in strains FST130-1 and FST130-2 was similar to that of FST130. Additionally, the growth and glucose utilization of strain FST130-1 were inhibited, which may be due to the highest inhibition efficiency of crRNARBS1 (Supplementary Figure S10B and C). These results indicate that the CRISPRi and TsCRISPRa systems based on dhfCas13X-Bhfq can simultaneously activate and repress the gene expression.
To further validate dhfCas13X-Bhfq as an effective metabolic engineering regulatory tool, we used the CRISPRi and TsCRISPRa systems to regulate the 2′-FL metabolic network (Figure 6F). 2′-FL is a human milk oligosaccharide that promotes the development of the nervous and immune systems in infants (39). De novo synthesis of 2′-FL in B. subtilis requires the heterologous introduction of α-1,2-fucosyltransferase and GDP-fucose synthesis pathway (40). Increasing 2′-FL production requires enhancing the 2′-FL synthesis pathway and redirecting metabolic flux from EMP (EmbdenMeyerhof-Parnas) pathways.
First, the genes manB, manC, gmd and wcaG from the E. coli K12 MG1655 and the gene futC from H. pylori were integrated into the B. subtilis 168 genome, resulting in strain HS, which achieved a 2′-FL titer of 112.1 mg/L (Figure 6G). Then, we used the TsCRISPRa system to activate the expression of the first gene in the 2′-FL synthesis pathway, manB. The promoter of the manB gene in strain BS12 was replaced with Switch102, Switch126, and Switch130, resulting in strains HS102, HS126 and HS130, respectively. Next, the expression cassettes Ptet-crRNA102, Ptet-crRNA126 and Ptet-crRNA130 were integrated into the genomes of strains HS102, HS126 and HS130, yielding strains HSC102, HSC126 and HSC130. Subsequently, plasmid pHT01-P43-Bhfq was transformed into strains HSC102, HSC126 and HSC130, resulting in strains HST102, HST126 and HST130. when the inducer aTC was not added, the 2′-FL production in strains HST102, HST126 and HST130 was lower than that in the original strain BS12 (Figure 6G). This may be because the manB promoter in strain BS12 was under the control of the constitutively strong promoter P43, whereas the inactivated Switch102, Switch126 and Switch130 promoters have very weak activity. When aTC was added, the 2′-FL production in strains HST102 and HST126 increased, but remained lower than that of the original strain BS12. However, the titer of 2′-FL in HST130 increased by 75.3% compared to strain BS12, reaching 189.9 mg/L (Figure 6G). The cell growth of HST102, HST126 and HST130 were similar to strain HS (Supplementary Figure S12A). This result further demonstrates that the TsCRISPRa system can indeed be used to activate gene expression and increase the production of metabolites of interest. Moreover, it shows that the activated Switch130 promoter has higher activity than the commonly used constitutively strong promoter P43.
Next, we similarly constructed a CRISPRi system targeting the pfkA gene to direct more metabolic flux towards the 2′-FL synthesis pathway, resulting in recombinant strains HST130-1, HST130-2 and HST130-3. Fermentation results showed that the titer of 2′-FL in HST130-2 increased by 21.3% compared to HST130 (Figure 6H). Moreover, the growth and glucose utilization of strain HST130-1 were also inhibited due to the highest inhibition efficiency of crRNARBS1 (Supplementary Figure S12B and C). We also observed that the expression level of the ManB protein in strain HST130-2 was significantly higher than in the strain HS, whereas the expression level of the PfkA protein was significantly lower compared to strain HS (Supplementary Figure S13). To further test the functionality of the constructed CRISPR system, we tested the system in a 2′-FL overproducing strain BS18 (40). As shown in Figure 6I, when using the switch130 to activate the expression of the gene manB, the titer of 2′-FL increased by 32%, reaching 2.9 g/L. Furthermore, when using the CRISPRi system to repress the expression of the pfkA gene, the titer of 2′-FL was further increased to 3.5 g/L (Figure 6J). This result further confirms that the CRISPR system constructed in this study is an effective tool for metabolic flux regulation. These results indicate that dhfCas13X-Bhfq is an effective dual-function gene expression regulation tool that can be used for metabolic engineering in microbial cell factories.
Discussion
Developing an efficient gene regulation tool for controlling metabolic flux is key to enhancing the synthesis efficiency of microbial cell factories. CRISPR-Cas systems, particularly based on Cas9 and Cas12, have been widely used for gene expression regulation and metabolic engineering (41–43). These systems can target DNA and regulate gene transcription. Creating an efficient CRISPR-Cas tool that targets RNA and regulates gene translation can enrich the toolkit for gene expression regulation. Moreover, in bacteria, most genes are clustered in operons, sharing a single promoter, making it challenging to regulate individual genes at the transcriptional level (44). Furthermore, coupling CRISPR-Cas systems for transcriptional and translational regulation can achieve multidimensional control of gene expression, enhancing the precision and efficiency of regulation (21). Currently, some RNA-targeting Cas13 systems have been used to inhibit gene translation (CRISPRi) in both eukaryotes and prokaryotes (19,45). However, only a few Cas13 systems have been designed to activate gene translation (CRISPRa) (22,46). These CRISPRa systems often involve complex operations and require specific fermentation processes and conditions.
In this study, we developed a series of tools based on hfCas13X that can inhibit and activate gene expression at the translational level. First, we tested the function of hfCas13X in B. subtilis and found that it significantly inhibited gene expression in a dose-dependent manner. As the expression level of hfCas13X increased, the inhibitory effect became more pronounced. Despite being a Cas13X variant with minimal collateral effects in mammalian cells, hfCas13X exhibited high collateral effects in B. subtilis, possibly due to the differences in genome architecture between prokaryotes and mammalian cells (47). By inactivating the nuclease activity of hfCas13X, we successfully eliminated its collateral effects while retaining its ability to inhibit mRNA translation by targeting the mRNA’s RBS. However, the inhibitory effect of dhfCas13X was significantly lower than that of hfCas13X and only effective when targeting the RBS region. Additionally, unlike DNA-targeting dCas9 or dCas12, RNA-targeting dhfCas13X required higher expression levels, which might consume more intracellular resources.
Activating gene expression is equally important for functions such as detecting the role of silent genes in cells or enhancing product synthesis in microbial cell factories. In eukaryotes, transcription activators fused to dCas9 proteins can activate target gene transcription by recruiting more RNA polymerase (48). Applying this strategy to prokaryotes, however, has been less effective, likely due to the less understood transcription activation mechanisms in prokaryotes. Otoupal et al. found that in E. coli, expressing crRNA alone could promote target gene translation, possibly due to RNA–RNA interactions stabilizing mRNA (22). Additionally, expressing dCas13RX could further enhance mRNA stability. In our study, we found that crRNA alone could not promote downstream gene translation, even when targeting the 5′-UTR RNA degradation tag. This might be because RNA-RNA interactions in B. subtilis require chaperone proteins (40). Like dCas13RX, co-expressing dhfCas13X and crRNA activated downstream gene expression, with higher expression levels yielding stronger regulatory effects. Furthermore, in eukaryotes, fusing translation IFs with dCas13 can activate target gene translation by promoting translation initiation (49). Otoupal et al. discovered that fusing dCas13RX with IF3 significantly enhanced CRISPRa activation in E. coli (22). In our study, fusing dhfCas13X with IFs did not improve CRISPRa activation. In contrast, fusing dhfCas13X with an RNA chaperone significantly boosted CRISPRa activation, confirming the importance of strengthening RNA–RNA interactions for RNA-targeted CRISPRa systems. Designing specific mRNA–crRNA pairs could further enhance CRISPRa activation.
In some dCas9-based activation systems, the sgRNA is fused with an RNA aptamer, and the transcription activator is fused to the corresponding ligand protein (50,51). This configuration allows the recruitment of more activators to the target gene, thereby activating downstream transcription. These researches focus on enhancing activator efficiency rather than sgRNA-target sequence interactions. In this study, we constructed a TsCRISPRa system based on a toehold switch, designing specific Trigger crRNAs targeting the toehold switch in the mRNA 5′-UTR to enhance mRNA–crRNA interactions. In this system, expressing the Trigger crRNA alone slightly activated mRNA translation, and the additional expression of dhfCas13X significantly enhanced activation by 21.2-fold. Further fusing dhfCas13X with Hfq greatly increased CRISPRa activation, demonstrating the importance of enhancing mRNA–crRNA–dhfCas13X interactions for RNA-targeted CRISPRa systems. We also validated that targeting the RBS with dhfCas13X-Bhfq could initiate CRISPRi functionality. This result shows that dhfCas13X-Bhfq can be used to construct both CRISPRa and CRISPRi systems, making it a convenient tool for dual-functional regulation in microbial cell factories. Using riboflavin and 2′-FL as examples, we demonstrated that dhfCas13X-Bhfq could be used for dual-functional metabolic network regulation. Additionally, most CRISPRa systems have weak activation effects on strong promoters, leading to lower maximum gene expression than strong promoter (13,14). The Switch130–Trigger–CRISPRa system constructed here, however, achieved higher expression levels than strong promoters, evidenced by increased fluorescence intensity and 2′-FL production. In this study, we achieved an activation fold of 43.2 by systematically optimizing the TsCRISPR system. During the construction of the TsCRISPR system, we observed that when the leakage expression of the Switch was low, the activation fold of the TsCRISPR system tended to be high, though the overall promoter activity was typically low. In contrast, when the leakage expression of the Switch was at a moderate level, its activity after regulation by crRNA and dhfCas13X was significantly higher. These findings suggest that when designing Switch-Pairs using computational tools (NUPACK and RBS caculator), different parameter settings can be employed to meet specific application scenarios. Screening more Switch–Trigger pairs may yield even higher expression CRISPRa systems.
Finally, we highlight the limitations of the CRISPRa and CRISPRi systems constructed here. First, targeting RNA requires the expression of more crRNA and dhfCas13X, potentially consuming more cellular resources. Second, while some proteins can assist dhfCas13X in processing pre-crRNA in mammalian cells, dhfCas13X seems unable to process pre-crRNA in B. subtilis, likely due to the absence or low expression of these assisted proteins in B. subtilis. This limitation means that targeting multiple sites simultaneously requires the independent expression of mature crRNA rather than the simpler crRNA array. Understanding the pre-crRNA processing mechanism of dhfCas13X in the future could simplify this process. Third, the TsCRISPRa system constructed here requires prior genome editing to insert switches upstream of the target genes, thus hindering direct regulation of native genes. Currently, constructing an efficient CRISPRa system to directly regulate native gene expression in bacteria remains a challenge, as the mechanisms of gene expression regulation in bacteria are not fully understood. The regulatory efficacy of such CRISPRa systems is influenced by multiple factors, including the targeting site of the crRNA. Even a single base pair variation in the crRNA targeting site can significantly impact the activation efficiency of the system. As a result, when targeting different native genes, it is crucial to screen for suitable crRNA targeting sites. Furthermore, these systems cannot prevent the leakage expression of genes prior to the initiation of regulation.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We thank Dr Wenwen Yu for helpful discussion.
Funding
National Natural Science Foundation of China [32200050, 32070085, 32021005]; Natural Science Foundation of Jiangsu Province [BK20221079]; Fundamental Research Funds for the Central Universities [JUSRP52019A, JUSRP121010, JUSRP221013]; National Key Research and Development Program of China [2020YFA0908300]; Jiangsu Basic Research Center for Synthetic Biology [BK20233003]. Funding for open access charge: National Natural Science Foundation of China [32200050].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments