





Abstract
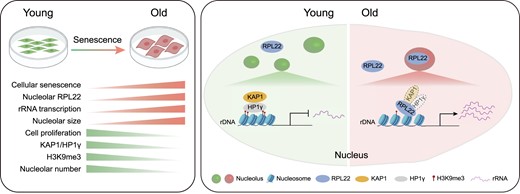
Dysfunction of the ribosome manifests during cellular senescence and contributes to tissue aging, functional decline, and development of aging-related disorders in ways that have remained enigmatic. Here, we conducted a comprehensive CRISPR-based loss-of-function (LOF) screen of ribosome-associated genes (RAGs) in human mesenchymal progenitor cells (hMPCs). Through this approach, we identified ribosomal protein L22 (RPL22) as the foremost RAG whose deficiency mitigates the effects of cellular senescence. Consequently, absence of RPL22 delays hMPCs from becoming senescent, while an excess of RPL22 accelerates the senescence process. Mechanistically, we found in senescent hMPCs, RPL22 accumulates within the nucleolus. This accumulation triggers a cascade of events, including heterochromatin decompaction with concomitant degradation of key heterochromatin proteins, specifically heterochromatin protein 1γ (HP1γ) and heterochromatin protein KRAB-associated protein 1 (KAP1). Subsequently, RPL22-dependent breakdown of heterochromatin stimulates the transcription of ribosomal RNAs (rRNAs), triggering cellular senescence. In summary, our findings unveil a novel role for nucleolar RPL22 as a destabilizer of heterochromatin and a driver of cellular senescence, shedding new light on the intricate mechanisms underlying the aging process.
Introduction
Cellular senescence is a state of irreversible growth arrest that results from a variety of stresses (1–6). Senescent cells accumulate across tissues during aging, ultimately leading to functional decline of organs and aging-related diseases (7–11). Conversely, clearance of senescent cells has been reported to efficiently attenuate pathological conditions and extend lifespan (12–15). As stem cells are characterized by self-renewal and regenerative properties that maintain tissue homeostasis (16,17), exhaustion of stem cells and functional deficiencies in stem cell properties have been recognized as drivers of aging (18–20). Within most if not all human organs and tissues, hMPC is a type of regenerative multipotent stem cell defined by its capability of differentiating into chondrocytes, adipocytes and osteoblasts in vitro (21–23). Consequently, hMPC senescence has been implicated as a culprit in tissue degeneration and proposed as a therapeutic target for geroprotection (24–26). Hence, alleviating hMPC senescence could be conducive for aging reprogramming and as an intervention strategy in aging-related diseases.
The ribosome is a molecular machine that translates mRNA into protein (27). It contains 4 rRNAs and 80 ribosomal proteins (RPs) and plays a fundamental role in the control of cell growth, survival and homeostasis in mammalian organisms. After translation in the cytoplasm, RPs are transported into the nucleus, where they assemble with rRNAs to form functional ribosome subunits. Each ribosome consists of two different subunits, a small one and a large one. The small ribosomal subunit (40S), composed of 18S rRNA and 33 RPs (28), recognizes and decodes messenger RNA (mRNA) by aminoacyl-transfer RNA (tRNA). The large ribosomal subunit (60S), consisting of 5S, 5.8S and 28S rRNA and 47 RPs, executes the formation of the peptide bond. The 40S and 60S subunits are transported into the cytoplasm to form the mature 80S ribosome (29). Evidence supports that ribosome biogenesis is altered in cancer development and control of skeletal muscle mass (30–33). Importantly, disordered ribosome biogenesis and accumulation of RPs are also known to be involved in aging (33–37). Yet, how machinery of ribosome biogenesis regulates cellular senescence remains unclear, especially in the context of human stem cells.
In the nucleus, the nucleolus is the primary site for ribosome biogenesis. The nucleolus is a specific nuclear organelle containing ribosomal DNA (rDNA), a highly repetitive DNA sequence also called nucleolar organizer regions (NORs) (38–40). In the process of ribosome biogenesis, rRNAs are transcribed from 47S rRNA genes and cleaved to form 5.8S, 18S and 28S rRNAs, while 5S rRNA is synthesized in the nucleoplasm (41). In general, the transcription activity of rDNA is regulated by chromatin state and epigenetic modifications, as well as nucleolar structure (42–44). Accumulating evidence indicated that loss of rDNA heterochromatin, rRNA activation, and nucleolar hypertrophy are biomarkers of cellular senescence (45–50).
CRISPR/Cas9-based genetic screening has become an established and powerful approach to uncovering regulatory networks in many biological processes (51–54). In this study, we performed a CRISPR-based LOF screen that targeted proteins associated with ribosomes in senescent hMPCs and identified that deficiency of RPL22 ameliorates hMPC senescence. We demonstrated that RPL22 is translocated into nucleolus, which results in degradation of KAP1 and HP1γ, followed by heterochromatin decondensation, expression of rRNA and alteration of nucleolar morphology, which ultimately leads to senescence. Overall, our analysis illustrates the regulatory function of RPL22 in rRNA transcription and nucleolar architecture during hMPC senescence, suggesting a potential target for intervention therapies in aging-associated disorders.
Materials and methods
CRISPR/Cas9-based screening in hMPCs
For our CRISPR screen, we first constructed a RAGs library containing single guide RNAs (sgRNAs) targeting ribosome-related genes and based on the lentiCRISPRv2 plasmid (Addgene # 52961) as previously described (55,56). Then, we obtained a lentivirus library and infected senescent hMPCs at a low multiplicity of infection (MOI) (0.3) to ensure that one cell receives no more than one sgRNA. At the same time, we set up a control in which hMPCs were transduced with non-targeting control (NTC) at the same MOI. After two days, the cells transduced with sgRNA were put under puromycin selection (1 μg/ml) (InvivoGen) for six days, after which the cells switched to fresh medium without puromycin and passaged until the control cells underwent growth arrest. We harvested cells at different time points and extracted the genomic DNA using TIANamp Genomic DNA Kit (TIANGEN), according to the manufacturer's protocol. Finally, the sgRNA library was constructed as previously described (53). The top enriched genes are listed in Supplementary Table S1.
Lentivirus packaging
HEK293T cells were co-transfected with lentiviral plasmids (including lentiviral overexpression plasmids, lentiCRISPRv2 plasmids or lentiviral shRNA plasmids), lentiviral vectors psPAX2 (Addgene # 12260) and pMD2.G (Addgene # 12259). The supernatant was collected at 24, 48 and 72 h, and filtered with a 0.45 μm membrane followed by centrifugation at 19 400 rpm for 2.5 h to collect lentiviral particles, which were resuspended with PBS and stored at -80°C.
Cell culture
Human embryonic stem cells (hESCs, Wild-type, Line H9, from WiCell Research) and RPL22−/− hESCs were cultured on mitomycin C (Selleck)-inactivated mouse embryonic fibroblast (MEF) feeders with hESC culture medium: 80% DMEM/F12 (Gibco), 20% Knockout serum replacement (Gibco), 2 mM GlutaMAX (Gibco), 0.1 mM non-essential amino acids (NEAA, Gibco), 55 μM β-mercaptoethanol (Invitrogen), 10 ng/ml bFGF (Joint Protein Central, JPC), and 1% penicillin/streptomycin (Gibco); hESCs were also cultured on Matrigel (BD Biosciences) with mTeSR medium (STEMCELL Technologies). hMPCs were cultured with culture medium containing 90% α-MEM medium with GlutaMAX (Gibco), 10% fetal bovine serum (FBS, Gibco), 0.1 mM NEAA, 1% penicillin/streptomycin and 1 ng/ml bFGF on Gelatin (Sigma)-coated plates. Human coronary artery endothelial cells (hCAECs) and human umbilical vein endothelial cells (hUVECs) were cultured with Endothelial Cell Growth Medium-2 BulletKit™ (Lonza, CC-3162).
Generation of RPL22-/- hESCs
CRISPR/Cas9-mediated gene editing was performed as previously described (57,58). In brief, sgRNA targeting the first exon of the RPL22 gene was cloned into the pCAG-mCherry-gRNA vector (Addgene # 87110). Before electroporation, H9-hESCs were treated with ROCK inhibitor Y-27632 (Selleck) for 8 h in mTeSR and dissociated into single cells with TrypLE Express (Gibco). 5 × 106 hESCs were co-electroporated with 7 μg pCAG-mCherry-sgRPL22 vectors and 14 μg pCAG-1BPNLS-Cas9-1BPNLS-2AGFP (Addgene # 87109) vectors by 4D-Nucleofector (Lonza). Subsequently, hESCs were cultured on Matrigel-coated plates treated with ROCK inhibitor Y-27632 in mTeSR. After 48 h, mCherry/GFP-double-positive hESCs were sorted (FACS, BD, Aria II). The deletion of RPL22 was confirmed by DNA sequencing and western blot analysis. The sgRNA sequence for RPL22 and primers for DNA sequencing are listed in Supplementary Table S2.
Generation and characterization of hMPCs
Differentiation of RPL22+/+ and RPL22-/- hMPCs was performed as described previously (59). Briefly, embryoid bodies (EBs) were generated from hESCs and transferred to Matrigel-coated plates with hMPCs differentiation medium containing 90% α-MEM medium with GlutaMAX, 10% FBS (Gibco), 0.1 mM NEAA, 1% penicillin/streptomycin, 10 ng/ml bFGF and 5 ng/ml TGF-β (PeproTech). After 10 days, confluent fibroblast-like cells were collected and transferred to Gelatin-coated plates with hMPCs culture medium. Then, CD73/CD90/CD105 triple-positive cells were sorted by FACS (BD, Aria II). Antibodies used for FACS included anti-CD73 (BD Biosciences, 550741), anti-CD90 (BD Biosciences, 555595) and anti-CD105 (eBioscience, 17-1057).
Methods for differentiation of hMPCs into adipocytes, chondrocytes and osteoblasts have been described previously (60,61). Here, we characterized adipocytes with Oil red O staining (Sigma-Aldrich), chondrocytes with toluidine blue staining (Sigma-Aldrich), and osteoblasts with Von Kossa staining (Genmed Scientifics, GMS80045.3).
Western blot analysis
1 × 106 cells were lysed with 1 × SDS lysis buffer (62.5 mM Tris–HCl, pH 6.8, 10% glycerol, 2% SDS and 2% β-mercaptoethanol) and denatured with thermomixer at 105°C for 10 min. Total protein was quantified with the BCA quantification kit (Dingguo Biotechnology, BCA02). 20 μg of total protein was subjected to SDS-PAGE and electro-transferred to PVDF membranes (Millipore). After blocking with 5% skim milk (powder from BBI Life Sciences) at room temperature for 1 h, the membrane was incubated with primary antibodies at 4°C overnight followed with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. The visualization and data processing were performed with ChemiDoc XRS system (Bio-Rad) and analyzed with ImageJ.
Primary antibodies used for western blot are anti-RPL22 (Proteintech, 25001-1-AP), anti-Lamin B1 (Abcam, ab16048), anti-LAP2 (BD Bioscience, 611000), anti-P21 (Abcam, ab188224), anti-P16 (BD Bioscience, 550834), anti-H3K9me3 (Abcam, ab8898), anti-H3 (Abcam, ab1791), anti-KAP1 (Abcam, ab22553), anti-HP1γ (Cell Signaling Technology, 2619), anti-β-Tubulin (Immunoway, YM3030) and anti-Flag (Sigma-Aldrich, F1804). HRP-conjugated secondary antibodies are goat-anti-mouse (ZSGB-BIO, ZB2305) and goat-anti-rabbit (ZSGB-BIO, ZB2301).
SA-β-Gal staining
SA-β-Gal staining was performed as described previously (62,63). In brief, cells were washed with PBS and fixed for 5 min by fixation buffer (2% formaldehyde and 0.2% glutaraldehyde in PBS) at room temperature. Then, fixed cells were stained by SA-β-Gal staining buffer at 37°C overnight. The percentages of SA-β-Gal-positive cells were quantified by ImageJ.
Clonal expansion assay
Clonal expansion assay was performed as described previously (64). In brief, 2 000 hMPCs were seeded in Gelatin-coated plates (Corning, 12-well), and cultured for approximately 10 days. Then, cells were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature, and stained with crystal violet (Biohao, C0520) for 1 h at room temperature. After washing with PBS, the images were scanned by Epson Perfection V370 Photo (EPSON). The relative cell density was quantified by ImageJ.
Immunofluorescence staining and microscopy analysis
Cells seeded on microscope coverslips were washed twice in PBS and fixed with 4% PFA for 15 min. After permeabilizing with 0.4% Triton X-100 for 15 min, and blocking with 10% donkey serum (Jackson ImmunoResearch Laboratories) for 1 h at room temperature, cells were incubated with primary antibodies at 4°C overnight, followed by washing and incubation with fluorescence-labeled secondary antibodies at room temperature for 1 h. Hoechst 33342 (Invitrogen) was used to visualize nuclei. Images were taken with a ZEISS LSM900 or a Leica SP5 confocal microscope. The percentage of positive cells or the intensity of signals were quantified by ImageJ.
Primary antibodies used for immunostaining are anti-OCT4 (Santa Cruz Biotechnology, sc-5279), anti-SOX2 (R&D, MAB2018), anti-NANOG (Abcam, ab109250), anti-TUJ1 (Abcam, ab78078), anti-SMA (ZSGB-BIO, ZM-0003), anti-FOXA2 (Cell Signaling Technology, 8186S), anti-RPL22 (Proteintech, 25001–1-AP), anti-Ki67 (ZSGB-BIO, ZM-0166), anti-H3K9me3 (Abcam, ab8898), anti-LAP2 (BD Bioscience, 611000), anti-Nucleolin (Santa Cruz Biotechnology, sc-8031), anti-53BP1 (Bethyl Laboratories, A300-273A) and anti-γH2AX (Millipore, 05–636). Secondary antibodies used for immunostaining are Alexa Fluor 488 donkey anti-mouse (Invitrogen, A21202), Alexa Fluor 568 donkey anti-goat (Invitrogen, A11057), Alexa Fluor 647 donkey anti-rabbit (Invitrogen, A31573) and Alexa Fluor 568 donkey anti-rabbit (Invitrogen, A10042).
Plasmid construction
For ectopic overexpression of RPL22, KAP1 and HP1γ, cDNAs were cloned from wild type hMPCs by PrimeSTAR HS DNA Polymerase (Takara, R044A) and then cloned into pLE4 vector (a gift from Tomoaki Hishida). The primer sequences used here are listed in Supplementary Table S2. RPL22ΔN9/C8, RPL22m88A and RPL22m13-16 were constructed by GenScript. The sequence information was verified by DNA sequencing.
For lentivirus-mediated CRISPR knock-out, sgRNAs targeting RPL22 were cloned into lentiCRISPRv2. The sequences of sgRPL22 are listed in Supplementary Table S2.
For lentiviral shRNA-mediated knock-down, shRNAs targeting KAP1 or HP1γ mRNAs were cloned into the pLVTHM vector (Addgene # 12247). The sequences of shRNAs are listed in Supplementary Table S2.
Enzyme-linked immunosorbent assay (ELISA)
For detection of IL-6 secretion, 1 × 105 hMPCs were seeded in Gelatin-coated 6-well plates (Corning), and cultured for 48 h. Then, the cell culture supernatant was collected and filtered with a 0.2 μm filter. Subsequently, the secretion level of IL-6 was measured according to the manufacturer's instructions (Biolegend, 430515).
Telomere length analysis
The telomere length was analyzed as described previously (65). Briefly, genomic DNA was extracted using TIANamp Genomic DNA Kit (TIANGEN, DP304). Then, quantified PCR (qPCR) was performed by Quant Studio 5 (Applied Biosystems). The primers for telomere length analysis are listed in Supplementary Table S2.
EdU staining analysis
DNA replication was gauged by EdU staining, following Beyotime's (C0071L) guidelines. hMPCs were exposed to 10 μM EdU in culture medium for 2–3 h. Cells were then fixed with 4% PFA for 15 min. A click reaction mixture (86% buffer, 4% CuSO4, 10% additive, and 1 μl Azide 488) was applied and incubated for 30 min in the dark at room temperature. After washing with PBS, nuclei were stained with Hoechst 33342. Analysis was completed using confocal microscopy and flow cytometry.
Chromatin immunoprecipitation (ChIP)
1 × 106 hMPCs were collected and cross-linked with 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature followed with 0.125M glycine (VWR International) for 10 min at 4°C. Next, cells were lysed for 10 min on ice and chromosomes were fragmented to a target peak size of 100–500 bp by S220 focused-ultrasonicator (Covaris). Protein-DNA mixture was incubated with antibody-conjugated Dynabeads Protein A (Thermo Fisher Scientific, 10006D) at 4°C overnight. Then, the protein-DNA cross-links were digested with proteinase K for 2 h at 68°C. DNA was purified with ethanol and used for qPCR analysis or sequencing. The primers used for ChIP–qPCR are listed in Supplementary Table S2.
For ChIP–seq, library construction was performed according to the manufacturer's instructions of KAPA Hyper Prep Kit (KAPA, KK8504) and sequenced through the DNBSEQ-T7 platform.
Antibodies used for ChIP: anti-RPL22 (Proteintech, 25001–1-AP), anti-KAP1 (Abcam, ab22553), anti-HP1γ (Cell Signaling Technology, 2619), anti-H3K9me3 (Abcam, ab8898) and anti-Flag (Sigma-Aldrich, F1804).
Northern blot analysis
Northern blot analysis was conducted according to standard protocols (66). In summary, 20 μg of total RNA, extracted with TRIzol Reagent (Ambion, 15596018), was combined with RNA loading buffer, denatured at 70°C for 5 min, and rapidly chilled on ice. The RNA was loaded onto a 1.2% agarose gel and subjected to electrophoresis at 120 V for 1 h in MOPS buffer (Beyotime, ST468-500ml). RNA transfer to a nylon membrane (Millipore) was achieved by capillary action in 20 × SSC buffer (3M NaCl, 0.3M Citric acid, pH 7.0–7.5) for 16 h. The RNA on the membrane was crosslinked at 120 mJ/cm² using an XL-1000 UV crosslinker (Spectronics Corporation). The membrane was pre-hybridized in DIG Easy Hyb solution (Roche, 1179695001) at 42°C for 4 h. Hybridization with DIG-labeled probes, prepared with the PCR DIG PROBE SYNTHESIS KIT (Roche, 11636090910) (65), occurred at 42°C for 20 h. After hybridization, the membrane underwent low and high stringency washes. It was then blocked with blocking reagent (Roche, 11096176001) for 4 h at room temperature before incubation with alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche, 11093274910) for 0.5 to 1 h. Visualization was completed with CDP star reagent (Roche, 12041677001) using the ChemiDoc XRS system. Probe sequences are detailed in Supplementary Table S2.
Agarose gel electrophoresis of rRNA
Total RNA was extracted from equal number of cells (1 × 106) with TRIzol Reagent and subjected to agarose gel electrophoresis. Then, the visualization and data processing of electrophoresis were performed with a ChemiDoc XRS system (Bio-Rad) and analyzed with ImageJ.
Surface sensing of translation (SUnSET) analysis
SUnSET analysis was performed as described previously (67). In brief, cells were collected after treatment with 2 μg/ml puromycin for 15 min. The negative control cells were treated with 100 μg/ml cycloheximide (CHX) for 5 min before 2 μg/ml puromycin treatment. Cells were subsequently harvested for western blot analysis. Total protein stained with coomassie brilliant blue was used as the loading control.
Co-immunoprecipitation (Co-IP) analysis
Co-IP analysis was performed as described previously (68). For exogenous Co-IP, HEK293T cells were transfected with expressing plasmids. After 72 h, cells were harvested and lysed in lysis buffer (40 mM HEPES, pH 7.5, 120 mM NaCl, 0.3% CHAPS, 1 mM EDTA, 1 mM PMSF and complete protease inhibitor cocktail (Roche, 4693159001) on a rotator for 2 h at 4°C. After centrifuging at 12 000 g at 4°C for 30 min, the supernatant was collected and anti-FLAG M2 Affinity Gel (Sigma-Aldrich, A2220) was added and incubated on a rotator at 4°C overnight. Subsequently, interacting proteins were eluted by FLAG peptides (Sigma-Aldrich, F3290) and western blot analysis was performed.
For endogenous Co-IP, RPL22+/+ and RPL22-/- hMPCs at the early passage (EP, P3) were collected and lysed as described above. Then, the supernatant was incubated with anti-RPL22 antibody at 4°C overnight. Subsequently, cell supernatant mixed with anti-RPL22 antibody was incubated with Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, sc-2003) at 4°C for 2 h. Finally, the interacting protein was eluted with 1 × SDS lysis buffer at 105°C for 10 min and western blot analysis was performed.
Liquid chromatography tandem-mass spectrometry (LC-MS/MS) analysis
Proteins obtained by Co-IP were subjected to SDS-PAGE gel electrophoresis and coomassie brilliant blue staining. After decolorization, gel bands containing the protein of interest were excised and subjected to dehydration (100% acetonitrile), reduction (10 mM DTT in 25 mM NH4HCO3 at 56°C for 45 min) and alkylation (10 mM DTT in 25 mM NH4HCO3 at 56°C for 45 min). The target protein was then digested by sequencing grade modified trypsin (Worthington) in 25 mM NH4HCO3 at 37°C overnight. The enzymatic reaction was then terminated with formic acid, and mass spectrometry was conducted by nano LC-Q EXACTIVE (Thermo Fisher Scientific) equipped with a nano-ES ion source (Proxeon Biosystems). Raw files were processed using MaxQuant software (version 1.3.0.5). The resulting peak list files were analyzed using Thermo Proteome Discoverer (version 1.4.0.288) based on the UniProt-proteome-human database. The required false discovery rate (FDR) at the peptide and protein level was set at 1%, and the minimum length of the obtained peptide was set at 7 amino acids. Each proteome requires at least one unique or razor peptide for protein identification.
Proteins not found in the control group but highly abundant in the RPL22 experimental group were identified as RPL22-interacting proteins. RPL22-interacting proteins are listed in Supplementary Table S3. GO cellular component (CC) analysis was performed on the identified proteins using Metascape.
Copy number variation (CNV) analysis
For CNV analysis, genomic DNA was extracted using TIANamp Genomic DNA Kit (TIANGEN, DP304). Then, library construction was performed and sequenced through Illumina NovaSeq 6000 platform.
Trim Galore (version 0.5.0) software (https://github.com/FelixKrueger/TrimGalore) was initially utilized to filter the reads in whole genome sequencing (WGS) data. Using Bowtie2 (version 2.2.9) software (69), the remaining superior reads were mapped to the human reference genome (hg19). The sam file was then converted into bam file using the Samtools (version 1.3.1) program (70). ReadCounter in HMMcopy_utils was used to count the number of reads for each 500-kb bin size. The R tool HMMcopy (version 1.28.1) was used for CNV analysis.
CRISPR screening analysis
MAGeCK (version 0.5.9.2) was used to calculate the CRISPR-based screening sequencing data (71). The data (reproducible data separated by spaces) was first trimmed and counted by the ‘count’ subcommand. Then, using the ‘test’ subcommand, we ran a statistical test on a supplied count table. Finally, we used the positive rate to rank the calculated findings and considered genes with P value <0.05 to be significant.
RNA–seq analysis
For RNA–seq, total RNA was extracted from cells with TRIzol Reagent. Then, library construction was performed according to the manufacturer's instructions of Next Ultra RNA Library Prep Kit for Illumina (NEB) and sequenced through the Illumina NovaSeq 6000 platform.
Trim Galore (version 0.5.0) was used for trim and quality control of RNA–seq data from RPL22-/- hMPCs. HISAT2 (version 2.0.4) (72) software was used to map clean data to the hg19 reference genome. HTSeq (version 0.11.0) (73) was used to calculate the number of reads mapped to each gene. DEseq2 (version 1.30.1) (74) was used to calculate differentially expressed genes (DEGs) with a criterion of an adjusted P value <0.05 and |log2 (fold change)| >0.5. RNA–seq raw data from RPL22-overexpressed hMPCs was trimmed by Trim Galore (version 0.6.7) software. Clean data was mapped to the hg19 reference genome by HISAT2 (version 2.2.1) software. The reads mapped to each gene were calculated using featureCounts (version 2.0.1) software (75). DEseq2 was used to calculate DEGs with a criterion of an adjusted P value <0.05 and |log2 (fold change) | >0.5. Metascape (76) was used for an enrichment analysis of Gene Ontology (GO) terms and pathways. The DEGs are listed in Supplementary Table S4. The DEGs of RNA-seq have been uploaded to the Aging Atlas and Regeneration Roadmap databases (77,78).
H3K9me3 ChIP–seq data analysis
Trim Galore was used for quality control and adapter removal on H3K9me3 ChIP–seq data. In order to explore the signals (RPKM, reads per kilobase per million mapped reads) in rDNA region, we reviewed the previous article (79) and assembled the rDNA region (GenBank: U13369.1) on chromosome 13 of hg19, which we named ‘hg19_plusrDNA’. Bowtie2 (version 2.2.9) was used to align the remaining reads to hg19_plusrDNA. To lessen the offset, we collected the identical number of reads for subsequent analysis (35 000 000 for H3K9me3 ChIP–seq). We used deepTools (version 2.5.4-2-5ee467f) to convert bam files to bw files, and generated heatmap files with deepTools before optimization with R.
Statistical analyses
All data are shown as the means ± SEMs. Two-tailed unpaired Student's ttest was performed by GraphPad Prism (version 8). P value <0.05 is considered statistically significant. *P <0.05; **P <0.01; ***P <0.001; ns: not significant.
Results
CRISPR/Cas9 screening identifies RPL22 as a driver of human stem cell senescence
To systematically investigate how genes related to ribosome biogenesis function during senescence, we performed a CRISPR/Cas9-based LOF screen in hMPCs. Firstly, we constructed a pooled library (RAGs library) that consisted of 995 sgRNAs targeting 332 genes related to ribosome synthesis, transport, and assembly (Figure 1A and Supplementary Figure S1A). Then, we transduced the RAGs library into replicative-senescent (RS) hMPCs with a multiplicity of infection (MOI) of 0.3 (56). Meanwhile, as a control, we transduced non-targeting control (NTC) sgRNAs into cells with the same MOI. After puromycin selection, we serially passaged the transduced RS hMPCs for 10 weeks, which is the time point when control cells undergo growth arrest (Figure 1A). To identify genes whose disruption prevents cellular senescence, we assessed the sgRNA presentation from cells collected at 2 weeks, 5 weeks, 8 weeks and 10 weeks by DNA sequencing, and observed that the enriched genes are not identical at different time points (Figure 1B and Supplementary Figure S1B). In addition, we repeated the pooled screening with an independent infection as a biological replicate. From these two screenings, we identified 9 markedly enriched genes that had alleviated or reverted hMPC senescence (Figure 1C).
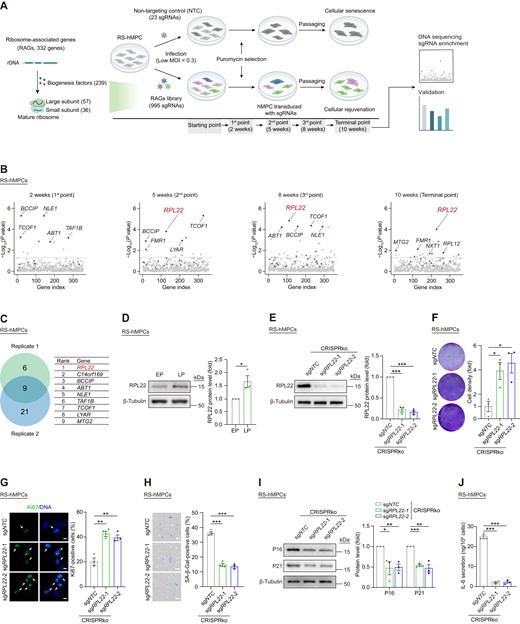
CRISPR/Cas9-based screening for ribosome-associated genes in promoting hMPC senescence. (A) Schematic of CRISPR/Cas9-based screening of RAGs library to identify senescence-promoting genes in RS hMPCs. (B) Enrichment levels were estimated for each gene at 2, 5, 8 and 10 weeks after infection of RAGs lentiviral library in RS hMPCs. The red dots indicated the top 5 enriched genes. The dark gray dots indicate non-targeting control (NTC) sgRNAs. (C) Venn diagram showing enriched genes (left) from two replicative screenings (replicate 1 at 10 weeks; replicate 2 at 12 weeks). The overlapped 9 genes were showed in the table (right). (D) Western blot analysis of RPL22 protein level in RS hMPCs at early passage (EP, P3) and late passage (LP, P13). β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (E) Western blot analysis of RPL22 protein level in RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (F) Clonal expansion analysis of RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (G) Immunostaining analysis of Ki67 in RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's ttest was performed. (H) SA-β-Gal staining of RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (I) Western blot analysis of P16 and P21 protein levels in RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (J) ELISA analysis of IL-6 secretion in RS hMPCs after CRISPR-mediated knock-out of RPL22 using lentivirus with two sgRNAs. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed.
Moreover, given that we observed an increased expression level of RPL22, the top hit, during hMPC senescence (Figure 1D), we chose to study the RPL22 gene, which encodes a ribosomal component belonging to the large 60S subunit (80). When we applied lentivirus-mediated CRISPR knock-out (CRISPRko) to deplete RPL22 in RS hMPCs (Figure 1E and Supplementary Figure S1C), we observed a resurgence in proliferative capacity, indicated by increased clonal expansion and a higher number of Ki67-positive cells (Figure 1F, G, and Supplementary Figure S1D, E). RPL22 deficiency also alleviated senescent traits, as evidenced by reduced senescence-associated β-Galactosidase (SA-β-Gal) activity, lower expression of senescence markers (P16, P21), and a decrease in senescence-associated secretory phenotype (SASP) factors, such as IL-6 (Figure 1H-J). Additionally, the absence of RPL22 resulted in fewer DNA damage foci and a reduction in nuclear size (Supplementary Figure S1F, G). Taken together, these results suggest that RPL22 might be a driver of aging, and that reducing RPL22 expression counteracts hMPC senescence.
RPL22 deficiency alleviates hMPC senescence
Next, we knocked out RPL22 in human embryonic stem cells (hESCs) via CRISPR/Cas9-based gene editing (Supplementary Figure S2A) using the sgRNA identified during screening that targeted the first exon of RPL22, causing the disruption of two alleles, and ultimate deletion of RPL22 (Supplementary Figure S2B-D). Relative to RPL22+/+ hESCs, the percentage of Ki67-positive cells and expression of pluripotency markers including NANOG, SOX2 and OCT4 were comparable in RPL22-/- hESCs (Supplementary Figure S2E, F). Moreover, we did not observe any differences in formation of the three germ layers in teratoma assays between RPL22-/- and RPL22+/+ hESCs (Supplementary Figure S2G) or in genome integrity based on karyotype and copy number variation (CNV) analysis (Supplementary Figure S2H, I). These observations suggest that RPL22 is dispensable for the maintenance of pluripotency and genomic stability in hESCs.
When we differentiated RPL22+/+and RPL22-/- hESCs into hMPCs (Figure 2A, B), both lines expressed typical hMPCs markers (CD73, CD90 and CD105) (Supplementary Figure S3A), and we did not detect any differences in genomic stability (Supplementary Figure S3B) or differentiation potential into adipocytes, chondrocytes and osteoblasts (Supplementary Figure S3C-E). In contrast to RPL22+/+ hMPCs, RPL22-/-hMPCs exhibited a robust growth rate upon serial passaging (Figure 2C). This was characterized by enhanced clonal expansion ability, increased numbers of Ki67-positive cells, and elevated DNA replication levels (Figure 2D–F). Moreover, RPL22 deficiency attenuated progressive senescence of hMPCs as evidenced by decreased level of SA-β-Gal and expression of P16 (Figure 2G, H). We also found increased expression of nuclear protein, Lamin B1 and lamina-associated polypeptide (LAP) 2, as well as reduced secretion of IL-6 in RPL22-/- hMPCs (Figure 2H–J). In addition, we observed longer telomeres, fewer DNA damage foci and smaller nuclear size in hMPCs with RPL22 deletion (Figure 2K–M). Furthermore, we performed RNA-sequencing to evaluate the molecular changes caused by RPL22knock-out. About three-tenths (1 731/5 912) of differentially expressed genes (DEGs) that were altered in RS hMPCs were reversed upon RPL22 depletion, including upregulated genes related to cell division, DNA replication and repair as well as downregulated genes associated with inflammatory cytokines (e.g., IL-18) (Figure 2N, O and Supplementary Figure S3F–H). Altogether, these results consolidate that loss of RPL22 delays cellular senescence of hMPCs.
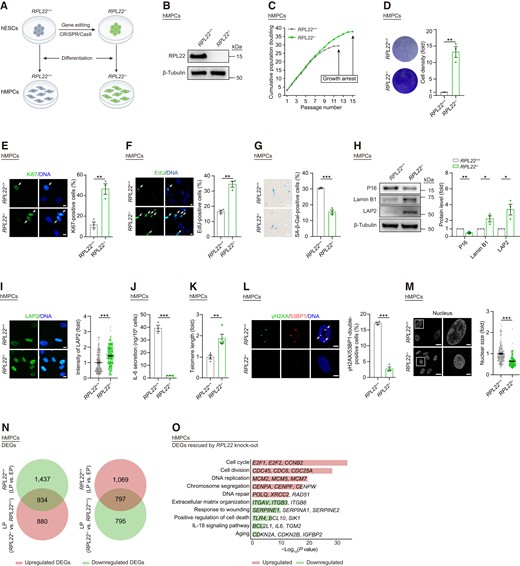
RPL22 deficiency alleviates hMPC senescence. (A) Schematic of generation of RPL22+/+ and RPL22-/- hMPCs derived from hESCs. (B) Western blot analysis of RPL22 protein level in RPL22+/+ and RPL22-/- hMPCs. β-Tubulin was used as loading control. (C) Growth curve analysis of RPL22+/+ and RPL22-/- hMPCs. (D) Clonal expansion analysis of RPL22+/+ and RPL22-/- hMPCs. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (E) Immunostaining analysis of Ki67 in RPL22+/+ and RPL22-/- hMPCs. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's ttest was performed. (F) EdU staining of RPL22+/+ and RPL22-/- hMPCs. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate EdU-positive cells. Two-tailed unpaired Student's ttest was performed. (G) SA-β-Gal staining of RPL22+/+ and RPL22-/- hMPCs. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (H) Western blot analysis of P16, Lamin B1 and LAP2 protein levels in RPL22+/+ and RPL22-/- hMPCs. β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (I) Immunostaining analysis of LAP2 in RPL22+/+ and RPL22-/- hMPCs. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 200 cells. Two-tailed unpaired Student's ttest was performed. (J) ELISA analysis of IL-6 secretion in RPL22+/+ and RPL22-/- hMPCs. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (K) Real-time quantitative PCR (qPCR) analysis of telomere length in RPL22+/+ and RPL22-/- hMPCs. The expression level was normalized to 36B4. Data are presented as the means ± SEMs. n = 4 technical replicates. Two-tailed unpaired Student's t test was performed. Data are representative of three independent experiments. (L) Immunostaining analysis of γH2AX and 53BP1 in RPL22+/+ and RPL22-/- hMPCs. Scale bar, 5 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate γH2AX and 53BP1-double-positive foci. Two-tailed unpaired Student's ttest was performed. (M) Nuclear size of RPL22+/+ and RPL22-/- hMPCs. Scale bar, 20 μm (left) and 5 μm (right). Data are presented as the means ± SEMs. n = 200 cells. Two-tailed unpaired Student's t test was performed. (N) Venn diagrams showing overlapped DEGs in both RS hMPCs (RPL22+/+ hMPCs, LP versus EP) and RPL22-/- hMPCs (LP versus RPL22+/+ hMPCs). (O) Bar plot showing GO terms and pathways associated with DEGs rescued by RPL22 knock-out. Downregulated DEGs in RS hMPCs that were reversed by RPL22 knock-out were colored in red. Upregulated DEGs in RS hMPCs that were reversed by RPL22 knock-out were colored in green.
Ectopic expression of RPL22 promotes cellular senescence in hMPCs
We then asked whether ectopic expression of RPL22 is sufficient to promote senescence. To this end, we performed lentivirus-mediated RPL22 overexpression into early passage wild-type hMPCs (young hMPCs) along with a lentiviral vector expressing luciferase (Luc) as control (Figure 3A). We found that RPL22 overexpression resulted in classical senescent phenotypes in young hMPCs, as revealed by compromised proliferative potential, and increased percentage of SA-β-Gal-positive cells (Figure 3B–D). Additionally, we observed induction of P16 expression and IL-6 secretion combined with shortened telomeres, increased DNA damage response, enlarged nuclear size, and decreased expression of Lamin B1 and LAP2 in young hMPCs upon RPL22 overexpression (Figure 3E–J). Consistent with the accelerating senescence phenotypes, RPL22 overexpression disrupted the transcriptional profile with downregulation of genes related to cell proliferation, which is opposite to RPL22-deficient hMPCs (Figure 3K, L and Supplementary Figure S3I). Collectively, these data show that RPL22 promotes human stem cell senescence.
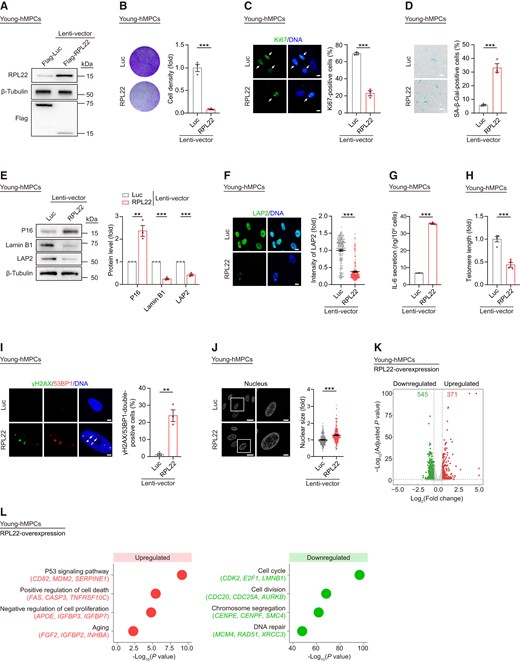
Overexpression of RPL22 accelerates hMPC senescence. (A) Western blot analysis of RPL22 protein level in young hMPCs transduced with lentiviruses expressing Flag-Luc or Flag-RPL22. β-Tubulin was used as loading control. (B) Clonal expansion analysis of young hMPCs transduced with lentiviruses expressing Luc or RPL22. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (C) Immunostaining analysis of Ki67 in young hMPCs transduced with lentiviruses expressing Luc or RPL22. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's t test was performed. (D) SA-β-Gal staining of young hMPCs transduced with lentiviruses expressing Luc or RPL22. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (E) Western blot analysis of P16, Lamin B1 and LAP2 protein levels in young hMPCs transduced with lentiviruses expressing Luc or RPL22. β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (F) Immunostaining analysis of LAP2 in young hMPCs transduced with lentiviruses expressing Luc or RPL22. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 200 cells. Two-tailed unpaired Student'st test was performed. (G) ELISA analysis of IL-6 secretion in young hMPCs transduced with lentiviruses expressing Luc or RPL22. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (H) Real-time quantitative PCR (qPCR) analysis of telomere length in young hMPCs transduced with lentiviruses expressing Luc or RPL22. The expression level was normalized to 36B4. Data are presented as the means ± SEMs. n = 4 technical replicates. Two-tailed unpaired Student's t test was performed. Data are representative of three independent experiments. (I) Immunostaining analysis of γH2AX and 53BP1 in young hMPCs transduced with lentiviruses expressing Luc or RPL22. Scale bar, 5 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate γH2AX and 53BP1-double-positive foci. Two-tailed unpaired Student's t test was performed. (J) Nuclear size of young hMPCs transduced with lentiviruses expressing Luc or RPL22. Scale bar, 20 μm (left) and 5 μm (right). Data are presented as the means ± SEMs. n = 200 cells. Two-tailed unpaired Student's t test was performed. (K) Volcano plot showing upregulated (red) or downregulated (green) DEGs in young hMPCs transduced with lentiviruses expressing RPL22 compared to Luc. (L) Dot plots showing GO terms and pathways associated with upregulated (red) and downregulated DEGs (green) in young hMPCs transduced with lentiviruses expressing RPL22 compared to Luc.
RPL22 induces rRNA expression during hMPC senescence
When we monitored global mRNA translation using surface sensing of translation (SUnSET) analysis (67), we did not observe clear differences between RPL22−/− hMPCs and RPL22+/+ hMPCs at early passage (EP) (Supplementary Figure S4A). In addition, we constructed a truncated RPL22ΔN9/C8 with nine and eight residues deleted from the N-domain and the C-domain, respectively, which was incapable of incorporation into ribosomes (81) (Figure 4A and Supplementary Figure S4B). Interestingly, transduction of RPL22ΔN9/C8 also caused a reduction in proliferative capacity and an increase in SA-β-Gal-positive cells when compared to Luc in RPL22−/− hMPCs (Figure 4B–D). These results indicate that the pro-senescence role of RPL22 is independent of its ribosomal functions.
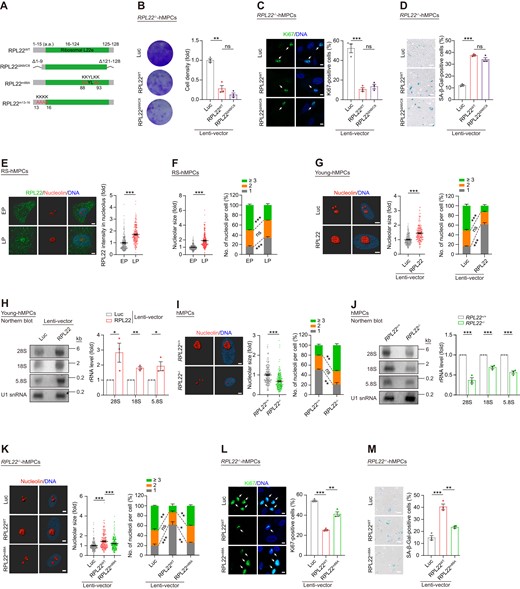
RPL22 induces rRNA transcription during hMPC senescence. (A) Schematic of RPL22WT, RPL22ΔN9/C8 with nine amino acid residues deleted at the N-terminal and eight amino acid residues deleted at the C-terminal of RPL22, RPL22m88A with four mutations (lysine at 88, 89, 92 and 93 to alanine), and RPL22m13-16 with four mutations (lysine at 13, 14, 15 and 16 to alanine). (B) Clonal expansion analysis of RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22 or RPL22ΔN9/C8. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (C) Immunostaining analysis of Ki67 in RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22ΔN9/C8. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's ttest was performed. (D) SA-β-Gal staining of RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22ΔN9/C8. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (E) 3D reconstruction of a Z-stack of RPL22 (green) and nucleolin (red) immunofluorescence images in RS hMPCs at early passage (EP, P3) and late passage (LP, P13). Scale bar, 5 μm. The intensity of RPL22 in nucleolus was quantified and presented as the means ± SEMs. n = 200 cells. Two-tailed unpaired Student's t test was performed. (F) The nucleolar size (left), and the percentage of cells with indicated nucleoli numbers (right) in RS hMPCs at early passage (EP, P3) and late passage (LP, P13). Data are presented as the means ± SEMs. n = 200 cells (left), or three biological replicates (right). Two-tailed unpaired Student's t test was performed. (G) 3D reconstruction of a Z-stack of nucleolin immunofluorescence images in young hMPCs transduced with lentiviruses expressing Luc or RPL22. The nucleolar size (middle) and the percentage of cells with indicated nucleoli numbers (right) were quantified. Scale bar, 5 μm. Data are presented as the means ± SEMs. n = 200 cells (left), or three biological replicates (right). Two-tailed unpaired Student's t test was performed. (H) Northern blot analysis of rRNAs (28S, 18S and 5.8S) levels in young hMPCs transduced with lentiviruses expressing Luc or RPL22. U1 snRNA was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (I) 3D reconstruction of a Z-stack of nucleolin immunofluorescence images in RPL22+/+ and RPL22-/- hMPCs at late passage (P10) (left). The nucleolar size (middle) and the percentage of cells with indicated nucleoli numbers (right) were quantified. Scale bar, 5 μm. Data are presented as the means ± SEMs. n = 200 cells (left), or three biological replicates (right). Two-tailed unpaired Student's t test was performed. (J) Northern blot analysis of rRNAs (28S, 18S and 5.8S) levels in RPL22+/+ and RPL22-/- hMPCs at late passage (P10). U1 snRNA was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (K) 3D reconstruction of a Z-stack of nucleolin immunofluorescence images in RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22m88A (left). The nucleolar size (middle) and the percentage of cells with different nucleolar number (right) were quantified. Scale bar, 5 μm. Data are presented as the means ± SEM. n = 200 cells (left), or three biological replicates (right). Two-tailed unpaired Student's ttest was performed. (L) Immunostaining analysis of Ki67 in RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22m88A. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's ttest was performed. (M) SA-β-Gal staining of RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22m88A. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed.
After synthesis in the cytoplasm, ribosomal proteins are imported into the nucleus and then transported to the nucleolus where they are assembled along with rRNAs (82). When we performed super-resolution microscopy, we found aggregation of RPL22 in the nucleolus in RS hMPCs at late passage (LP) (Figure 4E). Concurrently, the rRNAs levels were increased (Supplementary Figure S4C, D), accompanied by enlarged nucleolar size and fewer nucleoli (Figure 4F). Interestingly, these phenotypes were also observed when RPL22 was overexpressed in young hMPCs (Figure 4G, H and Supplementary Figure S4E, F). In RPL22−/− hMPCs, we instead observed lower levels of rRNAs by northern blot (Figure 4J and Supplementary Figure S4G, H), as well as reduced nucleolar volume and increased numbers of nucleoli (Figure 4I). Accordingly, we hypothesized that nucleolar RPL22 might regulate hMPC senescence by directly regulating rRNA expression and modulating nucleolar architecture. Significantly, the transcription of 45S pre-rRNA, which is the precursor to the 28S, 18S and 5.8S rRNAs, was diminished in RPL22-deficient hMPCs (83), but upregulated upon RPL22 overexpression in young hMPCs (Supplementary Figure S4I, J). Next, we constructed a lentiviral vector expressing an RPL22 mutant (RPL22m88A) (Figure 4A and Supplementary Figure S4K), which has been reported to abolish retention of RPL22 in the nucleoli (84). We then transduced RPL22m88A and RPL22WT into RPL22−/− hMPCs, and measured the transcription levels of rRNA, the nucleolar volume, and the number of nucleoli. We found that forced RPL22WT expression induced rRNA transcription, nucleolar expansion, consistent with a decrease in nucleolar number, which were not observed upon expression of RPL22m88A (Figure 4K and Supplementary Figure S4L). Meanwhile, RPL22m88A also failed to accelerate senescence, as opposed to RPL22WT in RPL22−/− hMPCs (Figure 4L, M and Supplementary Figure S4M). Additionally, an RPL22 variant (RPL22m13-16) that leads to cytoplasmic retention of RPL22 (84,85) similarly exhibited a loss of pro-senescence capacity (Figure 4A and Supplementary Figure S4N–Q). These results indicate that nucleolar accumulation of RPL22 induces rRNA expression and disrupts the nucleolar architecture in hMPC senescence.
RPL22 destabilizes and separates HP1γ and KAP1 from rDNA in senescent hMPCs
To discover mechanisms by which RPL22 regulates rRNA expression and cellular senescence, we expressed Flag-tagged RPL22 in HEK293T cells and performed co-immunoprecipitation (Co-IP) followed by mass spectrometry analysis (MS) (Supplementary Figure S5A). In line with RPL22’s function and location, nuclear and ribosome components were identified as RPL22-interacting proteins (Supplementary Figure S5B). Interestingly, we identified several new RPL22-interacting proteins associated with heterochromatin (Figure 5A and Supplementary Figure S5B). Among these, HP1γ, a heterochromatin adaptor protein, was previously reported to be involved in KAP1-mediated heterochromatin condensation through protein-protein interactions and binding to H3K9me3 marks (86,87). Consistently, we detected down-regulation of HP1γ, KAP1 and H3K9me3 in senescent hMPCs (Figure 5B). Next, we validated that HP1γ and KAP1 interact with RPL22 through exogenous and endogenous Co-IP assays (Figure 5C, D and Supplementary Figure S5C, D). Moreover, HP1γ, KAP1 and H3K9me3 expression were all affected by RPL22, as manifested by decreased protein levels in RPL22-overexpressing hMPCs (Figure 5E and Supplementary Figure S5E). Conversely, RPL22 knockout elevated the expression levels of these proteins (Figure 5F and Supplementary Figure S5F, G), an effect reversed by RPL22WT but not by RPL22m88A (Figure 5G). Furthermore, when we treated RPL22-transduced hMPCs with MG132, a proteasome inhibitor (88), we found that HP1γ and KAP1 degradation caused by RPL22 was reduced (Figure 5H). Taken together, these results support that RPL22 plays a role in destabilizing HP1γ and KAP1 during human stem cell senescence.
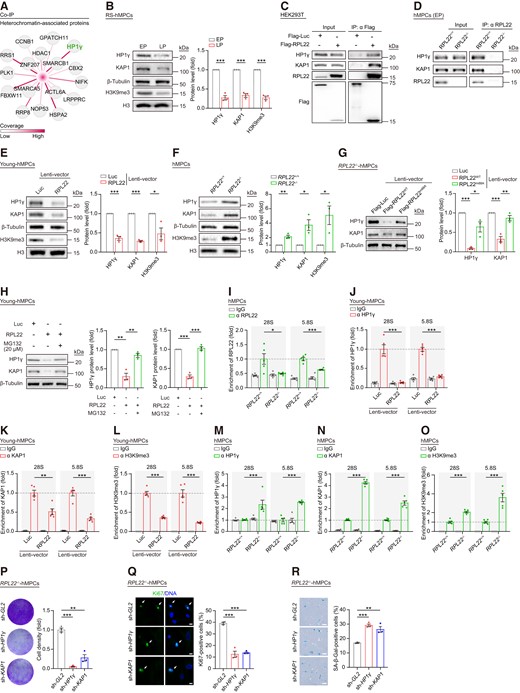
RPL22 binds rDNA and destabilizes HP1γ and KAP1 in senescent hMPCs. (A) Network diagram showing heterochromatin-associated proteins interacted with RPL22. The color key from white to red indicates coverage levels from low to high. (B) Western blot analysis of HP1γ, KAP1 and H3K9me3 protein levels in RS hMPCs. β-Tubulin was used as loading control for HP1γ and KAP1. Histone 3 (H3) was used as loading control for H3K9me3. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (C) Co-IP analysis of the exogenous interaction between RPL22 and HP1γ or KAP1 in HEK293T cells transduced with Flag-Luc and Flag-RPL22. (D) Co-IP analysis showing that endogenous RPL22 is interacted with HP1γ or KAP1 in RPL22+/+ and RPL22-/- hMPCs at early passage (EP, P3). (E, F) Western blot analysis of HP1γ, KAP1 and H3K9me3 protein levels in young hMPCs transduced with lentiviruses expressing Luc or RPL22 (E) and in RPL22+/+ and RPL22-/- hMPCs (F). β-Tubulin was used as loading control for HP1γ and KAP1. Histone 3 (H3) was used as loading control for H3K9me3. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (G) Western blot analysis of HP1γ and KAP1 protein levels in RPL22-/- hMPCs transduced with lentiviruses expressing Luc, RPL22WT or RPL22m88A. β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (H) Western blot analysis of HP1γ and KAP1 protein levels in young hMPCs transduced with lentiviruses expressing Flag-Luc or Flag-RPL22 after treatment with or without MG132 (20 μM for 12 h). β-Tubulin was used as loading control. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (I) Relative enrichment of RPL22 at rDNA regions in RPL22+/+ and RPL22-/- hMPCs. Data are presented as the means ± SEMs. n = 5 technical replicates. Two-tailed unpaired Student's ttest was performed. Data are representative of three independent experiments. (J–L) Relative enrichment of HP1γ (J), KAP1 (K) or H3K9me3 (L) at rDNA regions in young hMPCs transduced with lentiviruses expressing Luc or RPL22. Data are presented as the means ± SEMs. n = 5 technical replicates. Two-tailed unpaired Student's ttest was performed. Data are representative of three independent experiments. (M–O) Relative enrichment of HP1γ (M), KAP1 (N) or H3K9me3 (O) at rDNA regions in RPL22+/+ and RPL22-/- hMPCs. Data are presented as the means ± SEMs. n = 5 technical replicates. Two-tailed unpaired Student's t test was performed. Data are representative of three independent experiments. (P) Clonal expansion analysis of RPL22-/- hMPCs transduced with lentiviruses expressing sh-GL2, sh-HP1γ or sh-KAP1. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (Q) Immunostaining analysis of Ki67 in RPL22-/- hMPCs transduced with lentiviruses expressing sh-GL2, sh-HP1γ or sh-KAP1. Scale bar, 10 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. The white arrows indicate Ki67-positive cells. Two-tailed unpaired Student's t test was performed. (R) SA-β-Gal staining of RPL22-/- hMPCs transduced with lentiviruses expressing sh-GL2, sh-HP1γ or sh-KAP1. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed.
Because silenced rDNA is characterized by highly compact heterochromatin with H3K9me3 and HP1 proteins (49,89), we next sought to assess whether nucleolus-localized RPL22 could bind to rDNA regions. By performing chromatin immunoprecipitation followed by quantitative PCR (ChIP–qPCR), we found that RPL22 binds to 28S and 5.8S rDNA (Figure 5I and Supplementary Figure S5H), and that the occupancy is increased in senescent hMPCs (Supplementary Figure S5I), concomitant with lower levels of HP1γ, KAP1, as well as H3K9me3 at RPL22-binding sites (Supplementary Figure S5J–L). Overexpression of RPL22 in young hMPCs led to loss of HP1γ, KAP1 and H3K9me3 at RPL22-enriched rDNA (Figure 5J–L), whereas this was not the case when we replenished FLAG-tagged RPL22m88A, which is not located in nucleolus (Supplementary Figure S5M–P). Furthermore, RPL22 depletion resulted in higher levels of HP1γ, KAP1 and H3K9me3 at rDNA in hMPCs (Figure 5M–O and Supplementary Figure S5Q, R), suggesting the detachment of HP1γ and KAP1, and loss of H3K9me3 at rDNA are dependent on the accumulation of RPL22 in the nucleolus during human MPCs senescence.
Next, we repressed HP1γ and KAP1 separately via short hairpin RNA (shRNA) in RPL22−/− hMPCs (Supplementary Figure S5S,T). Suppression of either protein was associated with elevated levels of 28S and 18S rRNAs (Supplementary Figure S5U). In addition, both HP1γ and KAP1 inhibition phenocopied the accelerated senescence of RPL22−/− hMPCs with RPL22 overexpression, as reflected by compromised proliferation and increased proportion of SA-β-Gal-positive cells (Figure 5P–R). Collectively, these data demonstrate that the presence of RPL22 in the nucleolus promotes the degradation of HP1γ and KAP1, which leads to rDNA heterochromatin decondensation, rRNA upregulation and eventually hMPC senescence.
RPL22 depletion counteracts senescence in human cells
Finally, to explore whether RPL22 deficiency attenuates cellular senescence by suppressing rRNAs transcription more generally, we leveraged two different premature human MPCs models: Hutchinson-Gilford progeria syndrome (HGPS) human MPCs carrying the G608G mutation in the LMNA gene (HGPS hMPCs) and Werner syndrome human MPCs with mutations in the WRN gene (WS hMPCs) (90,91). Relative to wild-type hMPCs, rRNA expression was increased in both cell models (Supplementary Figure S6A, G). When we performed lentiviral delivery of CRISPR/sgRPL22 into HGPS hMPCs and WS hMPCs, we validated that RPL22 deficiency reduced rRNAs levels, and alleviation of senescent features (Figure 6A–D, Supplementary Figure S6B-F and Supplementary Figure S6H-L). Similarly, in senescent models induced by H2O2 treatment or ultraviolet irradiation (UV), the increased rRNAs levels, impaired proliferation ability, increased SA-β-Gal levels and loss of heterochromatin were restored by RPL22 knocking out (Figure 6E–H and Supplementary Figure S7A–H). Additionally, RPL22 deficiency also attenuated senescence of human primary MPCs from healthy aged adults (Figure 6I, J and Supplementary Figure S7I). Furthermore, the introduction of RPL22 accelerated cellular senescence in human coronary artery endothelial cells (hCAECs) and human umbilical vein endothelial cells (hUVECs), evidenced by a diminished capacity for clonal expansion coupled with an elevated percentage of SA-β-Gal-positive cells (Figure 6K–N). Collectively, these observations showcase that RPL22 is a potential target for attenuating senescence.
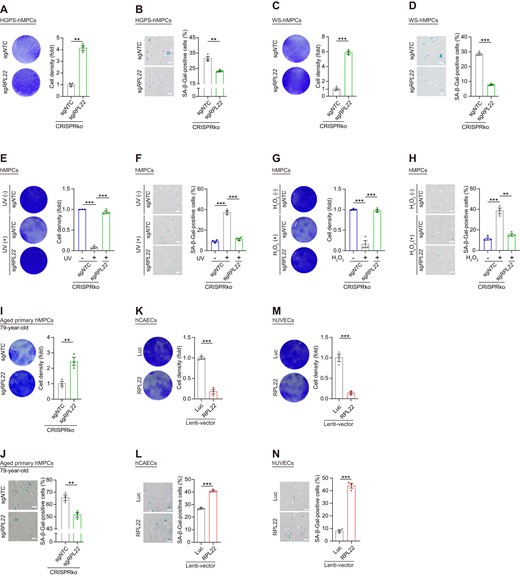
RPL22 depletion counteracts senescence of human cells. (A, C) Clonal expansion analysis of HGPS (A) and WS (C) hMPCs at late passage (P8) after lentivirus-mediated CRISPR knock-out with sgRPL22. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed. (B, D) SA-β-Gal staining of HGPS (B) and WS (D) hMPCs at late passage (P8) after lentivirus-mediated CRISPR knock-out with sgRPL22. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (E, G) Clonal expansion analysis of UV- (E) or H2O2- (G) induced hMPC senescence after lentivirus-mediated CRISPR knock-out with sgRPL22. Data are presented as the means ± SEM. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (F, H) SA-β-Gal staining of UV- (F) or H2O2- (H) induced hMPC senescence after lentivirus-mediated CRISPR knock-out with sgRPL22. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (I, K, M) Clonal expansion analysis of aged primary hMPCs (I) after lentivirus-mediated CRISPR knock-out with sgRPL22, hCAECs (K) and hUVECs (M) transduced with lentiviruses expressing Luc or RPL22. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's ttest was performed. (J, L, N) SA-β-Gal staining of aged primary hMPCs (J) after lentivirus-mediated CRISPR knock-out with sgRPL22, hCAECs (L) and hUVECs (N) transduced with lentiviruses expressing Luc or RPL22. Scale bar, 100 μm. Data are presented as the means ± SEMs. n = 3 biological replicates. Two-tailed unpaired Student's t test was performed.
Discussion
In this study, we performed a CRISPR/Cas9-based LOF screen in senescent hMPCs to systematically reveal the regulatory function of ribosome-associated genes during aging. For the first time, we identified RPL22 as a key driver of human stem cell senescence. Functionally, RPL22 accumulates in the nucleolus and binds to rDNA regions upon aging, which likely facilitates HP1γ and KAP1 degradation, concomitant with loss of H3K9me3 modification and the transcription of rRNAs, and onset of cellular senescence.
Abnormal ribosome biogenesis is associated with aging and aging-related diseases (33,37,92). Recently, decreased ribosomal activity and reducing RPs levels were reported to be linked with longevity in C. elegans, yeast and human (34,93–95). However, it remains an open question how ribosomal components regulate cellular senescence, especially in human stem cells. CRISPR screening is a powerful tool that has been widely used for identification of essential regulators in biological processes (51,53,55). To our knowledge, this is the first study to systematically identify ribosome-related genes involved in human stem cell senescence using CRISPR/Cas9-mediated LOF screening. Our results revealed that blocking of RPL22 attenuates hMPC senescence. Besides, we also identified other candidate genes that control senescence, such as C14orf169 and LYAR that await further investigations. Taken together, these efforts have opened the door for exploring how manipulating ribosome biogenesis might mitigate aging.
RPL22, incorporated into the 60S ribosomal subunit at a late stage of ribosome maturation, controls ribosome composition (96,97) and has been reported to participate in lymphocyte development and hematopoiesis (85,98,99). However, its function in human stem cells senescence has not yet been elucidated. Our data showed that RPL22 is upregulated and accumulates in the nucleolus during aging and that ectopic expression of RPL22 accelerates senescence. Conversely, deficiency of RPL22 conferred resistance to replicative senescence and stress-induced senescence, supporting that RPL22 actively promotes senescence. Interestingly, the deletion of RPL22 did not impact the overall translation process, suggesting a ribosome-independent role for RPL22 in modulating hMPC senescence. However, RPL22 might selectively influence the translation of specific mRNAs, a possibility that merits further exploration. Thus, our study has elucidated the unique interplay between the ribosomal protein RPL22, nucleolar heterochromatin regulation, and rDNA expression in hMPCs.
Aging-induced disarray in heterochromatin can reactivate repetitive sequences like rDNA (100). In our study, we discovered that nucleolar RPL22 interacts with rDNA repeats, inducing heterochromatin decondensation and promoting rRNA transcription. Conversely, RPL22 deficiency preserved rDNA heterochromatin and curbed rRNA expression. Mechanistically, the RPL22 nucleolus-aggregation reduced HP1γ and KAP1 protein levels, followed by decreased rDNA enrichment. Although we cannot exclude the possibility that RPL22 plays a role in the initial transcription of rRNA and the subsequent post-transcriptional processes. Interestingly, RPL22-mediated inhibition of HP1γ or KAP1 was reversed by a proteasome inhibitor, suggesting that RPL22 might promote proteasomal degradation of heterochromatin-related proteins, though the underlying mechanism awaits further investigations. However, our data validated that decondensation of heterochromatin and excessive rRNA expression is a key mechanism mediating the pro-senescence effect of RPL22.
Previous reports have indicated that RPL22 binds to CDK4 to induce senescence in human fibroblasts (100). However, our Co-IP/MS experiments did not identify CDK4 as an RPL22 partner, suggesting that RPL22 may have cell type-specific effects. Notably, in addition to heterochromatin proteins HP1γ or KAP1, we also identified proteins associated with mitochondrial homeostasis (e.g., TFAM), nuclear division (e.g., CDK13), and ribosome biogenesis (e.g., RPP40). These discoveries shed new light on the potential mechanisms through which RPL22 could impact hMPC senescence and merit further exploration. Consistent with our findings of reduced DNA damage signals following RPL22 deletion in hMPCs, the loss of KAP1 and HP1γ also leads to a DNA damage response (101,102). This implies that the RPL22-KAP1/HP1γ axis might play a role in DNA damage repair during hMPC senescence, warranting further investigation.
In sum, by leveraging a CRISPR/Cas9-mediated LOF screen for ribosome-associated genes in regulating human MPCs senescence, we discovered that the nucleolus-localized RPL22 functions as a heterochromatin destabilizer that stimulates rRNA transcription and ultimately promotes cellular senescence. Conversely, we showed that reducing RPL22 levels can ameliorate cellular senescence in diverse biological contexts, pointing to a key role for RPL22 in mediating cellular senescence, at least in human MPCs and human vascular endothelial cells. Consistent with data in the Aging Atlas showing that RPL22 is upregulated during aging in many organs (https://ngdc.cncb.ac.cn/aging/index) (77), our functional experiments exploring genetic strategies or inhibitors targeting RPL22 support further explorations of RPL22 as an intervention target in aging-related diseases. In addition, our study establishes a new correlation between nucleolar RPL22 and aging-associated nucleolar expansion (45,46), which could be considered a hallmark of cellular senescence.
Data availability
CRISPR-based screening, RNA–seq, H3K9me3 ChIP–seq and WGS data have been submitted to the Genome Sequence Archive in the National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation) of the Chinese Academy of Sciences, with accession number HRA005070. CRISPR-based screening data generated in this study have also been deposited in Gene Expression Omnibus (GEO) database with accession number GSE253231. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (103) via the iProX partner repository (104,105) with the dataset identifier PXD044052.
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We are grateful to Lei Bai, Luyang Tian, Qun Chu, Shangyi Qiao, Ruijun Bai, Shikun Ma, Xiangmei Jin, Jing Lu and Ying Yang for their administrative assistance; Feifei Liu and Jingyi Jia for their help in teratoma experiments; Jifeng Wang for assistance in LC–MS/MS; Junying Jia, Qing Meng and Shuang Sun for their assistance in the FACS experiments. We also acknowlodge the Biorender (https://www.biorender.com/) platform that was used for illustration.
Funding
This work was supported by National Key Research and Development Program of China [2020YFA0112200, 2022YFA1103700, 2022YFA1103800]; National Natural Science Foundation of China [81921006, 82125011, 92149301, 92168201, 92049304, 92049116, 32121001, 32200610, 82192863, 82122024, 82071588, 32100937, 82271600, 32000500, 32341001, 82330044, 82361148130, 82361148131, 82322025]; CAS Project for Young Scientists in Basic Research [YSBR-076, YSBR-012]; National Key Research and Development Program of China [2020YFA0804000, 2021YFF1201000, the STI2030-Major Projects-2021ZD0202400]; Informatization Plan of Chinese Academy of Sciences [CAS-WX2022SDC-XK14]; New Cornerstone Science Foundation through the XPLORER PRIZE [2021-1045]; Youth Innovation Promotion Association of CAS [2023092, 2022083]; CAS Youth Interdisciplinary Team, Beijing Municipal Public Welfare Development and Reform Pilot Project for Medical Research Institutes [JYY2023-13]; Excellent Young Talents Program of Capital Medical University [12300927]; Project for Technology Development of Beijing-affiliated Medical Research Institutes [11000023T000002036310]; Excellent Young Talents Training Program for the Construction of Beijing Municipal University Teacher Team [BPHR202203105]; Initiative Scientific Research Program, Institute of Zoology, Chinese Academy of Sciences [2023IOZ0102], CAS Special Research Assistant (SRA) Program, Young Elite Scientists Sponsorship Program by CAST [2023QNRC001, 2021QNRC001]. Funding for open access charge: Institute of Zoology, Chinese Academy of Sciences, Beijing, China. Key Research and Development Project of Hainan Province [2023ICAC-YANFA], the Science and Technology Platform Construction Project of Hainan Province [2023ICAC-YUNXING].
Conflict of interest statement. None declared.
References
Author notes
The first four authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments