





Abstract
Over the past five decades, DNA restriction enzymes have revolutionized biotechnology. While these enzymes are widely used in DNA research and DNA engineering, the emerging field of RNA and mRNA therapeutics requires sequence-specific RNA endoribonucleases. Here, we describe EcoToxN1, a member of the type III toxin-antitoxin family of sequence-specific RNA endoribonucleases, and its use in RNA and mRNA analysis. This enzyme recognizes a specific pentamer in a single-stranded RNA and cleaves the RNA within this sequence. The enzyme is neither dependent on annealing of guide RNA or DNA oligos to the template nor does it require magnesium. Furthermore, it performs over a wide range of temperatures. With its unique functions and characteristics, EcoToxN1 can be classified as an RNA restriction enzyme. EcoToxN1 enables new workflows in RNA analysis and biomanufacturing, meeting the demand for faster, cheaper, and more robust analysis methods.
Introduction
DNA restriction enzymes have been an enormous success in biotechnology for the past 50 years (1,2). DNA restriction enzymes are widely used in molecular biology research with the most prominent use in gene engineering, DNA mapping and sequencing (3). DNA restriction enzymes cleave double-stranded DNA (dsDNA) sequences at sequence-specific sites, which results in DNA fragments with a known sequence at each end. For RNA, that in contrast to DNA exists predominantly as single-stranded polynucleotides, the toolbox for sequence-specific cutting enzymes has been rather small. It contains either frequently cutting RNases (4,5) or RNases that depend on hybridization with short RNA or DNA guide oligos to generate local dsRNA/DNA hybrids for cutting (6,7).
RNA as a therapeutic has gained growing interest over the past decade, which also increases the demand for better and more robust analysis methods. Vaccines based on mRNA have been under development for viral infections for the past several years (8), and the industrial-scale production of mRNA and its use in vaccines received a boost in attention during the SARS-CoV-2 pandemic (9,10). These mRNA vaccines were the most effective remedy against the virus, triggering a significant development of this type of therapeutic (11), resulting in high demand.
The urgent need for effective vaccines has led to an adaptation of quality control tests to fast track safe and effective vaccines. As the world has moved to a post-pandemic state, it is ever more important to establish and standardize quality attributes connected to therapeutic mRNA and mRNA vaccines. Several critical quality attributes for mRNA therapeutics have been identified and are related to identity, content, integrity, purity, and safety (12). Integrity of mRNA, for example, comprises the ratio of intact versus fragmented mRNA, the ratio of 5′-cap versus uncapped 5′-termini, and the completeness of the poly(A) tail. To address these critical quality attributes, there is a growing need to develop new methods to analyze mRNA during manufacture to confirm its identity and integrity.
The 5′-cap is critical for stable and mature mRNA and is responsible for an efficient translation to protein (13). Depending on the manufacturing process, the 5′-capping can be applied during or after the in vitro transcription (IVT) step (14). The capping and the percentage of successfully capped material is critical for the further application of the mRNA. Capping of mRNA therefore needs to be thoroughly characterized and monitored. Today, mRNA is analyzed by liquid chromatography (LC), gel electrophoresis and mass spectrometry (MS) (15). Analyzing long RNA strands is, however, not practical for capping analysis and nearly all methods used today rely on the fragmentation of the mRNA into smaller pieces that can be analyzed more conveniently. The fragmentation approach should ideally result in a mixture which allows identification and discrimination of a 5′-capped fragment from a 5′-uncapped fragment that differs only by one nucleotide. The methods used today are based on: 1) RNase H: In this method a sequence-specific cut is generated close to the 5′ end via an RNA/DNA hybrid (6,7). 2) RNase A & RNase T: Here a DNA antisense oligo is used to protect the 5′ end while RNase A and RNase T indiscriminately digest the remaining mRNA into small pieces (5). A similar approach was described with the human RNase 4 (16). 3) Ribozymes: These catalytic RNAs bind and cleave mRNA at specific sites (17). 4) DNAzymes: Similar process to Ribozymes but based on catalytic DNA instead of RNA (18,19). 5) Complete digestion of the mRNA with a P1 3′ exonuclease and analyzing the remaining GpppN and pppN ratio (20). The first four methods require a DNA or RNA oligo either as a cofactor, a guide polynucleotide, or as a catalytic active fragment (Ribozyme and DNAzyme). They require one or several annealing phases of DNA or RNA oligos to the mRNA in a heating step of the heat labile mRNA up to 95°C. In addition, RNases and Ribozymes require magnesium ions for their function which also can cause unwanted degradation at elevated temperatures (21) (Supplementary Table S3).
The poly(A) tail length is another critical quality attribute of mRNA integrity that requires accurate monitoring. As is the case for the 5′ cap, the 3′-poly(A) tail also plays a critical role in maintaining the stability of mRNA. Therapeutic mRNA is either co-transcriptionally or post-transcriptionally modified with a poly(A) tail to enhance its half-life in vivo. For analysis of the integrity and length of the poly(A) tail, the tail is typically cleaved off using, for example, an RNase T1. Analysis can be performed with LC-MS (22) or gel filtration (23), but with a length of 100–150 nucleotides the analysis of the length and heterogeneity remains still a challenge. Extended poly(A) tail length can also indicate loop back-caused dsRNA formation (24).
EcoToxN1 (Escherichia coli ToxN, cluster 1 (25)) is a sequence-specific RNase of the type III toxin-antitoxin family (25,26) that recognizes a specific RNA pentamer sequence. It has the unique feature to cleave single-stranded RNA (ssRNA) with high precision at its recognition site resulting in two terminal nucleotides either carrying a 5′-hydroxy or a 2′-3′-cyclic-phosphate end (25,26). Its canonical recognition sequence is GAA↓AU with the cutting occurring after the third residue (25,26). Restriction enzymes are enzymes that restrict the replication of viral genomes. Since the type III TA systems do restrict the replication of an RNA virus (26), the type III toxins and EcoToxN1 could be classified as RNA restriction enzymes.
In this article, we describe EcoToxN1 as a novel tool that has the potential to simplify and enhance RNA analysis workflows with no need for guide RNA/DNA oligos, presence of Mg2+ or specificity-facilitating and precision-enhancing annealing steps as described elsewhere (7). At the same time this tool allows for improved resolution enabling accurate determination of the critical quality attributes like 5′-cap status, poly(A) tail completeness and identity of therapeutic RNA. EcoToxN1 shows high specificity and activity for cleavage of RNA substrates that contain the cleavage sequence for the enzyme. Furthermore, we propose several possible applications of this enzyme in manipulation of single-stranded RNA as applications in molecular biology. We also report the use of other homologues of EcoToxN1 with varying sequence recognition sites, increasing the repertoire of RNA restriction endoribonucleases for molecular biology applications, and we introduce a simple ‘Cut and PAGE’ strategy with EcoToxN1 that allows for fast and high-throughput analysis of RNA.
Materials and methods
Enzyme production and purification
The RNA restriction enzymes EcoToxN1 (pdb: 7D8O), BthToxN1 (4ATO) and EcoToxN5 were cloned together with their corresponding antitoxin into a pVB plasmid (Vectron Biosolutions AS, Trondheim, Norway). The antitoxin was kept under its natural promoter, while the toxin was inducible under a Pm promoter (27). A detailed description of the production and purification of EcoToxN1 can be found in the protocols described by Manikandan et al. (25,28). In short, after the lysis of the cells the toxin-antitoxin complex was purified via a HisTrap affinity chromatography. To improve the yield of free ToxN, the toxin was purified by an anion exchange chromatography step to separate the toxin from the remaining antitoxin. The purity of the protein was assessed via SDS PAGE analysis, and the absence of the antitoxin RNA was verified via urea-PAGE gels. Since both BthToxN1 and EcoToxN5 have an isoelectric point above 9 (in contrast to EcoToxN1 with an pI of 6.4), the anion exchange chromatography was replaced with a cation exchange chromatography for these two variants.
Mutagenesis to introduce EcoToxN1 recognition sites for 5′ cap analysis
Point mutations were introduced into the in vitro transcription plasmid using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific Inc) according to manufacturer's instructions. Primers were used in mutagenesis to insert EcoToxN1 cut site 13 nucleotides downstream of promoter in pGEM3Zf+ (MutET13). For capping purposes, an additional point mutation was performed to change the transcription start sequence from GGG to GGT. Primers are listed in Supplementary Table S1.
Sequence downstream of T7 promoter (in italics) of pGEM3Zf+:
TAATACGACTCACTATA_GGTCGAATTCGAA↓ATCGGTACCCGGGGATCCTCTAGAGTC*GAC
Resulting mRNA from construct above:
(5′cap)GGUCGAAUUCGAA↓AUCGGUACCCGGGGAUCCUCUAGAGUC
↓EcoToxN1 cleavage site
*HincII cleavage site
Plasmid linearization
Linearization was performed using 2 U of restriction enzyme per μg of plasmid, incubated at 37°C overnight. All samples were purified using PureLink™ PCR Purification Kit K310002 (Thermo Fisher Scientific Inc). Figure 2: pGEM3Zf + ETN13 GGT. Linearized using Hinc II (Thermo Fisher Scientific Inc) for a 40 nucleotide IVT-product. Figure 4 and S8 – pGEMEX-1. Linearized using NheI (Thermo Fisher Scientific Inc) for a 71 nucleotide IVT-product.
Figure S1. pGEMEX-1. Linearized using HincII (Thermo Fisher Scientific Inc) for a 912 nucleotide IVT-product.
RNA production
Synthetic RNA oligos were purchased from Metabion or IDT. With the exception for the poly(A)-oligo, the oligos contained a 3′-FAM label for visualization on denaturing PAGE gels. The oligos used in the main text are listed in Figure 1. The full list of all synthetic oligos used in this study is available in the Supplemental information.
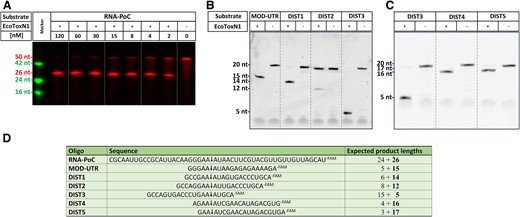
Sequence specific digestion of ssRNA with EcoToxN1. (A) Digestion of single stranded 50 nucleotide RNA-PoC with EcoToxN1. The oligo has a 3′ FAM label for visualization. The recognition site GAAAU is in the center of the oligo and results in a FAM labeled product of 26 nucleotides. EcoToxN1 cuts the 500 nM mRNA oligo in a dose dependent way with a ratio of 10:1 (RNA to EcoToxN1) sufficient for nearly complete digestion within 1 h at 15 °C. The marker deoxyribonucleotide oligos are TAMRA labeled and have the size of 42, 24 and 16 deoxyribonucleotides. Reaction condition: 500 nM of a synthetic RNA oligo of 50 bases was incubated with increasing amounts of EcoToxN1 (2–120 nM) at 15°C for 1 h in a buffer containing 25 mM Tris-HCl pH 7.5, 25 mM NaCl. (B, C) Positioning of the recognition site on a 20mer oligo. The recognition site for EcoToxN1 can be placed anywhere to cut the oligo. (D) Sequence of the oligos. The arrow indicates the cleavage site. The number in bold is the length of the FAM-labeled fragment that is detected on the gel. Dist2 oligo can form a hairpin loop that reduces the cleavage activity of EcoToxN1.
In-house RNA synthesis was performed using a standard IVT protocol. ArcticZymes T7 RNA polymerase (ArcticZymes Technologies, Tromsø, Norway) was used with Pyrophosphatase and RiboLock RNase inhibitor (Thermo Fisher Scientific Inc) in 50 μl reactions containing 40 mM Tris-HCl pH 8.0 (at 25°C), 2 mM Spermidine, 10 mM DTT, 6 mM MgCl2, 10 mM NTPs (Thermo Fisher Scientific Inc) and 1 μg linearized plasmid. RNA cleanup was performed using the NEB Monarch® RNA Cleanup Kit (New England Biolabs, USA).
RNA fragmentation
A typical EcoToxN1 reaction was performed as follows if not further specified in the figure descriptions: 500 nM of synthetic RNA oligos of 20 bases in length were incubated with 10 nM EcoToxN1 at 37°C for 10 min in a buffer containing 25 mM Tris-HCl pH 7.5, 25 mM NaCl. The reaction was stopped by adding 1:1 of a urea-PAGE loading buffer (95% formamide, 10 mM EDTA, bromophenol blue). The samples were loaded on a 7 M 20% urea-PAGE gel. The samples were visualized with a PharosFXTM Plus Molecular Imager (Bio-Rad, Hercules, California, USA) or a GelDocTM XR+ (Bio-Rad Hercules, California, USA) for detection of the FAM or TAMRA fluorophores. SYBR Gold staining was conducted with a 1:10000 dilution of SYBR Gold (Thermo Fisher Scientific Inc.) in MQ water before being visualized with a GelDoc™ XR+ .
Gel-based capping analysis
Purified RNA containing a 40-nucleotide long IVT-product was capped using Vaccinia Capping enzyme (New England Biolabs, USA), according to the manufacturer's protocol. For time-resolved analyses, 20 μl reactions were used containing ∼70 ng/μl RNA and 50% of recommended capping enzyme concentration (5 units). Reactions were stopped at the indicated time points using 2 mM EDTA followed by 5 min heat-inactivation at 70°C. After capping, no cleanup was performed prior to cutting with EcoToxN1.
Cleavage of capped and uncapped RNA was performed in 5 μl reactions, containing 3 μl (200 ng) capped or uncapped RNA, 1 μl EcoToxN1 (15 nmol) and 1 μl 5x cutting buffer (250 mM Tris-HCl pH 7.5 and 250 mM NaCl). Samples were incubated at 37°C for 10 min then mixed with 5 μl RNA formamide gel loading buffer prior to gel electrophoresis and visualization using a GelDoc™ XR+ (Bio-Rad Hercules, California, USA). Band intensity analysis was done using the Bio-Rad ImageLab software (ver. 4.1).
All the experiments have been repeated for their reproducibility and consistency.
Results
Sequence-specific RNases
EcoToxN1 (E. coli ToxN, cluster 1 (25)) is a sequence-specific RNase of the type III toxin-antitoxin family (25,26) that recognizes a specific pentameric RNA sequence (GAA↓AU). The enzyme has its maximum activity at ∼37°C but it is active at lower temperatures and thus sequence-specific cleavage analysis can be performed at room temperature or below (Figure 1 and Supplementary Figures S1, S2, S3A–C and S10). The endoribonuclease activity of EcoToxN1 can be detected in the absence of salt and with up to 200 mM NaCl present, however the activity drops significantly at NaCl concentrations above 100 mM. The maximum activity has been determined at 25 mM NaCl (Supplementary Figure S13). EcoToxN1 does not require an additional DNA or RNA oligo for its function, and it works in a Mg2+ free buffer. For optimal activity the concentration of Mg2+ and Mn2+ should be lower than 1 mM or free of uncomplexed Mg2+ and Mn2+. Both ions can function as efficient inhibitors for EcoToxN1. This inhibitory effect of Mg2+ and Mn2+ can be reversed with EDTA (Supplementary Figures S5 and S6). EcoToxN1 is not inhibited by standard RNase inhibitors such as RiboLock (Thermo Fisher Scientific Inc) which enables direct EcoToxN1 treatment of samples from IVT reactions that have been stopped with EDTA without any buffer exchange.
The pentameric recognition site of EcoToxN1 is versatile and can be placed anywhere in a 5′UTR (untranslated region), and even at the very ends of RNA. The enzyme does not require an overhang and cuts ssRNA that starts with the cleavage site. The shortest fragment that EcoToxN1 can produce is therefore three nucleotides long: GAA when cutting directly at the 5′ site (Figure 1C). However, secondary structures may impact the cutting efficiency of EcoToxN1 (Figure 1B), but the enzyme can tolerate agents that are weakening secondary structures of RNA such as urea and formamide (Supplementary Figure S10). Under certain conditions EcoToxN1 shows an altered sequence specificity which, according to DNA restriction enzyme terminology, could be referred to as ‘star activity’. We found that high EcoToxN1 concentration and a pH above 8.5 can induce ‘star activity’ (Supplementary Figure S9).
Capping-efficiency detection on urea-PAGE gel with EcoToxN1
Since no additional antisense oligos are needed for capping analysis with EcoToxN1, the analysis can be conducted using a simple urea-PAGE gel without the risk of antisense oligos interfering with the analyte. Figure 2 shows the simplicity of using RNA restriction enzymes such as EcoToxN1 in the analysis of capping efficiency. Figure 2A shows a urea-PAGE gel of an mRNA IVT construct that was either capped or not capped using the vaccinia capping enzyme (VCE) and either treated without or with EcoToxN1 for just 10 min at 37°C. The resolution of the untreated mRNA with 40 nucleotides for the uncapped or 41 nucleotides for the capped RNA is too poor to detect successful capping. However, after treatment with EcoToxN1, the band shift of the small fragment of 13 nucleotides uncapped compared to 14 nucleotides in the capped sample indicates a successful capping reaction with VCE. The capping analysis also works with co-transcriptional capped mRNA using capping analogues (Supplementary Figure S7A shows a 69 nt RNA and S7B shows a 1920 nt mRNA). The easy use and high resolution of employing EcoToxN1 in capping analysis allows for time-monitoring of a VCE capping reaction (Figure 2B and Supplementary Figure S14). The VCE reaction was stopped at indicated timepoints and the band intensities of the uncapped and capped fragment were analyzed. Under the chosen condition, in which we reduced the VCE concentration to 50% below the recommended concentration, the capping reaction reached 50% completeness after ∼12 min and >90% completeness after 60 min. This simple setup using the ‘Cut and PAGE’ principle allows optimization of capping reactions for mRNA production, such as for example reducing the time needed for a complete cap reaction or lowering the amount of expensive components such as the VCE enzyme to save costs.
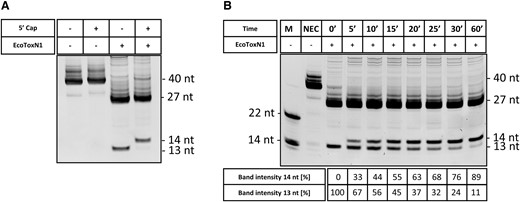
Capping analysis with EcoToxN1 of post-transcriptional (VCE vaccinia capping enzyme) capped mRNA. (A) An IVT of pGEM3Zf-ETN1 + 13 GGT was either capped in a VCE reaction or left uncapped, digested using the EcoToxN1 endoribonuclease. EcoToxN1 can cleave with high precision at its cleavage site and the capping efficiency can be determined. (B) Timed VCE capping experiments. With EcoToxN1 the capping efficiency can be easily monitored over time. For this gel, the measured ratios extracted from the band intensities are listed in the table below the figure. M = marker, NEC = no enzyme control. Marker oligoes are deoxyribonucleotides.
For practical reasons, fragments in the range of 10–25 bases are most suitable for band shift analysis on standard urea-PAGE gels. Longer fragments might not show a clear band shift since the difference in gel migration gets smaller with size. This is shown in Figure 2 where the 13–14 nucleotides are clearly separated (Figure 2A, B), whereas the 22 and 23 nucleotides from the experiment with the cap analogues are much closer (Supplementary Figure S7A and B). Longer gels or higher acrylamide concentration will improve the resolution for longer fragments. Smaller fragments on the other hand may be prone to poor staining with intercalating dyes.
RNA fingerprinting using EcoToxN1
Several other RNA analysis methods can be facilitated with EcoToxN1 such as RNA fingerprinting to determine the identity or integrity of mRNA. Like DNA fingerprinting with DNA restriction enzymes, RNA restriction enzymes such as EcoToxN1 exploit the different locations of the cleavage site in a ssRNA (Figure 3). In Figure 3 the two synthetic RNA oligos MOD-UTR and DIST1 are of identical length of 20 bases and would run at the same height in a urea-PAGE (MOD-UTR is based on the first 20 nucleotides of the 5′ UTR from mRNA1273/Moderna corona vaccine (29)). However, each has the EcoToxN1 cleavage site at a different position and can be undoubtedly identified after cleavage by their unique pattern. Furthermore, the band intensity can be used to estimate the composition of RNA in a mixture such as used in multivalent mRNA vaccines.
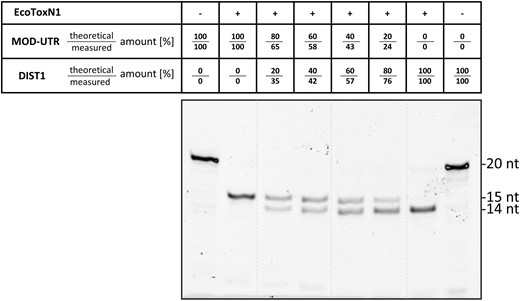
RNA Fingerprinting. Digestion of two 20 nucleotide RNA substrates simultaneously (MOD-UTR: GGGAA↓AUAAGAGAGAAAAGA−FAM and Dist1: GCCGAA↓AUAGUGACCCUGCA−FAM). The FAM labeled products differ by just one nucleotide resulting in product band of 15 and 14 nucleotides respectively. By exploiting the different location of the EcoToxN1 cleavage site the two substrates can be differentiated from each other and unambiguously identified. The qualitative ratio of the substrates in a mixture can be estimated from the band intensities in the gel. For this gel the theoretical mixing ratios as well as the measured ratios extracted from the band intensities are listed in the table above the figure. Only the product with the FAM label is visible. Reaction conditions: A total of 500 nM of synthetic RNA oligos of 20 bases were incubated with 12 nM EcoToxN1 for 10 min at 37°C in a buffer containing 25 mM Tris-HCl pH 7.5, 25 mM NaCl.
Influence of modified nucleotides on the performance of EcoToxN1
In vitro transcribed therapeutic mRNA is prone to nuclease degradation once injected into patients and the mRNA itself can also lead to innate immunogenicity. To avoid these effects, modified nucleotides are by default used in IVTs for mRNA vaccine production. Pseudouridine (Ψ) can be used to replace uridine in the IVT mRNA that leads to enhanced RNA stability and decreased anti-RNA immune response (30). Other common uridine variants used in mRNA are N1-methyl-pseudouridine (31) or 5-methoxy-uridine (32). EcoToxN1 has one uridine present in its canonical recognition sequence GAA↓AU. To test whether these uridine variants have an influence on the performance of EcoToxN1, IVTs with these modified uridine variants were treated with EcoToxN1 and analyzed on urea-PAGE gels. Generally, the results indicate that EcoToxN1 accepts modified uridine variants such as pseudouridine, N1-methyl-pseudouridine or 5-methoxy-uridine in the recognition site and correctly cuts the RNA oligo (Figure 4 and Supplementary Figure S8). Under the chosen conditions, the methoxy-uridine containing RNA seems to be cut with similar efficiency as the unmodified uridine RNA. RNA transcribed in presence of pseudouridine or N1-methyl-pseudouridine required however more enzyme and longer incubation time. The need for higher enzyme concentration and longer incubation time suggests a reduced activity of EcoToxN1 when encountering these uridine variants. However, even for the least active substrate, the N1-methyl-pseudouridine, >85% cleavage could be achieved. For analytic purpose and capping analysis, a complete cutting of the mRNA is not necessary since only the ratio of capped versus uncapped is determined.
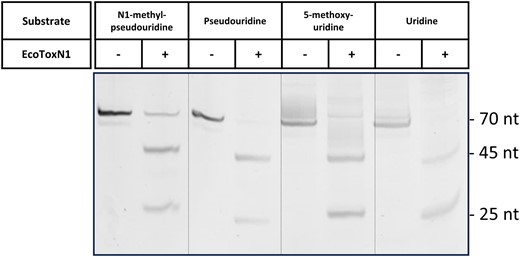
Modified uracil variants and their effect on the digestion with EcoToxN1. Cutting an IVT generated construct with EcoToxN1 that contains modified uracil. The 70 base nucleotide mRNA has the EcoToxN1 cleavage site after the 25 bases from the 5′-end resulting in two fragments of 25 and 45 bases respectively. The gel was stained with SYBRGold. Lane 1–2: IVT with N1-methyl-pseudouridine without and with EcoToxN1, lanes 3–4: IVT with pseudouridine, lanes 5–6: IVT with 5-methoxy-uridine, lanes 7–8: IVT with uridine. All the artificial uridine derivates are recognized and the sequence is cut by EcoToxN1 at pH 8.0. N1-methyl-pseudouridine shows a cleavage of more than 85% while pseudo-uridine, 5-methoxy-uridine and wt uridine are completely digested. The samples were digested with 190 nM EcoToxN1, 25 mM Tris-HCl, 25 mM NaCl, 3 mM EDTA, pH 8.0, 30 min at 37°C. The need for higher enzyme concentration and longer incubation time suggests a reduced processivity of EcoToxN1 when encountering these uracil variants.
Homologues of EcoToxN1
BthToxN1 (Bacillus Thuringiensis ToxN 1) is an additional toxin of the type III toxin-antitoxin system that has an alternative recognition site that is under investigation. BthToxN1 has recently been described to recognize the pentamer AAAAA (33). Interestingly, there is a discrepancy for BthToxN1 regarding the nature of its cleavage site. The crystal structure (PDB code 4ATO) of the toxin with its RNA antitoxin binds the hexameric sequence AAAAAA and shows cleavage after the third adenine (AAA↓AAA). In the cyclic heteromeric (three proteins, three RNAs) complex each protein binds two RNA antitoxins separated by the catalytic center of the toxin. The 5′ end and 3′ end of the RNA antitoxin contains (after cleavage with the toxin) three adenines each. This would also be in line with the EcoToxN1 mechanism (26). Our own experiments with synthetic RNA oligos indicate that an adenine hexamer is the preferred sequence, while an adenine pentamer shows significantly reduced cleavage activity. The proposed adenine pentamer is only cut if the first adenine as part of a hexameric sequence is replaced with a guanine (GAA↓AAA). Another sequence that is accepted by BthToxN1 is AAA↓ACA. Here, the fifth position tolerates a cytosine instead of the adenine. The activity on both noncanonical sequences (GAA↓AAA and AAA↓ACA) is however lower compared to the canonical AAA↓AAA sequence and could be classified as ‘star activity’ (Supplementary Figure S11). The fragments generated are in line with the results expected from the crystal structure and the cleavage happens after the third adenine in a hexameric AAA↓AAA sequence in the synthetic oligos. Previously published biochemical characterization data on the other hand indicated a cleavage after the first adenine in the pentameric A↓AAAA sequence (33).
EcoToxN5 (E. coli ToxN cluster 5) (25) is another RNA restriction enzyme of the type III toxin-antitoxin system with an alternative recognition site. EcoToxN5’s canonical sequence is AAAAUC, but it recognizes more general the sequence RAA↓AWC (R = A or G, W = A or U). (Supplementary Figure S12).
Poly(A) tail analysis with EcoToxN1 and EcoToxN5
EcoToxN1 and EcoToxN5 can be utilized for double digestion and RNA fingerprinting using two RNA restriction enzymes exploiting two different recognition sites. Figure 5 shows the double digestion of a synthetic RNA of 39 nucleotides. It contains the EcoToxN1 cleavage site after 10 nucleotides and a EcoToxN5 cleavage site after 23 nucleotides. Single digestion with EcoToxN1 leads to fragments of 10 and 29 nucleotides, single digestion with EcoToxN5 results in fragments of 16 and 23 nucleotides. Simultaneous double digestion with both RNA restriction enzymes leads to fragments of 10, 16 and 13 nucleotides. With its poly(A)-like recognition site, EcoToxN5 could be used as an elegant solution for the poly(A)-tail length analysis. Introducing a single cytosine in the poly(A) tail will generate a restriction site for EcoToxN5. This can allow to cut a short poly(A) RNA fragment from the 3′ end of the mRNA, that could be analyzed more conveniently compared to the average 100–150 nucleotides of a complete poly(A) tail which is described elsewhere in the literature (22,23).
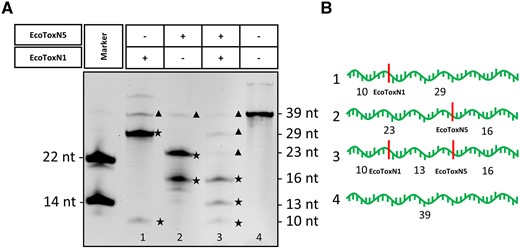
Double digestion of a synthetic RNA oligo with EcoToxN1 and EcoToxN5. (A) M: marker, 1: EcoToxN1 digestion, 2: EcoToxN5 digestion, 3: double digestion with EcoToxN1 and EcoToxN5, 4: No enzyme control. Both RNA restriction enzymes cut at their recognition site and produce the predicted product band pattern. Additional bands of 39 in lane 1 and 2 and 39, 29, 23 in lane 3 arise from undigested substrate and incomplete digestion. A contamination of the synthetic substrate as seen in the additional band of ∼60 nucleotides in lane 4 is responsible for the weaker unspecific bands. Asterisks★ indicate the products, triangle ▴undigested substrate and intermediate products. (B) Representation of the expected band pattern with the indicated cleavage sites Poly(A) oligo: AAAAAAAGAAAUAAAAAAAAAAAAACAAAAAAAAAAAAA. The cleavage sites for EcoToxN1 and EcoToxN5 are underlined. The double digestion with two RNA restriction enzymes was done at 37°C: 500 nM of a synthetic RNA oligo of 39 bases was incubated with 20 nM EcoToxN1 and 16 nM EcoToxN5 for 10 min in a buffer containing 25 mM Tris-HCl pH 7.5, 25 mM NaCl.
Discussion
Use of EcoToxN1 in analysis of RNA
We have produced and characterized EcoToxN1 as a sequence-specific RNA restriction enzyme for use in molecular biology or biotechnological applications. Out of the above-mentioned critical quality attributes for mRNA therapeutics, EcoToxN1 can help to simplify identity and integrity analysis of mRNA. In biotechnological applications, high specificity and high precision are a requirement for RNA assessment and in particular in capping analysis during therapeutic mRNA process development and production. The unique features of a mere single enzyme solution with EcoToxN1 (compared to the alternatives RNase H, Ribozyme or DNAzyme that depend on hybridization and RNA/DNA double strand formation to cut the RNA or P1 3′- exonuclease, MazF (4), hRNase 4 (16), RNase A and RNase T (5) that are frequent cutters and will shred the RNA into many pieces) allows for rapid, simple, and cost-efficient capping analysis, poly(A) tail length determination and RNA identity diagnostics. The only prerequisite for the use of EcoToxN1 and its homologues is that the recognition sites must be naturally present or introduced via mutations. The proposed simple ‘Cut and PAGE’ approach with EcoToxN1 is fast and reliable. It takes approximately 10 min to digest an mRNA sample for analysis and a complete setup including the pipetting, reaction time, PAGE and analysis of the results can be done in less than 2h. While Mg2+ and Mn2+ inhibit EcoToxN1, low concentrations can be tolerated, and the negative effects of these metals can be circumvented with EDTA (Figures S4–S6). EcoToxN1 shows good tolerance for up to 1 M urea, 1 M betaine and 1 M formamide (Supplementary Figure S10), compounds that could help to break secondary structures in ssRNA. Modified uridines (Figure 4 and Supplementary Figure S8), that are incorporated in mRNA for therapeutics, are recognized by EcoToxN1 as well. The result is a highly versatile enzyme that can be used in many analytical processes.
In terms of resolution, the use of RNA restriction enzymes in 5′-UTR analysis can detect other molecular weight changing modifications and variations in mRNA besides cap structures of mRNA. Detection of RNA initiation slippage (34,35) in suboptimal IVTs would be such an example. With the use of RNA restriction enzymes, we present an easy way to detect small molecular weight changes with hitherto unprecedented resolution. More sophisticated detection methods like LC-MS analysis can be applied to increase the resolution and details of the analysis even further.
The advantage of an RNA restriction enzyme, such as EcoToxN1, is that it achieves sequence specificity without the need of additional oligos (RNA or DNA). These ancillary oligos as well as DNAzymes or Ribozymes could otherwise interfere with the analysis such as showing up overlapping bands in gel electrophoresis.
Another benefit of a single enzyme approach with EcoToxN1 in an analytical method is that it can be used with an excess of the target mRNA. Other methods that have been described use a surplus of 1:5 or even 1:250 of DNA oligos, RNases or Ribozymes over the target mRNA (5–7,17) The simplicity of our solution answers the need for high-throughput optimization of industrial manufacturing processes and gives research a tool to verify the capping efficiency without the need for access to high-end instrumentation. It also potentially reduces the time-to-result by removing the need for annealing steps, reducing the incubation time, and eliminating purification steps before and after the nuclease treatment.
Type III bacterial toxin EcoToxN1 described here and its homologues EcoToxN5 and BthToxN1 have similar characteristics as DNA-restriction enzymes and can be classified as RNA-restriction enzymes with applications in mRNA analysis. There are, however, some decisive differences between RNA- and DNA-restriction enzymes. EcoToxN1 is a monomeric RNase, it cuts ssRNA, the pentameric recognition site (EcoToxN5 and BthToxN1 recognition sites are hexameric) is not a palindrome and the cleavage occurs within the recognition site. Mg2+ is not required for the activity. In contrast, the >3000 type II DNA-restriction endonucleases that have been discovered recognize short, mostly palindromic, sequences of 4–8 bp in dsDNA. They cleave dsDNA within or near to their recognition sequence and require Mg2+. The most common type II DNA restriction enzymes in use are homodimers (36).
To increase the flexibility and user-friendliness of RNA restriction enzymes, a set of RNA-restriction enzymes with a cleavability of a variety of different recognition sites is desirable (as it is the case for DNA restriction enzymes). Finding all variants might, however, be challenging. A pentameric recognition site for RNA-restriction enzymes has several advantages. In terms of frequency, one would expect to find the sequence on average once every ∼1000 bases in a random sequence. The coronavirus RNA genome (NCBI Reference Sequence: NC_045512.2), as an example, with ∼30 000 nucleotides, has 51 EcoToxN1 cleavage sites, roughly one every 600 nucleotides. For analytical purposes, this frequency is low enough that naturally occurring cleavage sites in a gene could be deleted through codon optimization without altering the function of the gene. In mRNA analysis that low frequency would statistically result in only a few fragments if any. The five-base recognition sequence is also short enough so that, with minimal alterations, the EcoToxN1 cleavage site can be introduced into the 5′-UTR or 3′-UTR of mRNA. There is an 80% chance that within a random sequence of 20 bases the pentameric recognition site of EcoToxN1 can be introduced by just 1–2 mutations. However, RNA-restriction enzymes that can recognize already existing cleavage sites in the 5′-UTR or 3′-UTR would be advantageous.
Use of EcoToxN1 in synthesis of RNA
In this publication we have focused primarily on analytical application of RNA restriction enzymes. Capping analysis of therapeutic mRNA is just one of many analysis applications where the sequence-specific endoribonucleases EcoToxN1, EcoToxN5 and BthToxN1 can make an impact (Figure 1 & 2). Poly(A)-tail analysis of the 3′ end of mRNA or RNA fingerprinting for the identification of different RNAs in a mixture based on specific fragmentation pattern (Figures 3 and 5) are just a small selection of analytic applications that these enzymes and its relatives can enable.
Besides their shown relevance in analytical applications, RNA restriction enzymes could open new roads and generate new and bold ideas for additional applications in the growing therapeutic RNA world. Albeit outside the scope of this publication, interesting ideas regarding new production methods of different RNA species are briefly discussed.
EcoToxN1 could be used in the manufacturing processes of RNA enabling novel and unique workflows. In production of RNA variants, EcoToxN1 could advance and simplify manufacturing of anti-sense-oligos (ASO), in situ probes, aptamer, ribozymes, siRNA, or miRNA. These short RNA pieces can be amplified as multiple copies in long IVT constructs separated by EcoToxN1 cleavage sites, creating long concatemers of RNA. Digestion of these long transcripts can result in an increase in production yield. Rolling circle transcription that could theoretically yield infinitely long IVTs of concatemers could be especially interesting to be combined with RNA restriction enzymes such as EcoToxN1.
Circular RNAs (circRNA) have gained a lot of attention in the past years due to their increased stability compared to linear mRNA (37–39). CircRNA construction can be enabled by combining EcoToxN1 with, for example, an RtcB ligase (40). RtcB ligase accepts 3′-phosphate groups as substrate for ligation. Concatemeric RNA transcripts divided by EcoToxN1 recognition sites can be cleaved uniformly resulting in single RNA units ready for ligation with RtcB ligase. A combination of an alkaline phosphatase and a standard RNA ligase such as T4 RNA Ligase 1 or R2D ligase (41) could substitute for RtcB as well.
The bacterial toxin–antitoxin systems are a noteworthy class of enzymes. The type III toxin–antitoxin systems and its representatives: EcoToxN1, EcoToxN5, BthToxN1, as described in this paper, along with the type II toxin–antitoxin systems consist of a set of toxins that are often RNases and show remarkable sequence and substrate specificity (42–44). The MazEF from the type II toxin–antitoxin systems, as an example, has been under intense investigations (44,45). The focus of applications of toxin-antitoxin systems has been so far in the field of antibacterial or antiviral strategies and possible applications in anti-cancer therapy (46). Biotechnological applications such as counter selection, containment control, in vivo bioluminescent imaging, single protein production, and targeted killing of pathogenic bacteria in microbiota are other applications of toxin–antitoxin systems (46,47). The true strength of the RNA restriction enzymes such as EcoToxN1 and its homologues could be in applications connected to RNA analysis and possibly RNA manufacturing.
Earlier attempts to generate sequence-specific RNases by fusing RNA-binding domains to RNA cleavage domains did not slice within the recognition sequence and could not cut very precisely (48).
While several RNA analytical methods have been developed for molecular biology applications, there is a demand for the introduction of alternative, orthogonal and simpler approaches. The simple ‘Cut and PAGE’ strategy that is enabled by the single-enzyme method with RNA restriction enzymes such as EcoToxN1 is better suited for fast and high-throughput quality assurance in RNA vaccines and RNA therapeutics research and development. Clearly, the possibilities of combining RNA restriction with other ways of nucleic acid analysis such as LC–MS are given and described copiously in the scientific literature (6,49).
Sequence-specific DNA restriction enzymes have been the work horse of molecular biotechnology for over 50 years. With the rise and the growing field of mRNA therapeutics and production, demands for novel enzyme tools that can manipulate and analyze RNA will follow. EcoToxN1 can simplify many of the existing workflows in RNA analysis and production and, like in the case with DNA restriction enzymes, might trigger the development of new ideas and applications for RNA restriction enzymes in the future.
Data availability
All data are available in the main text or the supplementary materials. Sequences of the ToxN RNA restriction enzymes are based on the published crystal structures PDB code: 4ATO (BthToxN1) UniProt ID: Q3YN09 and 7D8O (EcoToxN1) UniProt ID: A0A8M0FGQ7. The sequence of EcoToxN5 is listed in the supplemental information (Supplementary Table S1) Sequence ID: WP_112017384.1. The sequences of the RNA oligos and plasmids for IVTs are listed in the material and method section and supplemental information (Supplementary Table S2). Requests for ToxN prototypes can be sent to ArcticZymes Technologies ASA. Email: [email protected].
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We would like to thank Jeremy Gillespie for the critical comments. Graphical abstract was created with BioRender.com.
Author contributions: U.R. & B.K.S.: conceived and conceptualized the work., U.R.: Principal Investigator; planned, supervised and executed the experiments; analysis and interpretation of the results; purified the proteins; performed and analyzed the experiments for RNA digestion with EcoToxN1, EcoToxN5 and BthToxN1; wrote the manuscript., SEG: performed EcoToxN1 RNA digestion experiments and kinetics; IVT with cap analogues. E.W.M.: purified and characterized EcoToxN5 and BthToxN1, SS: EcoToxN1 RNA digestion experiments; mutagenesis of IVT plasmids; performed the IVT and IVTs with cap analogues and the modified uridine nucleotides. Capping with VCE. M.S.: provided guidance and contributed to writing and editing., F.C. & F.J.P.: Biophysical characterization of capping efficiency with urea-PAGE. A.M. & Y.P.: Enzyme kinetics assays with quenching substrates, contributed to writing and editing. F.S.: mutagenesis of IVT plasmids; IVT with cap analogues, enzyme kinetic assays with quenching substrates., O.L.: provided guidance, allocated resources, contributed to writing and editing. B.K.S.: planned the experiments; analysis and interpretation of results; administration of the work, wrote the manuscript. All authors have read the manuscript and agreed with its content.
Funding
This work was supported by the Research Council of Norway, Innovation Project for the Industrial Sector 2023 [346796]. Funding for open access charge: the Research Council of Norway [346796].
Conflict of interest statement. The authors declare the following competing interests: U.R. and B.K.S. are the inventors on the patent application related to RNA sequence-specific restriction enzymes; U.R., S.E.G., E.W.M., S.S., F.S., A.M., Y.P., O.L. and B.K.S. are employees of ArcticZymes Technologies ASA; ArcticZymes is a manufacturer and vendor of enzymes used in this study. This affiliation does not affect the authors’ impartiality. M.S., F.C. and F.J.P. have no conflicts of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments