





Abstract
Formamidopyrimidine (Fapy•dG) is a major lesion arising from oxidation of dG that is produced from a common chemical precursor of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-OxodGuo). In human cells, replication of single-stranded shuttle vectors containing Fapy•dG is more mutagenic than 8-OxodGuo. Here, we present the first data regarding promoter dependent RNA polymerase II bypass of Fapy•dG. 8-OxodGuo bypass was examined side-by-side. Experiments were carried out using double-stranded shuttle vectors in HeLa cell nuclear lysates and in HEK 293T cells. The lesions do not significantly block transcriptional bypass efficiency. Less than 2% adenosine incorporation occurred in cells when the lesions were base paired with dC. Inhibiting base excision repair in HEK 293T cells significantly increased adenosine incorporation, particularly from Fapy•dG:dC bypass which yielded ∼25% adenosine incorporation. No effect was detected upon transcriptional bypass of either lesion in nucleotide excision repair deficient cells. Transcriptional mutagenesis was significantly higher when shuttle vectors containing dA opposite one of the lesions were employed. For Fapy•dG:dA bypass, adenosine incorporation was greater than 85%; whereas 8-OxodGuo:dA yielded >20% point mutations. The combination of more frequent replication mistakes and greater error-prone Pol II bypass suggest that Fapy•dG is more mutagenic than 8-OxodGuo.
Introduction
DNA modifications (lesions) produced by oxidizing or alkylating agents can compromise information transfer by inhibiting polymerase activity and/or increasing the frequency at which nucleotides are misincorporated (1,2). Replication errors can contribute to disease development, including cancer (3). The effects of a considerable number of DNA lesions on replication have been examined (4). When RNA polymerase II (Pol II) encounters a DNA lesion inhibition or misinformation transfer can result in truncated or mutated protein. The latter is referred to as transcriptional mutagenesis (5), which contributes to tumorigenesis in non-proliferating and proliferating cells (6–9). In addition to cancer, transcriptional mutagenesis is associated with a variety of diseases, including Alzheimer's and Parkinson's diseases (5,7,10–17). Compared to replication, the effects of individual DNA lesions on transcription are much less well characterized. Herein, we describe the effects of the two major lesions formed from oxidation of the most readily oxidized native nucleotide, 2′-deoxyguanosine (dG) on transcription by Pol II. Experiments on N6-(2-deoxy-α,β-d-erythro-pentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy•dG), and for comparison 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-OxodGuo) (18) were carried out in HeLa nuclear lysate and HEK 293T cells. Analyses were carried out using the competitive transcription and adduct bypass (CTAB) assay developed by Wang and a modified version of Essigmann's restriction endonuclease and post-labeling (REAP) method (19,20). Transcriptional bypass of 8-OxodGuo has been reported (5,13,21–25). The experiments reported here enable a side-by-side comparison of these two related lesions under identical conditions.
8-OxodGuo and Fapy•dG emanate from a common intermediate (1, Scheme 1) (26–31). Formation of the former is favored under O2-rich conditions. However, Fapy•dG is produced in equal or greater amounts under O2-deficient conditions. Fapy•dG has been determined to be more mutagenic than 8-OxodGuo in human cells (32–38). The unusual ability of Fapy•dG to exist as a mixture of configurational isomers that even equilibrate within crystalline DNA contributes to the promutagenicity of the lesion (32,39). Cleavage of the 5-membered ring component of the purine enables Fapy•dG to adopt multiple conformational isomers. In contrast, the promutagenicity of 8-OxodGuo is rationalized by population of the syn-conformation (Scheme 1) and utilization of its Hoogsteen face to direct misincorporation of dA and subsequent primer extension (40,41).
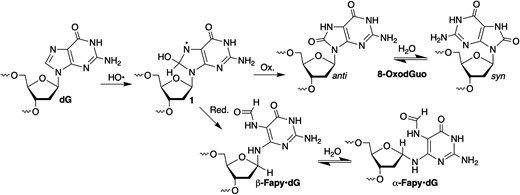
Competitive formation of Fapy•dG and 8-OxodGuo.
Similar to DNA polymerases, adenosine incorporation opposite 8-OxodGuo is the most frequent misinsertion event catalyzed by Pol II (22,24,42). However, incorporation of U and to a lesser extent G, has also been observed (24). 8-OxodGuo-induced transcriptional mutagenesis by Pol II has also been characterized in mammalian cells when the lesion is in the template strand or present as the corresponding nucleoside triphosphate (13,43,44). The effects of Fapy•dG on Pol II activity are limited to primer dependent kinetic analyses of transcriptional bypass within a miniscaffold that revealed the contribution of the α- and β-configurational isomers (45). Although Pol II inserts C opposite either Fapy•dG anomer more rapidly than A, G or U, all 4 nucleotides are incorporated, and the primer is extended past the lesion in the template. The accessory protein, TFIIS helps repair promutagenic insertions. In this report, we provide the first description of the effects of Fapy•dG on Pol II transcription in HeLa cell lysates and HEK 293T cells.
Materials and methods
General materials and methods
[γ-32P]-ATP was obtained from Perkin Elmer Health Sciences Inc. (Shelton, CT). Oligonucleotides were synthesized using an ABI 394 oligonucleotide synthesizer at Johns Hopkins University (JHU) or purchased from Integrated DNA Technologies (Coralville, IA). Oligonucleotides containing Fapy·dG were synthesized following previously established protocols (46,49), purified via 20% denaturing polyacrylamide gel electrophoresis, and characterized using ESI or MALDI-TOF MS. The 5′-cyanoethyl phosphoramidites for thymidine (T), N,N-dimethylformamidine-2′-deoxyguanosine (dGdmf), and other requisite reagents for oligonucleotide synthesis were obtained from GLEN Research (Sterling, VA). N-acetyl 2′-deoxycytidine 5′-cyanoethyl phosphoramidite (dCAc) and Universal UnyLinker™ Support were acquired from Chemgenes (Wilmington, MA). The 5′-cyanoethyl phosphoramidite of N-Pac-2′-deoxyadenosine was synthesized as previously described (46). Disodium 2-carbamoyl-2-cyanoethylene-1,1-dithiolate trihydrate for methyl deprotection was prepared following established methods (47). The pTGFP-T7-Hha10 shuttle vector, XPC and CSB double knockout HEK293T cells were gifts from Dr. Yinsheng Wang. All other materials were purchased from New England Biolabs, unless otherwise specified.
Preparation of lesion-containing vectors for CTAB assay
The preparation of a lesion-containing vector was carried out based on an established method (19). pTGFP-T7-Hha10 plasmid was digested by incubating with Nt.BstNBI (10 U/μl, 22.5 μl) in 1 × NEB buffer 3.1 (total volume 500 μl) at 55°C for 2 h. The final concentration of the plasmid was 600 ng/μl. To completely nick the plasmid DNA, a second portion of Nt.BstNBI (10 U/μl, 22.5 μl) was added to the reaction mixture and incubated at 55°C for 4 h. To analyze whether the restriction digestion was complete, a portion of the mixture (2 μl) was electrophoresed in a 1% (wt/vol) agarose gel (80 V for 90 min) in the presence of ethidium bromide (1 μg/ml). The restriction digest mixture was extracted with phenol:chloroform:isoamyl alcohol solution (25:24:1, 500 μl), while gently shaking by hand for ∼5 min. The mixture was centrifuged at 15 000 g for 10 min at 4°C. The aqueous layer was precipitated with ethanol (1.25 ml) and sodium acetate (3 M, 50 μl) at − 20°C for 2 h, and then centrifuged at 15 000 g for 60 min at 4°C. The DNA pellet was gently washed with 70% (vol/vol) ethanol (500 μl) and then centrifuged at 15000 g for 5 min at 4°C. The supernatant was removed, the DNA pellet was dried, and then resuspended in sterile ddH2O (100 μl). To remove the excised fragment, a 25-mer complementary oligonucleotide (10 μM, 5′-d(CAT CGA CTC CCG AAT AGC CCG CCA T)) in 1 × NEB buffer 3.1 (total volume 500 μl) was added. The reaction mixture was heated at 85°C for 5 min, and then slowly cooled to room temperature. The gapped plasmid was purified by removing the newly formed 25-mer duplex with an Amicon Ultra spin filter (0.5 ml, 100 K MWCO) using four changes of TE buffer (10 mM Tris·HCl pH 8.0, 1 mM EDTA, 400 μl). This process was repeated 4 times. The resulting gapped plasmid was stored at -80°C until use. The 25-mer synthetic oligonucleotide (5′-ATG GCG XGC TAT TCG GGA GTC GAT G-3′, X = Fapy·dG, 8-Oxo·dG or dG) was phosphorylated by incubating the oligonucleotide (10 μM) with T4 PNK (0.25 U/μl), ATP (1 mM) in 1 × T4 PNK buffer (total volume 20 μl) at 37°C for 2 h. The phosphorylated oligonucleotide solution (150 pmol) was mixed with gapped plasmid (15 pmol) in 1 × T4 ligation buffer (total volume 120 μl), heated to 90°C for 5 min, and slowly cooled to room temperature. The resulting annealed vector was incubated with T4 DNA ligase (8000 U) in 1 × T4 ligation buffer (total volume 200 μl) in a thermal cycler for 20 h, alternating between 10°C for 30 s and 30°C for 30 s. The mixture was electrophoresed in a 1% (wt/vol) agarose gel in the presence of ethidium bromide (1 μg/ml) in 1 × TAE (40 mM Tris–acetate pH 8.6, 1 mM EDTA) buffer at 80 V for 90 min. The band corresponding to the supercoiled plasmid DNA was visualized using a long-wavelength (365-nm) UV light source and excised with a scalpel. The supercoiled plasmid DNA was eluted from the gel using a QIAquick gel extraction kit according to the manufacturer's instructions. The recovered plasmid DNA was stored at − 80°C until use. All vectors were submitted for sequencing (Supplementary Table S1).
Preparation of HeLa nuclear extract
HeLa cells were grown to 80–90% confluency in a 150 mm cell culture dish with DMEM. The culture medium was removed, the cells were washed with PBS (20 ml), scraped into fresh PBS (10 ml), and transferred to conical tubes. The cells were centrifuged for 10 min at 1850 g, and the supernatant was discarded. The cells were resuspended in 5 packed cell volumes (pcv) of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 0.5 M DTT) and centrifuged for 5 min at 1850 g. The subsequent steps were performed at 4°C. After discarding the supernatant, the cells were resuspended in 3 pcv of hypotonic buffer and incubated on ice for 20 min. The cells were homogenized with a Dounce homogenizer (tight pestle) until 75% of the cells were lysed (20–30 strokes). Cell lysis was monitored by trypan blue staining. Nuclei were pelleted by centrifugation at 3300 g for 15 min, and the supernatant containing cytoplasmic extract was discarded. The nuclei were resuspended in 0.5 packed nuclei volume (pnv) of low-salt buffer (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 M DTT) and stirred with a magnetic stir bar. An equal volume of high salt buffer (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.84 M KCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 M DTT) was added dropwise to the suspension, which was then stirred for 40 min and centrifuged for 30 min at 25000 g. The resulting supernatant was dialyzed against at least 50 volumes of dialysis buffer (20 mM HEPES pH 7.9, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 M DTT). The nuclear extract was centrifuged again at 25 000 g for 20 min, aliquoted, frozen in liquid nitrogen, and stored at –80°C until used. The concentration of protein (measured using a Bradford assay) in the prepared HeLa nuclear lysate was 3.5 μg/μl.
In vitro transcription assay
The lesion-bearing or lesion-free control plasmids were premixed with the competitor vector at a molar ratio of 3:1 (lesion : competitor). The plasmid mixture (500 ng) was incubated with NotI (5 U), in BSA (100 μg/ml) and 1 × NEB buffer 3.1 (total volume 20 μl) at 37°C for 1 h. The enzyme was deactivated by heating the reaction solution at 70°C for 20 min. The resulting linearized vector was purified using the E.Z.N.A. Cycle Pure kit (Omega Bio-Tek, Norcross, GA), and then eluted in elution buffer (30 μl) according to the manufacturer's instructions. The mixture of linearized vectors (50 ng) was incubated with nuclear extract (3.5 μg/μl, 3 μl), rNTP (2.5 mM each, 4 μl), RNase inhibitor (40 U/μl, 0.25 μl) in 1 × dialysis buffer with 3 mM MgCl2 (total volume 20 μl) at 30°C for 2 h. The reaction was terminated by adding EDTA (0.2 M, 2 μl).
Cellular transcription assay
The cellular transcription was conducted in accordance with a previously described protocol (48) HEK293T wild type, XPC-, or CSB-knockout cells were seeded in 24-well plates with DMEM medium (500 μl) and incubated at 37°C with 5% CO2 until they reached 40–60% confluency. The lesion-bearing or lesion-free plasmid was premixed with the competitor vector at a molar ratio of 3:1 (lesion/competitor). The premixed vectors (50 ng total) were then combined with a carrier vector (450 ng, self-ligated pGEM-T) in a total volume of 2 μl. Subsequently, the mixture (2 μl) was transfected into cells using LipofectamineTM 3000 reagent (0.75 μl) and P3000TM Reagent (1 μl) following the manufacturer's instructions. To investigate the effect of hOGG1, SU0268 (10 μM) was incubated with cells for 2 h before transfection. Cells were harvested 24 h after transfection.
Purification and RT-PCR amplification of mRNA
mRNA resulting from in vitro or cellular transcription was purified using E.Z.N.A. Total RNA kit I (Omega Bio-Tek, Norcross, GA) following the manufacturer's protocol and eluted with 50 μl nuclease free water. The purified RNA (50 μl) was combined with 10 × DNase I buffer (5 μl) and DNase I (2 U/μl, 1 μl). The reaction was incubated at 37°C for 30 min. The mRNA was purified following the manufacturer's guidelines for DNA-free™ DNA Removal Kit (Thermo Fisher Scientific, Waltham, MA). The residual mRNA (50 μl) was subjected to a second treatment of DNase I (2 U/μl, 1 μl) and 10 × DNase I buffer (7.5 μl) and purified. The DNA-free mRNA (6.6 μl) was combined with the primer (10 μM, 0.4 μl; 5′-d(TCGGTGTTGCTGTGAT)) in a sterile, RNase-free microcentrifuge tube. The tube was heated at 70°C for 5 min and rapidly chilled on ice. The annealed mRNA (7 μl) was then mixed with M-MLV reverse transcriptase (200 U/μl, 0.4 μl), dNTP (10 mM each, 0.5 μl), and RNase inhibitor (40 U/μl, 0.1 μl) in the presence of the commercial 1 × M-MLV reaction buffer (total volume 10 μl). The reaction was incubated at 42°C for 1.5 h and then at 75°C for 15 min. Control experiments were conducted, either without mRNA or M-MLV reverse transcriptase, to ensure no DNA contamination was present. For PCR, the reverse transcription reaction mixture (1 μl) was combined with primers (10 μM × 1 μl each, 5′-d(CTAGCGGATGCATCGACTC) and 5′-d(GGCCGCTCTCGTCGCTCTC)), dNTP (10 mM, 1 μl), and Phusion® High-Fidelity DNA Polymerase (2 U/μl, 0.3 μl) in 1 × Phusion HF buffer (total volume 50 μl). The PCR cycle consisted of the following steps: Step 1: 98°C for 30 s; Step 2 (repeated for 35 cycles): 98°C for 10 s, 60°C for 30 s, 72°C for 15 s; Step 3: 72°C for 5 min. Finally, an aliquot of the PCR reaction (3 μl) was analyzed using a 2% agarose gel and the rest of the PCR reaction was purified using the E.Z.N.A. Cycle Pure kit following the manufacturer's instructions. The concentration of PCR product was determined by measuring the absorbance at 260 nm using a Nanodrop 2000c.
Analysis of RT-PCR product
The purified PCR product (50 ng) was mixed with NcoI (10 U/μl, 0.5 μl) and SAP (1 U/μl, 0.5 μl) in 1 × NEB buffer 3.1 (total volume 10 μl). The reaction was incubated at 37°C for 1 h and subsequently heated at 70°C for 20 min. Following this, the entire reaction mixture was mixed with T4 PNK (10 U/μl, 0.5 μl), ATP (100 μM, 0.1 μl) and [γ-32P]ATP (10 μCi/μl, 0.5 μl) in 1 × NEB buffer 3.1 (total volume 15 μl). The kinase reaction was incubated at 37°C for 1 h and then heated at 65°C for 20 min. Subsequently, the resulting solution was combined with SfaNI (2 U/μl, 1 μl) in 1 × NEB buffer 3.1 (total volume 20 μl), and incubated at 37°C for 1.5 h. The reaction was halted by adding 2 × gel loading buffer (20 μl, 45% formamide, 0.1% bromophenol blue, 0.1% xylene cyanol, and 5 mM EDTA). An aliquot (7 μl) of the sample was separated by 30% native PAGE (0.4 mm x 40 cm × 32 cm) in 0.5 × TBE buffer at 40 W for 5 h. Following electrophoresis, the gel was wrapped and exposed to a storage phosphor screen for >12 h. Finally, the phosphor screen was scanned using a phosphorimager. The 16-mer and 12/13-mer bands were quantified to calculate bypass efficiencies and mutation frequencies.
REAP assay
The RT-PCR product (50 ng) was digested with Cac8I (0.5 U/μl) and SAP (0.05 U/μl) in 1 × NEB buffer 3.1 (total volume 10 μl) at 37°C for 3 h. The enzymes were deactivated by heating the reaction mixture at 70°C for 20 min. The reaction mixture was then supplemented with ATP (1 μM), [γ-32P]-ATP (0.67 μCi/μl), T4 PNK (0.33 U/μl) in 1 × NEB Buffer 3.1 (total volume 15 μl) and incubated at 37°C for 1 h. T4 PNK was deactivated by heating at 65°C for 20 min. Subsequently, NsiI (0.1 U/μl) was added to the mixture in 1 × NEB buffer (total volume 20 μl) and incubated at 37°C for 2 h. The reaction was quenched by adding 20 μl of formamide loading buffer. The mixture was separated by 20% denaturing PAGE (0.4 mm × 40 cm × 32 cm) in 1 × TBE and run at 50 W for 4 h. The target region for the 21 nt fragment was excised, crushed, and eluted with a solution (200 μl) containing NaCl (0.2 M) and EDTA (1 mM) at 37°C overnight with shaking. After removing the gel particles through filtration, the filtrate was desalted twice using a 1 ml Sephadex G25 (pre-equilibrated with ddH2O, 3 × 200 μl) spin column, and then dried under vacuum. The residue was resuspended in nuclease P1 (20 U/μl) solution in commercial 1 × nuclease P1 reaction buffer (total volume 5 μl) and incubated at 37°C overnight. Before use, the PEI cellulose TLC plate was soaked in water for 2 min and dried overnight. An aliquot (0.5 μl, ∼3000 cpm) of the nuclease P1 reaction was spotted on the TLC plate, which was then immersed in a tank containing saturated (NH4)2HPO4 (200 ml, pH 5.8). Saturated (NH4)2HPO4 was prepared by mixing an excess of the salt (250 g) with 200 ml ddH2O and shaking (250 rpm) overnight at room temperature, followed by adjusting the pH of the supernatant to ∼5.8 with H3PO4. After the solvent front reached the top (18 h), the plates were air-dried and exposed to a storage phosphor screen for 16 h.
Results and discussion
Constructs for transcription experiments using HeLa cell nuclear lysates and in HEK 293T cells were prepared using the pTGFP-T7-Hha10 vector as described by Wang (Supplementary Figure S1) (19). The oligonucleotide containing Fapy•dG (ESI-MS, Supplementary Figure S16) was synthesized, as previously described (49). All other oligonucleotides, including that containing 8-OxodGuo (MALDI-TOF MS, Supplementary Figure S17) were synthesized using standard methods and commercially available reagents. CTAB experiments were carried out in HeLa cell nuclear lysates or HEK 293T cells. The experiments provided the percent bypass, as well as the percentages of point mutations and deletion products (19). All data are the average ± standard deviation of at least three replicates.
Combining the CTAB and REAP assays for identifying the nucleotide incorporated opposite a DNA lesion
A strength of the CTAB assay is the ability to determine transcription bypass efficiency on a template containing a DNA lesion and quantitatively identifying the nucleotide incorporated opposite the lesion via a single native PAGE experiment (19,50). Briefly, the outcome of polymerase bypass is quantitatively determined using restriction enzymes to produce a short 5′-32P-oligonucleotide (13mer) containing the position at which the nucleotide of interest is present. Coadministration of a control vector comprised of native nucleotides that produces a final product 3 nts longer enables one to determine the bypass efficiency. The mutation frequency is determined by quantifying the four possible progeny oligonucleotides that are separated from one another by native PAGE. Alternatively, the 13mers can be analyzed by mass spectrometry.
Access to a suitable mass spectrometer can be a limitation in some environments, and separating all four possible 13mer oligonucleotides from one another can be challenging. Consequently, we adapted the CTAB strategy to quantify the nucleotides incorporated opposite the DNA lesions using elements of the REAP assay (Figure 1, Supplementary Figure S2) (20). REAP utilizes thin layer chromatography (TLC) to quantitatively determine the mixture of nucleotides. A judicious choice of oligonucleotide sequence and restriction endonuclease enables one to selectively 32P-label the 5′-hydroxyl group of the nucleoside of interest in an oligonucleotide that is isolated via denaturing PAGE. Following nuclease digestion, the mononucleotides are separated using polyethylenimine cellulose TLC plates and quantified via a phosphorimager.
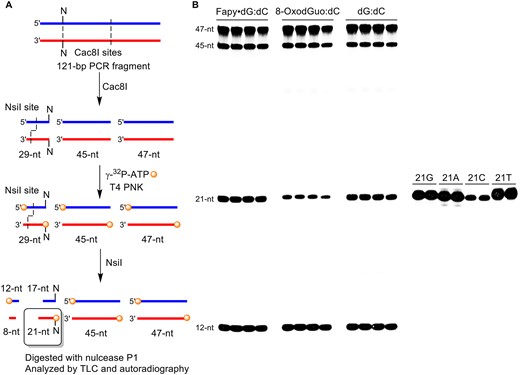
Cleavage of 121-bp RT-PCR product for REAP assay. (A) Four radiolabeled fragments are formed during the digestion by Cac8I and NsiI: 12-nt, 21-nt (target), 45-nt and 47 nt. (B) Representative gel showing the efficient restriction digestion of 121-bp fragment.
Fortuitously, the position within the pTGFP-T7-Hha10 vector used for the promoter dependent CTAB assay where the nucleotide of interest is positioned is a cleavage site for Cac8I. Cac8I recognizes the 5′-GCNNGC (N = A, C, G or T) sequence and cleaves between the two unspecified nucleotides (N). Fapy•dG or 8-OxodGuo are positioned (5′-GCG XGC; X = Fapy•dG, 8-OxodGuo) to yield cleavage at the 5′-position of the nucleotide(s) present in the progeny. Having chosen Cac8I to enable radiolabeling the nucleotide(s) of interest, the reverse primer was chosen to optimize the RT-PCR product length (Supplementary Figure S3). The PCR amplification product obtained by positioning the reverse primer as described (526 bp) in the original CTAB description encompasses a significant portion of the GFP coding region within the pTGFP-T7-Hha10 vector and contains 7 Cac8I sites (19). The reverse primer was chosen to reduce the complexity of the digestion product mixture (2 Cac8I sites, Figure 1) but produce a PCR product long enough (121 bp) to ensure efficient amplification and facile purification (Supplementary Figure S4). To produce a unique 5′-32P-labeled oligonucleotide (21mer) containing the nucleotide of interest at its 5′-terminus, the mixture obtained from Cac8I digestion was 5′-32P-labeled and then treated with NsiI (Figure 1A). The viability of this strategy was demonstrated in HeLa cell nuclear lysates on shuttle vectors containing Fapy•dG:dC, 8-OxodGuo:dC or dG:dC base pairs (Figure 1B). In each instance, the four anticipated oligonucleotide products were observed. In addition, 21mer standards exhibited the same migrations regardless of which native nucleotide was present at the position of interest, indicating that the method is not biased towards a particular result. Finally, the radiolabeled 21mer DNA was isolated via the crush and soak method. Following digestion with nuclease P1, the 5′-32P-dNMPs were analyzed by TLC, side-by-side with independently prepared standards (20).
Promoter dependent transcription in HeLa cell nuclear lysates
A template containing Fapy•dG:dC does not block bypass by Pol II (Table 1, Supplementary Figure S5). In contrast, Fapy•dG:dA modestly blocks Pol II. Transcription is approximately one-half as efficient compared to a dG:dC containing shuttle vector, and is almost 3-times less efficient than when the lesion is opposite dC (Table 1). The strong dependence of bypass efficiency on opposing nucleotide in the shuttle vector could reflect the difference in populations of Fapy•dG configurational isomers in the duplexes. Structural data indicate that Fapy•dG predominantly adopts the β-configuration opposite dC; whereas the lesion exists as a dynamic mixture of anomers when opposed by dA, even in the crystalline state (32). Primer dependent Pol II kinetic data describing extension past a Fapy•dG:N (N = A, G or U) obtained using a miniscaffold also indicate that both anomers of the lesion are present (45). The relative bypass efficiencies can be attributed to this structural difference, provided Pol II is more tolerant of β-Fapy•dG in the template strand than α-Fapy•dG. This possibility is supported by kinetic measurements carried out on a miniscaffold in which a mixture of α- and β-Fapy•dG is unpaired in a template. Pol II incorporates C opposite one of the anomers, presumably the β-isomer, more than 40-times faster than opposite the other isomer (45).
Promoter dependent Pol II transcription in HeLa cell nuclear lysatea
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 140.2 ± 9.4 | 5.6 ± 1.1 | 8.0 ± 1.7 |
| Fapy•dG:dA | 46.4 ± 1.6 | 21.8 ± 0.4 | 17.1 ± 0.6 |
| 8-OxodGuo:dC | 66.1 ± 0.1 | 4.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 65.3 ± 3.8 | 11.5 ± 0.3 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 140.2 ± 9.4 | 5.6 ± 1.1 | 8.0 ± 1.7 |
| Fapy•dG:dA | 46.4 ± 1.6 | 21.8 ± 0.4 | 17.1 ± 0.6 |
| 8-OxodGuo:dC | 66.1 ± 0.1 | 4.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 65.3 ± 3.8 | 11.5 ± 0.3 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected
Promoter dependent Pol II transcription in HeLa cell nuclear lysatea
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 140.2 ± 9.4 | 5.6 ± 1.1 | 8.0 ± 1.7 |
| Fapy•dG:dA | 46.4 ± 1.6 | 21.8 ± 0.4 | 17.1 ± 0.6 |
| 8-OxodGuo:dC | 66.1 ± 0.1 | 4.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 65.3 ± 3.8 | 11.5 ± 0.3 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 140.2 ± 9.4 | 5.6 ± 1.1 | 8.0 ± 1.7 |
| Fapy•dG:dA | 46.4 ± 1.6 | 21.8 ± 0.4 | 17.1 ± 0.6 |
| 8-OxodGuo:dC | 66.1 ± 0.1 | 4.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 65.3 ± 3.8 | 11.5 ± 0.3 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected
Primer dependent experiments using a miniscaffold indicated that 8-OxodGuo does not block Pol II, although the effect of an opposing nucleotide was not examined (24). Promoter dependent assays revealed that 8-OxodGuo modestly blocked rat Pol II (23). 8-OxodGuo was a stronger block to human Pol II, but the effect was overcome in the presence of transcription elongation factor IIS (21,22). In the quantitative, promoter dependent assay employed here, 8-OxodGuo modestly blocks transcription (Table 1, Supplementary Figure S4). The effect is independent of whether the lesion is opposite dC or dA, even though the lesion may exist in the syn-conformation when paired with the latter (40,41). The lack of an effect of mispairing dA with 8-OxodGuo on Pol II bypass is consistent with some DNA polymerases (51,52).
Point mutation levels obtained upon bypass of both lesions were dependent upon the opposing nucleotide (Table 1, Supplementary Figure S6). When paired with dC, ∼5% point mutations were observed using the CTAB assay when either Fapy•dG or 8-OxodGuo was present in the template. The levels of mutations increased significantly, almost 4-fold in the case of Fapy•dG, when either lesion was paired with dA in the DNA. The levels of point mutations observed using the REAP assay were similar to these values, with modest increases observed when either lesion is opposite dC (Supplementary Figure S6). The mutation frequencies measured using REAP were within experimental error of those determined using the CTAB assay (Table 1) when Fapy•dG or 8-OxodGuo was paired with dA. Adenosine incorporation was the dominant mutation detected for transcriptional bypass of Fapy•dG or 8-OxodGuo. However, low levels of uridine (≤0.6%) and guanine (≤0.3%) incorporation were also detected (Supplementary Figure S6). Single nucleotide deletions were produced at comparable levels as point mutations upon Fapy•dG bypass but were not detected following 8-OxodGuo transcription (Table 1).
Promoter dependent transcription in HEK 293T cells
Fapy•dG and 8-OxodGuo transcription in cells yielded some similar observations as HeLa lysate experiments but also differences (Figure 2, Table 2, Supplementary Figure S7). One similarity was that single nucleotide deletions were only detected when Fapy•dG was bypassed. However, the levels of deletions when dC or dA were paired with the lesion were within error of one another and were significantly lower than those observed in nuclear lysate (Table 1). It is not possible to determine from these experiments why Fapy•dG bypass in HEK 293T cells results in fewer single nucleotide deletions. However, it is possible that one or more (properly folded) proteins are present in different quantities within the (HeLa) nuclear lysates and cells.
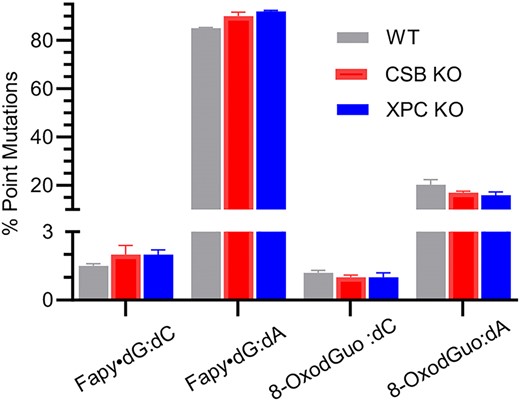
Percent point mutations from promoter dependent transcription of Fapy•dG and 8-OxodGuo in wild type and CSB or XPC knockout HEK 293T cells determined using the CTAB assay. (For statistical analysis see Supplementary Figure S15.)
Promoter dependent Pol II transcription in HEK 293T cellsa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 96.6 ± 5.3 | 1.5 ± 0.1 | 1.3 ± 0.2 |
| Fapy•dG:dA | 90.0 ± 2.4 | 85.1 ± 0.3 | 1.6 ± 0.3 |
| 8-OxodGuo:dC | 89.4 ± 5.5 | 1.2 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 87.4 ± 4.4 | 20.3 ± 2.1 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 96.6 ± 5.3 | 1.5 ± 0.1 | 1.3 ± 0.2 |
| Fapy•dG:dA | 90.0 ± 2.4 | 85.1 ± 0.3 | 1.6 ± 0.3 |
| 8-OxodGuo:dC | 89.4 ± 5.5 | 1.2 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 87.4 ± 4.4 | 20.3 ± 2.1 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
Promoter dependent Pol II transcription in HEK 293T cellsa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 96.6 ± 5.3 | 1.5 ± 0.1 | 1.3 ± 0.2 |
| Fapy•dG:dA | 90.0 ± 2.4 | 85.1 ± 0.3 | 1.6 ± 0.3 |
| 8-OxodGuo:dC | 89.4 ± 5.5 | 1.2 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 87.4 ± 4.4 | 20.3 ± 2.1 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 96.6 ± 5.3 | 1.5 ± 0.1 | 1.3 ± 0.2 |
| Fapy•dG:dA | 90.0 ± 2.4 | 85.1 ± 0.3 | 1.6 ± 0.3 |
| 8-OxodGuo:dC | 89.4 ± 5.5 | 1.2 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 87.4 ± 4.4 | 20.3 ± 2.1 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
Bypass of any of the four lesion-containing shuttle vectors was at most slightly blocked in HEK 293T cells. While the point mutation levels resulting from transcriptional bypass of Fapy•dG:dC and 8-OxodGuo:dC duplexes were still within error of one another, the misincorporation levels declined from ∼5% in HeLa cell nuclear lysate to slightly more than 1% in HEK 293T cells (Figure 2, Table 2). The point mutation levels measured in the REAP assay were slightly greater (<2%) and still within experimental error of one another (Supplementary Figure S8). Transcription was more error-prone when either lesion was paired with dA and the increase in overall mutation frequencies were greater in cells than observed when reactions were carried out in nuclear lysate. This was especially true for Fapy•dG:dA, for which point mutations accounted for 85% of bypass events in the CTAB experiment. The point mutation levels measured in the REAP assay were slightly higher and only mutations resulting from adenosine incorporation were detected (Table 3). It is not possible to parse why point mutations increased more when Fapy•dG or 8-OxodGuo was paired with dA in HEK 293T cells than in nuclear lysates. However, the either environment a mispair within DNA (e.g. Fapy•dG:dA) gives rise to greater error-prone bypass than when the nonmutagenic base pair (e.g. Fapy•dG:dC) is present. In addition, transcriptional bypass of Fapy•dG opposite dA is more mutagenic than 8-OxodGuo opposite this nucleotide.
The effect of hOGG1 inhibition on adenosine incorporation by Pol II in HEK 293T cells determined using the REAP assaya
| % Adenosine Incorporated | ||
|---|---|---|
| Duplex | – Inhibitor | + Inhibitor |
| Fapy•dG:dC | 1.9 ± 0.2 | 25.8 ± 1.4 |
| Fapy•dG:dA | 88.9 ± 0.8 | 91.0 ± 1.6 |
| 8-OxodGuo:dC | 1.7 ± 0.3 | 7.3 ± 1.2 |
| 8-OxodGuo:dA | 25.3 ± 1.8 | 26.0 ± 1.4 |
| % Adenosine Incorporated | ||
|---|---|---|
| Duplex | – Inhibitor | + Inhibitor |
| Fapy•dG:dC | 1.9 ± 0.2 | 25.8 ± 1.4 |
| Fapy•dG:dA | 88.9 ± 0.8 | 91.0 ± 1.6 |
| 8-OxodGuo:dC | 1.7 ± 0.3 | 7.3 ± 1.2 |
| 8-OxodGuo:dA | 25.3 ± 1.8 | 26.0 ± 1.4 |
aData are the ave. ± std. dev. of three replicates.
The effect of hOGG1 inhibition on adenosine incorporation by Pol II in HEK 293T cells determined using the REAP assaya
| % Adenosine Incorporated | ||
|---|---|---|
| Duplex | – Inhibitor | + Inhibitor |
| Fapy•dG:dC | 1.9 ± 0.2 | 25.8 ± 1.4 |
| Fapy•dG:dA | 88.9 ± 0.8 | 91.0 ± 1.6 |
| 8-OxodGuo:dC | 1.7 ± 0.3 | 7.3 ± 1.2 |
| 8-OxodGuo:dA | 25.3 ± 1.8 | 26.0 ± 1.4 |
| % Adenosine Incorporated | ||
|---|---|---|
| Duplex | – Inhibitor | + Inhibitor |
| Fapy•dG:dC | 1.9 ± 0.2 | 25.8 ± 1.4 |
| Fapy•dG:dA | 88.9 ± 0.8 | 91.0 ± 1.6 |
| 8-OxodGuo:dC | 1.7 ± 0.3 | 7.3 ± 1.2 |
| 8-OxodGuo:dA | 25.3 ± 1.8 | 26.0 ± 1.4 |
aData are the ave. ± std. dev. of three replicates.
The decreased point mutation frequency for Fapy•dG:dC and 8-OxodGuo:dC duplexes in cells compared to cell lysates could be attributed to hOGG1 initiated base excision repair (53). hOGG1 incises 8-OxodGuo almost 3000-times faster when the lesion is opposite dC than paired with dA. The selectivity for DNA containing Fapy•dG is smaller, but still almost 50-fold faster when dC is opposite the lesion compared to dA. The contribution of hOGG1 to Fapy•dG and 8-OxodGuo mutagenesis was examined by carrying out the experiments in the presence of SU0268, a potent inhibitor of the glycosylase (Tables 3, 4, Figure 3, Supplementary Figures S9, S10) (54). hOGG1 inhibition had no effect on transcriptional bypass efficiency of either lesion, whether it was paired with dC or dA (Tables 2, 4). The levels of single nucleotide deletions were also unaffected. However, inhibiting hOGG1 gave rise to higher levels of point mutations when either lesion was paired with dC (Figure 3, Tables 3, 4). The effects of inhibiting hOGG1 on 8-OxodGuo transcriptional bypass are qualitatively consistent with report by Doetsch who observed significant increases in error-prone transcriptional bypass of 8-OxodGuo in mouse embryonic fibroblasts (MEFs) in which OGG1 had been knocked out (13). In contrast, Boiteux reported that 8-OxodGuo in the transcribed strand was still rapidly repaired when OGG1 was knocked out in MEFs (55). Importantly, these authors proposed that transcription coupled repair protected the cell from 8-OxodGuo. These observations were consistent with another study on 8-OxodGuo transcription in MEFs in which OGG1 and Cockayne Syndrome Group B protein (CSB), a protein involved in transcription coupled-NER, were knocked out (56). Both proteins were shown to play a role in protecting against 8-OxodGuo transcriptional mutagenesis.
Promoter dependent Pol II transcription in HEK 293T cells in the presence of a hOGG1 inhibitora
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 103.6 ± 10.7 | 23.5 ± 1.8 | 1.2 ± 0.2 |
| Fapy•dG:dA | 94.2 ± 10.2 | 88.4 ± 1.3 | 0.8 ± 0.2 |
| 8-OxodGuo:dC | 86.7 ± 16.3 | 4.9 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 98.0 ± 13.1 | 22.2 ± 1.6 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 103.6 ± 10.7 | 23.5 ± 1.8 | 1.2 ± 0.2 |
| Fapy•dG:dA | 94.2 ± 10.2 | 88.4 ± 1.3 | 0.8 ± 0.2 |
| 8-OxodGuo:dC | 86.7 ± 16.3 | 4.9 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 98.0 ± 13.1 | 22.2 ± 1.6 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
Promoter dependent Pol II transcription in HEK 293T cells in the presence of a hOGG1 inhibitora
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 103.6 ± 10.7 | 23.5 ± 1.8 | 1.2 ± 0.2 |
| Fapy•dG:dA | 94.2 ± 10.2 | 88.4 ± 1.3 | 0.8 ± 0.2 |
| 8-OxodGuo:dC | 86.7 ± 16.3 | 4.9 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 98.0 ± 13.1 | 22.2 ± 1.6 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 103.6 ± 10.7 | 23.5 ± 1.8 | 1.2 ± 0.2 |
| Fapy•dG:dA | 94.2 ± 10.2 | 88.4 ± 1.3 | 0.8 ± 0.2 |
| 8-OxodGuo:dC | 86.7 ± 16.3 | 4.9 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 98.0 ± 13.1 | 22.2 ± 1.6 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
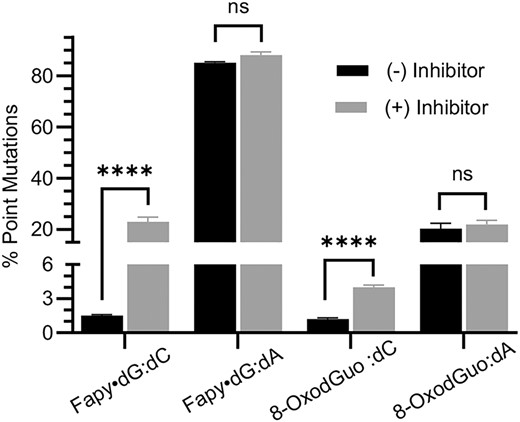
Effect of hOGG1 inhibition on percent point mutations from promoter dependent transcription of Fapy•dG and 8-OxodGuo in wild type HEK 293T cells determined using the CTAB assay. ns, no significant difference; ****P < 0.0001.
Point mutation levels were comparable in the CTAB (Figure 3, Tables 2, 4) and REAP (Table 3) assays. Adenosine incorporation was the only mutation detected using the REAP assay. The mutation frequency increase for Fapy•dG when hOGG1 was inhibited was significantly greater than that of 8-OxodGuo. Point mutations from Fapy•dG bypass increased 13- and 15-fold, depending upon which assay was used. In contrast, both assays indicated that inhibiting hOGG1 resulted in ∼4-fold increase in mutations. Inhibiting hOGG1 had no effect on point mutation frequency when either lesion was paired with dA. The effects of hOGG1 inhibition on mutation frequency as a function of opposing nucleotide is consistent with the expectation that the enzyme selectively incises Fapy•dG or 8-OxodGuo opposite dC versus dA. Kinetics experiments indicate that it is unlikely that hOGG1 incises Fapy•dG more efficiently than 8-OxodGuo (53). Therefore, we propose the larger increase in point mutations when Fapy•dG is present in the template is due to greater error-prone Pol II bypass of this lesion in this sequence.
The CTAB and REAP assays were also used in conjunction with CRISPR/Cas9 HEK 293T knock-out cells (50) to determine the contribution of nucleotide excision repair (NER) in transcriptional bypass of Fapy•dG and 8-OxodGuo (Figure 2, Tables 5, 6, Supplementary Figures S11–S15). Transcriptional bypass efficiency of 8-OxodGuo or Fapy•dG was unaffected in cells lacking CSB (Tables 2, 5). Point mutations were analyzed using the CTAB and REAP assays (Figure 2, Supplementary Figure S15). For some DNA lesion constructs statistical differences in point mutation levels were observed between wild type and CSB XPC knock out cells. However, the differences were very small and not necessarily consistent between the CTAB and REAP assays. Consequently, we believe that CSB plays at most a modest role in protecting against mutations resulting from Fapy•dG:dA bypass and none when Pol II encounters Fapy•dG:dC or 8-OxodGuo paired with dC or dA. The absence of an effect on 8-OxodGuo transcription is consistent with experiments reported by Doetsch who observed no increase in mutagenesis in cells in which CSB was knocked out (13). Like those reported by Doetsch, our experiments were inconsistent with reports by Boiteux and Sarasin who observed that CSB played a role in 8-OxodGuo repair (55,56). Comparable observations were made when transcription was examined in cells in which XPC, a protein involved in global genome NER, were carried out. (Figure 2, Supplementary Figure S15, Table 6) (57). Although individual differences were detected, overall deleting XPC had no effect on transcriptional bypass of either lesion It is notable that the experiments described in this study were carried out on lesions within a single flanking sequence and only the CMV promoter. It is possible that different results could be obtained using other promoters and/or different local sequences (56).
Promoter dependent Pol II transcription in HEK 293T cells lacking CSBa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 119.6 ± 4.3 | 2.3 ± 0.4 | 1.2 ± 0.2 |
| Fapy•dG:dA | 105.6 ± 0.9 | 90.3 ± 1.7 | 0.8 ± 0.1 |
| 8-OxodGuo:dC | 83.1 ± 0.4 | 1.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 79.6 ± 4.4 | 17.4 ± 0.6 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 119.6 ± 4.3 | 2.3 ± 0.4 | 1.2 ± 0.2 |
| Fapy•dG:dA | 105.6 ± 0.9 | 90.3 ± 1.7 | 0.8 ± 0.1 |
| 8-OxodGuo:dC | 83.1 ± 0.4 | 1.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 79.6 ± 4.4 | 17.4 ± 0.6 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected
Promoter dependent Pol II transcription in HEK 293T cells lacking CSBa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 119.6 ± 4.3 | 2.3 ± 0.4 | 1.2 ± 0.2 |
| Fapy•dG:dA | 105.6 ± 0.9 | 90.3 ± 1.7 | 0.8 ± 0.1 |
| 8-OxodGuo:dC | 83.1 ± 0.4 | 1.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 79.6 ± 4.4 | 17.4 ± 0.6 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 119.6 ± 4.3 | 2.3 ± 0.4 | 1.2 ± 0.2 |
| Fapy•dG:dA | 105.6 ± 0.9 | 90.3 ± 1.7 | 0.8 ± 0.1 |
| 8-OxodGuo:dC | 83.1 ± 0.4 | 1.9 ± 0.1 | n.d. |
| 8-OxodGuo:dA | 79.6 ± 4.4 | 17.4 ± 0.6 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected
Promoter dependent Pol II transcription in HEK 293T cells lacking XPCa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 127.6 ± 5.9 | 2.1 ± 0.2 | 1.1 ± 0.2 |
| Fapy•dG:dA | 115.1 ± 3.2 | 92.6 ± 0.4 | 0.7 ± 0.2 |
| 8-OxodGuo:dC | 99.5 ± 10.7 | 1.8 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 88.6 ± 12.0 | 16.8 ± 1.3 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 127.6 ± 5.9 | 2.1 ± 0.2 | 1.1 ± 0.2 |
| Fapy•dG:dA | 115.1 ± 3.2 | 92.6 ± 0.4 | 0.7 ± 0.2 |
| 8-OxodGuo:dC | 99.5 ± 10.7 | 1.8 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 88.6 ± 12.0 | 16.8 ± 1.3 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
Promoter dependent Pol II transcription in HEK 293T cells lacking XPCa
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 127.6 ± 5.9 | 2.1 ± 0.2 | 1.1 ± 0.2 |
| Fapy•dG:dA | 115.1 ± 3.2 | 92.6 ± 0.4 | 0.7 ± 0.2 |
| 8-OxodGuo:dC | 99.5 ± 10.7 | 1.8 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 88.6 ± 12.0 | 16.8 ± 1.3 | n.d. |
| Duplex | % Bypass | % Point mutation | % Deletion |
|---|---|---|---|
| Fapy•dG:dC | 127.6 ± 5.9 | 2.1 ± 0.2 | 1.1 ± 0.2 |
| Fapy•dG:dA | 115.1 ± 3.2 | 92.6 ± 0.4 | 0.7 ± 0.2 |
| 8-OxodGuo:dC | 99.5 ± 10.7 | 1.8 ± 0.2 | n.d. |
| 8-OxodGuo:dA | 88.6 ± 12.0 | 16.8 ± 1.3 | n.d. |
aData are the ave. ± std. dev. of 3 replicates. n.d. = not detected.
Summary
The effects of the two major products resulting from oxidation of dG on promoter dependent Pol II transcription were compared in nuclear lysate and in human cells. To our knowledge, this is the first report on the effects of Fapy•dG on transcription in nuclear lysate or cells. Neither Fapy•dG or 8-OxodGuo significantly blocks transcription in these environments. Low levels of adenosine are misincorporated in transcripts when either lesion is paired with dC, particularly in HEK 293T cells. Fapy•dG:dC bypass also yields small amounts of single nucleotide deletions; whereas none were observed from 8-OxodGuo. The higher frequency of deletions upon transcriptional bypass of Fapy•dG was also observed upon replicative bypass of these lesions in HEK 293T cells (34,36). hOGG1 inhibition increases the adenosine incorporation frequency when the lesions are bypassed by Pol II. The increase is significantly greater when Fapy•dG is present in the template strand, suggesting that error-prone bypass of this lesion is inherently greater than that of 8-OxodGuo. The relative mutation frequencies in the transcription experiments are consistent with single strand shuttle vector replication experiments in which Fapy•dG is more promutagenic than 8-OxodGuo upon in HEK 293T cells (34,36).
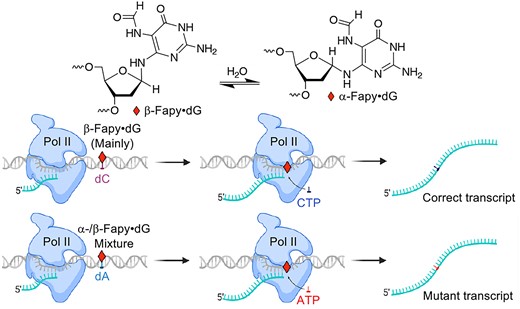
Pol II bypass of Fapy•dG or 8-OxodGuo is significantly more error-prone when either lesion is paired with dA (Scheme 2). Error-prone transcriptional bypass of Fapy•dG:dA is extremely high (>80%). Interaction of multiple stereoisomers (configurational and conformational) (32) of the lesions with Pol II and base excision repair efficiency could contribute to these outcomes. The general trend would be observed if hMYH repair is less efficient in the HEK 293T cells than that initiated by hOGG1. The activity of hMYH on Fapy•dG has not been reported. However, 8-OxodGuo:dA is repaired 4-fold more rapidly by MutY from E. coli than a comparable duplex containing Fapy•dG (58). If less efficient Fapy•dG:dA repair is carried forward in HEK 293T cells, it could contribute to the ∼4-fold greater point mutation frequency observed upon transcriptional bypass of this DNA than 8-OxodGuo:dA.
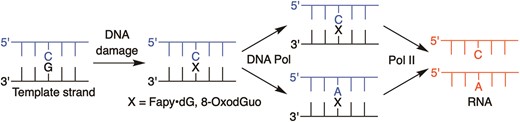
DNA and RNA polymerase bypass of Fapy•dG and 8-OxodGuo.
Error-prone Pol II bypass may also be inherently higher for Fapy•dG:dA substrates than 8-OxodGuo:dA. When Pol II encounters 8-OxodGuo opposite dA the lesion is more likely in the syn-conformation than when paired with dC. In this situation the Hoogsteen face of 8-OxodGuo will be presented to the enzyme, resulting in greater A incorporation (40). When Fapy•dG is paired with dA, the enzyme is likely to interact with roughly equal amounts of the α- and β-configurations of the lesion (32). Experiments in a miniscaffold indicate that Pol II incorporates A opposite one Fapy•dG anomer as rapidly as it does C (45). In contrast, Pol II greatly prefers to incorporate C opposite Fapy•dG when the lesion is paired with dC. Hence, which Fapy•dG stereoisomer the enzyme encounters could contribute significantly to determining the nucleotide incorporated opposite either lesion, giving rise to more error prone transcriptional bypass of Fapy•dG:dA base pairs than when dC is paired with the lesion. Studies on Fapy•dG and 8-OxodGuo replication in HEK 293T cells indicate that the former is more likely to yield products in which the lesion base pairs with dA (Scheme 2) (34,36). When combined with the data reported here, it is plausible that Fapy•dG formation will ultimately result in greater transcriptional mutagenesis than 8-OxodGuo in cells.
Data availability
The data underlying this article are available in the article, in its online supplementary material and from the corresponding author.
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We are grateful for support by NIH Grants GM-131736 and ES-027558 to MMG and CA-217809 to ETK. We thank Professor Yinsheng Wang for providing the pTGFP-T7-Hha10 vector, XPC and CSB double knockout HEK293T cells, and for helpful discussions. We are grateful to members of the Greenberg lab for assistance with cell culture experiments (Xuanhe Jiang, Dr Tingyu Wen) and oligonucleotide synthesis (Dr Haozhe Yang).
Funding
National Institutes of Health [GM-131736 and ES-027558 to MMG, CA-217809 to ETK]. Funding for open access charge: National Institutes of Health [GM-131736].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments