





Abstract
In vertebrates, the BRCA2 protein is essential for meiotic and somatic homologous recombination due to its interaction with the RAD51 and DMC1 recombinases through FxxA and FxPP motifs (here named A- and P-motifs, respectively). The A-motifs present in the eight BRC repeats of BRCA2 compete with the A-motif of RAD51, which is responsible for its self-oligomerization. BRCs thus disrupt RAD51 nucleoprotein filaments in vitro. The role of the P-motifs is less studied. We recently found that deletion of Brca2 exons 12–14 encoding one of them (the prototypical ‘PhePP’ motif), disrupts DMC1 but not RAD51 function in mouse meiosis. Here we provide a mechanistic explanation for this phenotype by solving the crystal structure of the complex between a BRCA2 fragment containing the PhePP motif and DMC1. Our structure reveals that, despite sharing a conserved phenylalanine, the A- and P-motifs bind to distinct sites on the ATPase domain of the recombinases. The P-motif interacts with a site that is accessible in DMC1 octamers and nucleoprotein filaments. Moreover, we show that this interaction also involves the adjacent protomer and thus increases the stability of the DMC1 nucleoprotein filaments. We extend our analysis to other P-motifs from RAD51AP1 and FIGNL1.
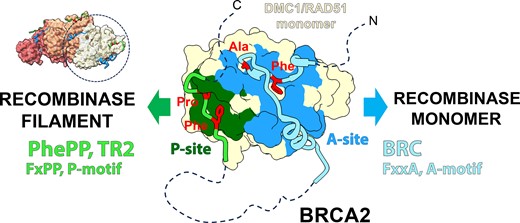
Introduction
BRCA2 is essential for homologous recombination (HR) in somatic cells (1,2) and during meiosis (3–9). This function is attributed to its interaction with the strand exchange proteins (‘recombinases’): the ubiquitous RAD51 (10,11) and the meiosis-specific DMC1 (12–16). BRCA2 facilitates the loading of RAD51 and DMC1 on single-strand DNA (ssDNA) produced by end resection during HR. Thus, it controls the formation of helical nucleoprotein filaments (17), which catalyze homology search and strand exchange—the key events in HR. BRCA2 acts as an HR mediator (18), helping to evict the competing ssDNA-binding proteins and to direct filament formation to the correct substrate. Less detail is known about BRCA2 in meiosis, e.g. whether BRCA2 coordinates both recombinases directly or affects DMC1 indirectly via its effect on RAD51 (13,19). We engineered a mouse with an internal deletion of Brca2 exons 12–14, the region we called ‘the meiotic domain of BRCA2’ because only meiotic functions were abolished (20). In the females, we found that DMC1 foci became undetectable, but RAD51 foci remained. Thus, BRCA2 indeed directly controls DMC1 and has a dedicated domain for it, lost in our mouse strain. The deleted region contains the so-called ‘PhePP motif’ (Figure 1), which was previously shown to bind DMC1 (14), but whose relevance was not clear (20,21). We confirmed that PhePP preferentially binds to DMC1 rather than RAD51 (14,20), but the details of the interaction and why it is required for DMC1 function remained unresolved. In this study we aimed to address this using structural and biochemical methods.
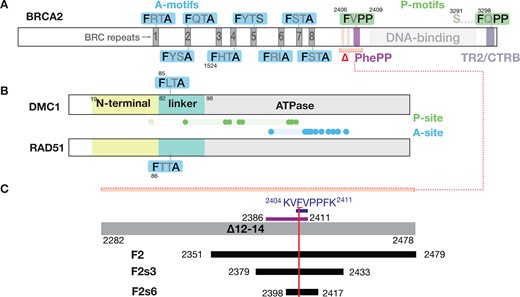
Definition of recombinase-binding motifs. The figure depicts schematically the motifs and domains of (A) BRCA2, and (B) the recombinases DMC1 and RAD51. We refer to the conserved FxPP (green) and FxxA (blue) motifs as P- and A-motifs, and regions on the recombinases binding to P- and A-motifs (dots: residues defining the sites) as P- and A-sites, as detailed in the results section. Residue numbers indicated for the domain boundaries and key amino acid positions relate to human proteins. Region deleted in the Brca2Δ12–14 mouse (Δ) is indicated by a red bracket in panel A; it contains the two HSF2BP-binding sites (light pink) (68,69) and the DMC1-binding PhePP domain (violet). (C) Scheme of the BRCA2 fragments used in this study. The region deleted in the Brca2Δ12–14 mouse (grey bar) and the fragments defined by Thorslund et al.(14) experimentally (’PhePP domain’, violet bar) and from consensus (‘PhePP motif’, dark blue) are shown at the top. The recombinant and synthetic peptides we used are shown in black. The vertical red bar marks the position of F2406. To avoid coining new terms, we will refer to the BRCA2 region we study, encoded by exon 14 and binding DMC1, as the ‘PhePP domain’, and the set of conserved positions within it as the ‘PhePP motif’, although the region boundaries and position set will be adjusted compared to the original definition by Thorslund et al. (14) based on the new data we present.
The human BRCA2 PhePP domain contains one of the BRCA2 FxPP motifs, further named P-motifs. It was discovered in vitro (14), but not studied further, except in a mouse model that did not reveal its involvement in meiotic HR (21). It shares a critical phenylalanine residue with the much better characterized FxxA motif of the BRC repeats, further named A-motif, which represents a conserved feature of BRCA2 in eukaryotes and binds RAD51 and DMC1 (Figure 1A). Other motifs with a FxxP sequence were detected in BRCA2 from vertebrates, invertebrates, plants and fungi (14). In Brh2, an ortholog of BRCA2 from fungus Ustilago maydis and a crucial model (22–24), such motifs act synergistically with BRCs, leading to suggestion of similarity and redundancy between the motifs FxxA and FxxP (25,26). The FxxP motifs interact with RAD51 (DMC1 is absent from Ustilago maydis) thus contributing to DNA repair proficiency (25). In vertebrates, a motif called TR2 (27), CTRD (28) or CTRB (29) that binds to RAD51 was detected in the C-terminal disordered region of BRCA2. It has a stabilizing effect on RAD51-DNA complexes, counteracting BRCs (30–32). Research on this C-terminal site was focused on S3291, which can be phosphorylated by CDKs at the G2-M transition (27,29–34). Phosphorylation abolishes the stabilizing effect, suggesting an HR control mechanism. In addition to BRCA2-S3291, the adjacent highly conserved BRCA2-F3298 is essential for the TR2-RAD51 interaction, and the surrounding conserved sequence matches the P-motif consensus (29). Thus, whereas our Brca2Δ12–14 mouse model supports a meiotic function for the interaction between the BRCA2 P-motif encoded by exon 14 and DMC1, another BRCA2 P-motif encoded by exon 27 binds to RAD51, suggesting that interactions between P-motifs and recombinases may share structural and functional properties (20).
The determinants of the A-motif interaction with RAD51 were studied in a fusion protein hBRC4∼RAD51-ATPase domain (35), to overcome the tendency of purified human RAD51 to aggregate during crystallization attempts. The crystal structure of this fusion protein revealed that, in the absence of the linker region of RAD51, the invariant phenylalanine from the A-motif of BRC4 occupies a hydrophobic pocket, which is a docking site for I26 from the bacterial RecA linker region in the RecA filaments (36); this interaction mediates RecA self-oligomerization and filament formation, providing an explanation to the filament-disruptive effect of the BRCs (36,37). In vertebrate RAD51 and DMC1, the I26 residue from the linker region of bacterial RecA corresponds to a phenylalanine in a conserved A-motif (Figure 1B). The competition between the A-motif of the BRC repeats and that of the recombinase linker was further supported by the analysis of the X-ray structure of an archaeal RAD51 heptamer (37), the cryo-EM structure of RAD51 nucleofilaments (38) and the X-ray structure of Leishmania RAD51-ATPase domain in complex with BRC (39). The BRC repeat exhibits conserved residues outside of its A-motif (40–42), including a second hydrophobic patch that sometimes contains a phenylalanine and contributes to RAD51 binding, but it is both less conserved and less studied. Two seminal studies showed that, whereas BRC interacts with monomeric RAD51, TR2 only binds to RAD51 in oligomeric state suggesting an overlap or interaction between interfaces (30,31).
No structure of BRC-DMC1 exists. DMC1 behaves somewhat differently from the other recombinases (17) because it has higher propensity to form rings rather than filaments (43–46). It was crystallized several times as an octamer (44–46), either in its full-length or its N-terminally truncated form. However, even if crystallized as a full-length protein, its 3D structure was defined only from residue I81, suggesting that the N-terminal region from aa 1 to aa 80 is flexible. Cryo-EM structures of DMC1 bound to ssDNA or dsDNA were also reported (47,48), in which the N-terminal region is observed and contributes to the oligomerization interface. In all these 3D structures, the invariant F85 from the DMC1 linker region is buried in a hydrophobic pocket of the ATPase domain of the adjacent monomer, thus mediating both self-oligomerization and filament formation.
Other BRC-like A-motifs have been spotted in proteins such as RECQL5 (49), MCM9 (50), FIGNL1 (51) that bind RAD51, while a motif similar to PhePP but with a tryptophane in place of phenylalanine was described in the RAD51-binding protein RAD51AP1 and shown to bind DMC1 (52,53). No structural description of the interfaces between the A- and P-motifs of these proteins and recombinases is available.
Here, we describe the crystal structure of the complex between full-length human DMC1 and the BRCA2 fragment T2398-H2417, containing the PhePP motif. This structure revealed that rather than binding to the hydrophobic pocket used by the invariant phenylalanine of the BRC motifs and the self-oligomerization region of the recombinases, the phenylalanine of the PhePP motif uses a distinct, non-overlapping binding interface. Through mutagenesis we determined the importance of the interactions beyond the FxPP consensus. We suggest that there are two distinct classes of recombinase-binding motifs, defined by the nature of the residue in the fourth position of the four-residue core motif. The new binding mechanism explains why the P-motifs identified in PhePP and TR2 can stabilize recombinase nucleoprotein filament, while the A-motifs of the BRC repeats disrupt and remodel it.
Materials and methods
Protein Expression and Purification
DMC1 and its variants (F89A, F85A) were purified exactly as in our paper describing the Brca2Δ12–14 mouse model (20). Constructs for bacterial expression of human his-TEV-DMC1 (pAZ379 wild-type, pAZ454 F89A, pAZ435 F85A) were engineered by subcloning DMC1 coding sequence into pETM-11 vector (EMBL) using Gibson assembly. Rosetta2 DE3 pLysS Escherichia coli strain was transformed with the construct and plated on selective (50 μg/mL kanamycin, 30 μg/mL chloramphenicol) LB-agar plates. Ten colonies were used to inoculate 200 ml selective LB media, the culture was grown overnight at 37°C with shaking and used to inoculate 9 L selective LB media. The 9 L culture was grown at 37°C till OD600 reached 0.6–0.8, expression was induced by adding IPTG to 0.2 mM final concentration. Cells were cultured for another 3 h at 37°C, collected by centrifugation and frozen. The pellet was thawed in two volumes of lysis buffer (3 M NaCl, 100 mM Tris pH 7.5, 10% Glycerol, 0.5 mM EDTA, 5 mM β-mercaptoethanol, protease inhibitors (Roche)) and sonicated on ice (10 pulses of 10 s). The lysate was incubated overnight at 4°C on a rotator, and then cleared by centrifugation at 58 540 × rcf for 45 min at 4°C. Cleared supernatant was supplemented with 5 mM imidazole and incubated with 5 ml washed (500 mM NaCl, 25 mM Tris pH 8, 10% Glycerol, 0.5 mM EDTA, 1 mM DTT) Ni-NTA beads for 1 h at 4°C on a rotator. The beads were washed with 20 and 50 mM imidazole (two times 10 ml for each wash). Bound protein was eluted with 400 mM imidazole, fractions analyzed by electrophoresis and pooled, supplemented with TEV protease and dialyzed overnight against 2 l of 150 mM NaCl, 10% glycerol, 50 mM Tris pH 8, 5 mM β-mercaptoethanol. Protein was further purified using 5 ml HiTrap heparine and CaptoQ columns.
Crystallization of DMC1/PhePP complex and structure determination
Crystallization assays of DMC1/PhePP complexes were performed at 20°C in the High Throughput Crystallization (HTX) laboratory (EMBL Grenoble). The complex was prepared by mixing the protein DMC1 with the PhePP peptide at a molar ratio of 1:2.5. The final protein concentration was at 7.4 mg/ml. Crystallization screens were performed by sitting drop vapor diffusion, mixing 100 nl of sample with 100 nl of reservoir solution. Several crystallization conditions were identified within several days and the crystals were grown for a month. They were prepared for X-ray diffraction experiments using the CrystalDirect harvesting and processing robot (54). The crystals were flash-frozen in liquid nitrogen without cryoprotection. The best crystals were obtained using a crystallization solution of 0.1 M MES at pH 6 or 2.4 M of sodium formate at pH 6. They diffracted up to 3.46 Å of resolution. X-ray diffraction data were collected on the MASSIF-1 beamline at ESRF synchrotron (Grenoble). Diffraction data were indexed, integrated and scaled using XDS (55). The crystal structure was determined by molecular replacement with MOLREP (56) using the human DMC1 ATPase domain crystal structure (PDB code 6R3P, Dunce, J.M. & Davies, O.R., unpublished) and the AlphaFold2 predicted structure of the N-terminal domain as search models. Refinement was performed with BUSTER-2.10.4 (57) using TLS groups, NCS restraints and target geometrical LSSR (58) restraints from the higher resolution structure (PDB code 6R3P). Inspection of the density map and manual rebuilding were performed using COOT (59). Molecular graphic images were generated using ChimeraX (60). In the final model, DMC1 chain A was modeled from Q28 to V62, from L65 to T271 and K286 to K339, the DMC1 chain B was modeled from I81 to T271 and P284 to K339, the PhePP peptide C was modeled from P2402 to S2414, and the PhePP peptide D was modeled from V2405 to P2409. Crystallographic data and refinement parameters are summarized in Table S1. The crystal structure was deposited at the Protein Data Bank as 8QQE.
Isothermal titration calorimetry (ITC)
Analysis was performed using a VP-ITC Calorimeter (GE Healthcare). Purified bacterially expressed (DMC1, BRCA2-F2s3) or synthetic (BRCA2-F2s6) peptides were used. Experiments were performed at 20°C and duplicated; interactions with F2s3-F2406A and TR2 were also tested at 10°C. The buffer was 25 mM Tris buffer, pH 7.5, 100 mM NaCl and 5 mM β-mercaptoethanol. 10 μM of DMC1 in the cell was titrated with 100 μM of BRCA2 peptide in the injection syringe; 10 μl of the syringe volume were injected every 210 s. Data recording were performed using the Origin 7.0 software provided by the manufacturer.
GST pull-down assay
GST pull-down assays were performed exactly as in our paper describing the Brca2Δ12–14 mouse model (20). Bacterial expression constructs for expression of his-GST-TEV-tagged BRCA2 fragments and their amino acid substitution variants were engineered using Gibson assembly in pETM-30 vector (EMBL) and sequence-verified. Constructs were transformed into Rosetta2 DE3 pLysS E. coli expression strain. Two ml of selective LB (50 μg/ml kanamycin, 30 μg/ml chloramphenicol) media was inoculated, the culture was grown overnight at 37°C with shaking, diluted to 10 ml, further incubated till OD600 reached 0.6–0.8, induced with 0.2 mM IPTG, grown for additional 3 h at 37°. Cells were collected by centrifugation, pellet was resuspended in 1 ml NETT+DP buffer (NaCl 100 mM, 50 mM Tris–HCl pH 7.5, 5 mM EDTA pH 8, Triton X100 0.5%, freshly supplemented with protease inhibitors (Roche), 1 mM DTT, 1 mM PMSF) and sonicated (10 × 5 s on, 10 s off). Lysate was transferred to Eppendorf minicentrifuge tubes and cleared by centrifugation (30 min at 4°C). Supernatant was mixed with 20 μl GSH-Sepharose beads (GE Healthcare 17-5132-01) and incubated overnight at 4°C. Beads were collected by centrifugation (500 rcf, 2 min, 4°C), washed with NETT+ buffer (NaCl 100 mM, Tris–HCl pH 7.5 50 mM, EDTA pH 8 5 mM, Triton X100 0.5%, freshly supplemented with protease inhibitors (Roche)), and incubated with DMC1 protein solution (∼2 μg in 1 ml NETT+) for 1.5 h at 4°C. Prior to incubation, bead suspension was vortexed, a 40 μl aliquot was collected as input, mixed with 12 μl 5 × sample buffer (50% glycerol, 250 mM Tris–HCl pH 6.8, 10% SDS, 0.5% bromophenol blue, 0.5 M β-mercaptoethanol) and denatured for 5 min at 95°C. After incubation with DMC1, beads were washed three times with NETT+ buffer and incubated with 25 μl of Laemmli sample buffer for 5 min at 95°C to elute bound proteins. Samples (5 μl of eluate) were run on a 13% SDS-PAGE, transferred to PVDF membrane, blocked (5% skim milk in PBS-T (PBS with 0.05% Tween-20)) and immunodetected with a mixture of rabbit anti-RAD51 polyclonal (1:20 000 (61)) and mouse anti-GST monoclonal (1:5000, B-14 Santa Cruz, sc-138) antibodies overnight. After washes, the membranes were incubated with fluorescently labelled secondary antibodies (anti-mouse CF680 (Sigma #SAB460199), anti-rabbit CF770 (Sigma #SAB460215), washed (5 × 5 min PBS-T) and scanned using Odyssey CLx imaging system (LI-COR).
ATPase activity assay
Non-radioactive colorimetric malachite green ATPase activity assay was performed in 96-well microtiter plates in triplicates. Prior to the experiment, all glassware was thoroughly rinsed with ultrapure milli-Q water to remove all traces of phosphate. Solution AM was prepared by adding 17.25 ml fuming HCl to 25 ml milli-Q water, then slowly dissolving 2.1 g ammonium heptamolybdate·4H2O (Sigma-Aldrich A7302) and adjusting the volume to 50 ml with milli-Q water. Solution MG was prepared by dissolving 67.5 mg malachite green (Sigma-Aldrich M6880) in 100 ml and then adjusting to 150 ml milli-Q water. Solution MGAM was prepared by mixing 150 ml MG and 50 ml AM for at least 30 min and filtering through 0.22 μm PES or cellulose acetate filter. The solution was stored at 4°C in the dark and used in multiple experiments. Before the experiment, an aliquot of MGAM solution was complemented with Tween-20 (0.01% final). The reaction was performed by adding 5 μl of 0.8 mM ATP solution every 15 min to a tube containing 1 μM DMC1 or RAD51 in 20 μl final reaction buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1.5% glycerol, 2 mM DTT, 2 mM MgCl2, 100 ng/μl acetylated BSA, 50 μM (nucleotides) phiX174 ssDNA). Samples were incubated at 37°C for the different time points. The reaction was stopped by adding the samples to 100 μl MGAM-Tween solution. Two min later 12 μl of 34% sodium citrate (Sigma-Aldrich 71405) solution was added. The absorbance at 620 nm was read 20–40 min later using Spectramax iD3 instrument (Molecular Devices). Standard phosphate solutions were in concentration range 0–160 μM.
AlphaFold calculations
Initial structure predictions were obtained using AlphaFold2 as implemented in the ColabFold version 1.3.0. Multiple sequence alignments were obtained from the MMSeqs2 MSA server in the mode: ‘UniRef + Environmental’. For ColabFold, all default parameters were kept, notably model_type: ‘AlphaFold2-ptm’ and rank_by: ‘plddt’, except for num_recycles that was set to 18. No Amber relaxation or any manual interventions were applied to the models. All predictions were repeated using a similar protocol on the Integrative Bioinformatics platform of I2BC (https://bioi2.i2bc.paris-saclay.fr). For each complex, a series of five to fifteen models were calculated and the five best models were analyzed using the delivered heatmap and lDDT plots, as well as the pTM- and ipTM scores (62,63).
Biolayer interferometry (BLI)
Biomolecular interactions between DMC1 and biotinylated peptides BRCA2 F2s6: Biotin-GSG-TTGRPTKVFVPPFKTKSHFH-NH2, BRCA2 F2s6-F2406A: Biotin-GSG-TTGRPTKVAVPPFKTKSHFH-NH2, BRCA2 TR2: Biotin-GSG-VSPAAQKAFQPPRSSGTKYE-NH2, FiGNL1 P-motif: Biotin-GSG-RSRGILGKFVPPIPKQDGGE-NH2; RAD51AP1 P-motif: Biotin-GSG-SAESKKPKWVPPAASGGSRS-NH2, BRCA2 BRC1: Biotin-GSG-SNHSFGGSFRTASNKEIKLSEHNIKKSKMFFKDIEE-NH2; BRCA2 BRC4: Biotin-GSG-IKEPTLLGFHTASGKKVKIAKESLDKVKNLFDEKEQ-NH2 (synthesized by Proteogenix and Genecust) have been measured by BLI using an Octet RED96 instrument (FortéBio) and Streptavidin (SA) biosensors. The measurements were made at 25°C. The biosensors were hydrated for 20 min in 25 mM Tris pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol buffer (the same buffer as for the DMC1 protein). For the binding assays, the biotinylated peptides were immobilized on Streptavidin sensors by immersing the sensors with 0.25 μg/ml peptide solutions. No binding of DMC1 was observed in the binding assays without loaded biotin peptides. The SA biosensors were washed with the buffer before the incubation (300 s) with DMC1 at different concentrations to record the association processes. The kinetic assays were performed at different ranges of concentrations of DMC1, using 2-fold serial dilution, as indicated. Dissociation processes were measured by immersing the sensors in the buffer for 600 sec. 5 cycles of 10 s incubation in the regeneration buffer (1 M NaCl) followed by 10 s incubation in the assay buffer (25 mM Tris, 100 mM NaCl, 5 mM β-mercaptoethanol, 0.05% Tween 20) were made before each baseline for every kinetic experiment. Most Kd were measured in the steady-state mode.
The interaction between DMC1 filaments (formed on a biotinylated ssDNA of either 60 nt or 100 nt, as indicated) and different peptides (F2s6, F2s6-F2406A, F2s6-K2404E, F2s6-K2413E, F2s6-R2401A-K2404A and F2s6-K2411A-K2413A) was also tested by BLI. The peptides were synthesized by Proteogenix and the ssDNA by Integrated DNA Technologies (IDT). The sequence of the 100 nt ssDNA is: Biotin-AATTCTCATTTTACTTACCGGACGCTATTAGCAGTGGGTGAGCAAAAACAGGAAGGCAAAATGCCGCAAAAAAGGGAATAAGGGCGACACGGAAATGTTG. The peptide sequences are: F2s6: Ac-TTGRPTKVFVPPFKTKSHFH-NH2; F2s6-F2406A: Ac-TTGRPTKVAVPPFKTKSHFH-NH2; F2s6-R2401A-K2404A: Ac-TTGAPTAVFVPPFKTKSHFH-NH2; F2s6-K2411A-K2413A: Ac-TTGRPTKVFVPPFATASHFH-NH2; F2s6-K2404E: Ac-TTGRPTEVFVPPFKTKSHFH-NH2; F2s6-K2413E: Ac-TTGRPTKVFVPPFKTESHFH-NH. The experiments were conducted at 25°C. The biosensors were hydrated in 10 mM Tris, pH 8, 50 mM NaCl, 5 mM β-mercaptoethanol, 2 mM CaCl2 and 2 mM ATP. A solution of 40 nM of ssDNA (in the same buffer) was used to load the SA sensors and were then incubated with a 2 μM DMC1 solution (in the same buffer + 0.05% Tween20) during 30 min. After a short dissociation step and a baseline, the kinetic assays were performed at different concentrations of peptides using a two-fold serial dilution (20, 10, 5, 2.5, 1.25 μM). The Kd values were measured in the steady-state mode.
Electrophoretic mobility shift assay (EMSA)
EMSA protocols differed somewhat between two laboratories. For experiments shown in Figure 5E and Supplementary Figures S2C and S7B, the following oligonucleotides were annealed at 90°C for 10 min to make ds AF-647 90-mer. For ssDNA substrate, only the AF-647-90 mer was used. AF-647-SK3-90fw AATTCTCATTTTACTTACCGGACGCTATTAGCAGTGGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGT; SK3-RC unlabeled ACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCCACTGCTAATAGCGTCCGGTAAGTAAAATGAGAATT. For experiments shown in Supplementary Figure S2C, 5 nM DNA (ss or ds molecules) was incubated with DMC1 (0–4 μM) in a 20 μl reaction volume with final buffer composition of (50 mM Tris pH 7.5, 150 mM NaCl, 1% glycerol, 2 mM CaCl2, 1 mM ATP, 1 mM DTT). Samples were incubated at 37°C for 15 min, then 2 μl of 50% glycerol was added. Samples were analyzed on a 5% PAGE gel (80:1 acrylamide/bisacrylamide) at 4°C in 0.5 × TB. PAGE gel was pre-run for 1 h 150 V, then samples were loaded and run for another 2 h at 150 V in 0.5 × TB at 4°C. The gel was scanned on a Typhoon imager using the Cy5 channel. Experiments shown in Figure 5E, S7B were performed by incubating for 30 min at 37°C a reaction containing 2 μM DMC1 (wild-type or F85A or F89A variants) 2 μM PhePP F2s3, 6 μM (nucleotides) AF-647–90 ssDNA substrate, 50 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM CaCl2, 2 mM ATP, 1% glycerol, 1 mM DTT in the final volume of 20 μl. After incubation, the reactions were placed on ice, glycerol was added to 5%, and all was loaded on a 0.8% 1 × TB agarose gel. The gel tank was kept on an ice bath during the run for 1.5 h at 80 V.
For EMSAs shown in Figure 6G, DMC1 filaments were formed by incubating 6 μM of a single stranded (ss) DNA (100 nucleotides (nt)) or a double stranded (ds) DNA (200 bp) labeled with Cy5 with 2 μM DMC1 (1 protein per 3 nt) in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM CaCl2, 2 mM ATP and 1 mM DTT, during 20 min at 37°C. The effect of the F2s3 or F2s6 peptide was tested by preincubating it with DMC1 just before the filament assembly reaction following various DMC1:peptide stoichiometries (1:0.5, 1:1, 1:2 for F2s3; 1:1, 1:2, 1:4, 1:8 for F2s6) in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl and 2 mM CaCl2. The F2s3 F2406A mutant peptide was preincubated with DMC1 in a 1:2 stoichiometry. The F2s6-K2404E, F2s6-K2413E, F2s6-R2401A-K2404A and F2s6-K2411A-K2413A mutant peptides were preincubated with DMC1 in a 1:4 stoichiometry. Finally, the effect of peptides on the DNA substrates in absence of DMC1 was also tested by incubating 12 μM of each peptide with 6 μM DNA in a buffer containing 10 mM Tris–HCl pH7.5, 50 mM NaCl, 2 mM CaCl2, 2 mM ATP and 1 mM DTT during 20 min at 37°C. Protein–DNA complexes were fixed with 0.01% glutaraldehyde 5 min at room temperature. The reaction products were run on a 1% agarose gel in 0.5 × Tris acetate EDTA at 80 V during 45 min at 4°C, then analyzed using a Typhoon imager (GE Healthcare Life Science).
Tailed DNA substrate
DNA with a 3′ ss overhang was made as follows: using the URA3 gene from Saccharomyces cerevisiae as template DNA, an 810-bp polymerase chain reaction (PCR) fragment was produced using primer URA3 (GAAGGAAGAACGAAGGAAGGAGC) which was 5′ phosphorylated and primer BIO 5′ (TTTCCCGGGGGGCCCGGGTTCTATACTGTTGACCC) which was 5′ biotinylated. The PCR product was purified on a GFXTM column (GE Healthcare) and incubated with 1 mg of streptavidin-coated magnetic beads (Dynabeads M-270, Invitrogen) for 2 h at 37°C in 400 μl of BW buffer (5 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl). Using a magnetic particle concentrator, the immobilized DNA was washed two times with 100 μl of BW buffer. Subsequently, the DNA was washed once with 100 μl of 1× λ exonuclease buffer (NEB). The DNA bound to the beads was made ss by λ exonuclease digestion of the strand with 5′ phosphate (5 U/mg DNA, NEB). The reaction was carried out at 37°C for 1 h. Immobilized ssDNA was washed with 100 μl of BW buffer. Subsequently, the DNA was washed once with 100 μl of 1× Sequenase buffer (USB) and hybridized with oligonucleotide primer DR3 (GGTCCCAAAATTTGTTTACTAAAA), which anneals at position 290–313 along the ss PCR product. Primer DR3 was extended for 30 min at 37°C in a reaction containing SequenaseTM DNA polymerase Version 2.0 (USB) in 100 μl of 1 × Sequenase buffer supplemented with 200 mM dNTPs. The resulting DNA with 505 bp ds region and 289-nt 3′ overhang was washed with 100 μl of BW buffer, released from streptavidin-coated magnetic beads by digestion with SmaI (Roche) and purified on a GFX column.
Scanning force microscopy (SFM)
DMC1 (30 nM) was incubated in buffer containing: 30 mM HEPES–KOH pH 7.2, 150 mM NaCl, 1 mM CaCl2 and 1 mM DTT for 10 min and deposited onto freshly cleaved mica. After 10 s the mica surface was washed with MilliQ water and dried with a stream of filtered air. DMC1-tailed DNA complexes were formed in 20-μl reactions containing 3.75 μM DNA, 2.5 μM DMC1, 30 mM HEPES–KOH pH 7.2, 150 mM NaCl, 1 mM CaCl2 1 mM ATP and 1 mM DTT. Reaction mixtures were incubated at 37°C for 10 min, diluted 15-fold in deposition buffer (10 mM HEPES–KOH (pH 7.5) and 10 mM MgCl2) and deposited on freshly cleaved mica. After 10 s, the mica was washed with water and dried in a stream of filtered air. When the effect of the presence of PhePP on filament formation of DMC1 was studied, DMC1 was first preincubated with PhePP or PhePP-F2406A on ice for 10 min (6-fold molar excess of peptide) and then DMC1-tailed DNA complexes were formed in 20-μl reactions containing 1 μM DMC1, 3.75 μM DNA, 30 mM HEPES–KOH pH 7.2, 150 mM NaCl, 1 mM CaCl2 1 mM ATP and 1 mM DTT. Reaction mixtures were incubated at 37°C for 10 min, diluted 10-fold in deposition buffer (10 mM HEPES–KOH (pH 7.5) and 10 mM MgCl2) and deposited on freshly cleaved mica. After 10 s, the mica was washed with water and dried in a stream of filtered air. RAD51–tailed DNA complexes were formed in 20-μl reactions containing 3.75 μM DNA, 1.25 μM RAD51, 50 mM Tris–HCl (pH 7.5), 2 mM CaCl2, 1 mM ATP, 1 mM DTT and 30 mM KCl. Reaction mixtures were incubated at 37°C for 10 min, diluted 15-fold in deposition buffer (10 mM HEPES–KOH (pH 7.5) and 10 mM MgCl2) and deposited on freshly cleaved mica. After 10 s, the mica was washed with water and dried in a stream of filtered air. Images were obtained on a NanoScope IV SFM (Digital Instruments; Santa Barbara, CA) operating in tapping mode in air with a type J scanner. Silicon Nanotips were from AppNano (Santa Clara, CA).
Transmission electron microscopy (TEM)
DMC1 filaments were formed in the same conditions as for the EMSA. 2 μM DMC1 were preincubated with 2 μM F2s3 or 8 μM F2s6 peptides in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl and 2 mM CaCl2, then 6 μM (nucleotides) ssDNA (100 nt), 1 mM DTT and 2 mM ATP were added to the reaction and incubated 20 min at 37°C. The effect of F2s3 (1:1 DMC1:F2s3 stoichiometry) and F2s6 (1:4 DMC1:F2s6 stoichiometry) on DMC1 filament stability was tested by measuring their length using TEM. Concretely, for statistical analysis of filament length, positive staining combined with a TEM dark-field imaging mode were used: 1 μl of the reaction was quickly diluted 20 times in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl and 2 mM Cacl2. During one minute, a 5 μl drop of the dilution was deposited on a 600-mesh copper grid previously covered with a thin carbon film and pre-activated by glow-discharge in the presence of amylamine (Sigma-Aldrich) (64,65). Grids were rinsed and positively stained with aqueous 2% (w/v) uranyl acetate, dried carefully with a filter paper. To better observe the effect of peptides on filament architecture, the sample was also spread using negative staining and observed in bright-field. In that way, a drop of the reaction was directly deposited on a carboned copper grid pre-activated with a glow discharge (plasma).
TEM grids were observed in the annular dark-field mode in zero loss filtered imaging or in canonical bright-field using a Zeiss 902 transmission electron microscope. Images were captured at a magnification of 85 000× with a Veleta CCD camera and analyzed with iTEM software (both Olympus Soft Imaging Solution). For the quantifications, the length of filaments was measured on at least two independent experiments with a total of at least 75 molecules measured.
Results
BRCA2 PhePP binds DMC1 via a novel interface
In our study describing the Brca2Δ12–14 mouse model (20), we demonstrated that DMC1 but not RAD51 binds with a detectable affinity to the BRCA2 region that is encoded by exon 14. We sequentially narrowed down the binding domain to the GST-tagged BRCA2 fragments F2 (2351–2479), F2s3 (2379–2433) and F2s6 (2398–2417) that all contain the conserved PhePP P-motif (Figure 1C). We also previously found by ITC that DMC1 interacts directly with F2s6 and that F2406, the phenylalanine from the PhePP domain, is required for binding (20). To further describe the mechanism of the interaction, we first used AlphaFold to predict the interfaces between DMC1 and the BRCA2 fragment F2s3, and, for comparison, between DMC1 and BRC4. We obtained high score models for both complexes (interface scores ipTM (62) above 0.7) (Figure 2A; Supplementary Figure S1A). In the models of DMC1 bound to PhePP (F2s3) the predictions did not place F2406 into the deep highly conserved hydrophobic pocket that is occupied by F1524 of BRC4 and F86 of the linker region of the adjacent monomer (35,37,44,46) in the crystal structures of RAD51. They systematically positioned F2406 in an alternative hydrophobic pocket located in the ATPase domain, 15 Å away from the previously described pocket. AlphaFold produced very low score models of DMC1 bound to F2s3 mutant F2406A, supporting the importance of this phenylalanine for the DMC1–PhePP interaction (Supplementary Figure S1A). It produced models with a decreased precision on the local structure of the peptide (lDDT) when the size of the complex was increased (larger BRCA2 fragment, two DMC1 monomers), and no new contact site between DMC1 and BRCA2 could be identified (Supplementary Figure S1B). Finally, in the models of the complex between full-length DMC1 and BRC4, the predictions placed F1524 in the expected hydrophobic pocket identified in RAD51 (35,37,39), also occupied by F85 of the linker region of the adjacent monomer in the crystal structures of DMC1 (44,46) (Figure 2B).

Structure of the DMC1–PhePP complex. (A) AlphaFold models of the DMC1–PhePP interaction. The BRCA2 peptide is colored as a function of the lDDT score per residue. Side chains of the phenylalanine and proline defining the P-motif are displayed in sticks. DMC1 is colored in cyan in its N-terminal region (from M1 to E82), pale green in its linker region (P83 to M97) and dark green in its ATPase domain (V98 to E340). (B) AlphaFold model of the DMC1-BRC4 interaction, displayed as in (A). (C) Crystal structure of the octameric DMC1 (blue, green and wheat) bound to the BRCA2 peptide PhePP (F2s6, red). The blue protomers are observed from Q28 to K339 (one protomer is distinguished by being colored in green), whereas the wheat protomers are observed from I81 to K339. In all protomers, a single loop from A272 to either D283 or K285 is not detected. (D) Focus on the interactions between a wheat DMC1 protomer (surface view) and the adjacent green DMC1 protomer (cartoon view) as well as the red BRCA2 peptide from P2402 to S2414 (cartoon view). The side chains of the BRCA2 peptide are shown in red sticks. BRCA2 residues that are more than 30% buried upon DMC1 binding are marked (red labels correspond to residues more than 80% buried). The side chain of DMC1 F89 (green protomer), located in front of BRCA2 K2413, is displayed in dark green sticks and labeled. In the insets, the positions of the BRCA2 peptides in the X-ray structure (red) and in the AlphaFold models 1 and 2 (blue) are compared by superimposing the interacting DMC1 protomers.
A detailed analysis of the AlphaFold models of DMC1 interacting with PhePP revealed that two different models have high (ipTM > 0.7) and medium (ipTM > 0.5) interface scores, respectively (Figure 2A; Supplementary Figure S1A). In both models, the central FVPP motif is buried in the same region of the ATPase domain of DMC1, but in model 1, the N-terminal region of DMC1 (V6-A8) forms an additional β-sheet with the BRCA2 residues preceding the core PhePP motif (K2404-F2406). To experimentally test these models, we first produced active untagged full-length human DMC1 (Supplementary Figure S2A–D) and analyzed the complexes between DMC1 and either BRCA2 F2s6 or BRCA2 F2s3 by ITC (Supplementary Figure S2E), which resulted in similar micromolar affinities for both peptides (Kd = 3.0 ± 0.5 μM for F2s3 and 3.4 ± 1.3 μM for F2s6). Therefore, we co-crystallized DMC1 with the BRCA2 peptide F2s6 and determined the structure of this complex by X-ray crystallography at a resolution of 3.46 Å. A dimer consisting of two DMC1-F2s6 complexes was observed in the asymmetric unit, which was assembled into a ring-shaped octamer using the crystallographic symmetries (Figure 2C), as also reported for human DMC1 alone (44,45). The dimer comprises two monomers observed from F21 to K339 (chain A, blue or green in Figure 2C) and I81 to K339 (chain B, wheat in Figure 2C): the chain A monomer exhibits a homogeneously positioned and folded N-terminal domain (between Q28 and L80), and both monomers show a structured linker region (between I81 to V98) and an ATPase domain (between F99 and K339). Density corresponding to the N-terminal domain is characterized by high B-factors (225–295 A2), compared to that of the linker and ATPase regions (150–225 A2), revealing that the position of the observed N-terminal domain is still fluctuating in the crystal (Supplementary Figure S3A). In the ring-shaped octamer, the DMC1 protomers interact mainly through their linker and ATPase regions, as previously reported. The N-terminal domain of one protomer (chain A) interacts through a positively charged surface with a negatively charged surface of the ATPase domain of the adjacent protomer (chain B, Supplementary Figure S3B). Two salt bridges were identified in our crystal structure, between chain A K49 and chain B D204, as well as chain A K74 and chain B E212. Thus, the N-terminal domain contributes to the intermolecular interactions in the DMC1 octamer.
A BRCA2 F2s6 peptide is bound to each DMC1 protomer. Its F2406 is buried in a hydrophobic groove on the outer surface of the DMC1 octamer (Figure 2C). The peptide bound to the DMC1 chain A (green/blue) protomer is defined from BRCA2 V2405 to P2409 (chain D), including the FVPP motif, and its density is partial due to the crystal packing. Its position overlaps with that of an equivalent peptide bound to another protomer (Supplementary Figure S3C). The peptide bound to the DMC1 chain B (wheat) protomer is defined from BRCA2 P2402 to S2414 (chain C, Figure 2D). Its position relative to the wheat protomer is close to that of the peptide bound to the blue protomer (Supplementary Figure S3D). It is located as predicted by AlphaFold in model 1, which is the best scoring model, but the additional β-sheet formed by the DMC1 N-terminus in this model is not observed in the crystal structure (Figure 2D, insets). At the interface, BRCA2 residues F2406 and P2409 are almost totally buried (>80%), whereas residues K2404, P2408, F2410 and K2413 are substantially buried (more than 30%), upon interaction with the wheat colored DMC1 protomer. K2413 is close to F89 of the green colored DMC1 protomer: their interaction could also help to stabilize the DMC1 octamer (Figure 2D).
Binding extends beyond the core FxPP motif and engages the DMC1 N-terminal and linker domain
To support our structural description of the interface between the BRCA2 PhePP domain and DMC1 (Figures 3A and B), we used GST pull-down assays. We determined which positions in and around the FVPP motif contribute to the interaction, focusing on those that are evolutionarily conserved: pairs of positively charged residues on either side of FVPP: R2401·K2404 and K2411·K2413, and F2410, which is as strictly conserved as F2406 (Figure 3B). As the F2s6 peptide used for crystallography is too short (20 aa) for efficient purification from E. coli, we used longer fragments tagged with GST for these experiments, first F2 and later F2s3. Alanine substitutions at F2406 and P2409 in the core FXPP motif consistently abolished the efficiency of co-precipitation of DMC1 with the GST-tagged F2 and F2s3 fragments, while the effect of substitutions at the positively charged positions was more subtle (Figure 3C, Supplementary Figure S4A and B). Double substitution of the C-terminal pair (K2411A·K2413A) in F2 or F2s3 had a strong effect, abolishing binding almost as effectively as the F2406A and P2409A substitutions, whereas single substitutions had mild (K2411) or no (K2413) impact. Single R2401A and K2404A substitutions decreased binding only in experiments with the larger GST-F2 fragment, which is unstable when produced in bacteria and thus present at much lower concentrations than F2s3 during the pull-downs. Substituting the invariant F2410 for alanine had no effect on the binding. F2410 is only partially (40% to be compared to 80% in the case of F2406 and 100% in the case of P2409) buried at the interface and seems to play no role in the binding. Going back to our crystal structure, we identified that F2406 is close to DMC1-G153 and G154, K2404 is close to DMC1-E213 and to the N-terminal domain of the adjacent protomer, whereas K2413 is close to DMC1-D182 and D186 as well as F89 in the adjacent DMC1 protomer (Figure 3A). Using a set of amino acid substitutions in DMC1, we found that G153R, G153E, G154Q and E213K substitutions disrupted or abolished the interaction, while F89A, D186K, D182K substitution did not affect it (Figure 3D, Supplementary Figure S4C).
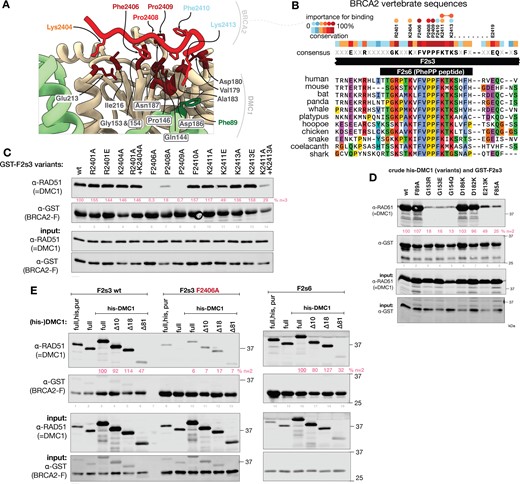
Validation of the DMC1–PhePP binding interface. (A) Crystal structure of the interface between DMC1 and the BRCA2 PhePP (F2s6) peptide, colored as in Figure 2D. In BRCA2, only side chains buried upon binding are displayed, and their label is colored as a function of their contribution to binding, as defined in panels B. In DMC1, buried sidechains are dark-red and labeled, DMC1-F89 forming cation-π interaction with BRCA2-K2413 is green, residues conserved in RAD51 are boxed. (B) Alignment of the central region of human BRCA2 F2s3 (2379–2433) sequence with homologous vertebrate BRCA2 sequences, conservation score and consensus sequence. Conserved positions tested by alanine substitution are displayed above the conservation plot. Circles above indicate the importance of the position for the interaction. (C) GST pull-down between BRCA2-F2s3 substitution variants and purified recombinant DMC1. Replicates are shown in Supplementary Figure S4B. Bound DMC1 band intensities normalized to the wild-type GST-F2s3 lane is shown in pink. (D) GST pull-down of DMC1 variants with GST-F2s3 BRCA2 fragment. A replicate is shown in Supplementary Figure S4C. Bound DMC1 band intensities normalized to the wild-type lane is shown in pink. (E) Interaction between BRCA2–PhePP (fragments F2s3 and F2s6, GST-tagged) and DMC1 (purified or in crude bacterial lysate, with or without his-tag) and its truncation variants, determined by GST pull-downs. A replicate is shown in Supplementary Figure S4D. Bound DMC1 band intensities normalized to the full-length his-DMC1, is shown in pink.
We also explored the contribution of the interactions with the N-terminal region of DMC1. In AlphaFold model 1, in addition to the DMC1 ATPase domain, the interaction with F2s6 also involved the first 18 amino acids of DMC1, including a small DMC1 motif V6-A8 forming a β-sheet with K2404-F2406 of BRCA2 (Figure 2A). This region of DMC1 is rich in acidic charged side chains (8 out of 18) and is located before the folded DNA binding domain of DMC1 (residues Q28-I81). It is not visible in the crystal structure. To determine if it contributes to binding, we engineered three DMC1 truncation variants, lacking 10, 18 or 81 N-terminal amino acids. Only the largest deletion missing both the acidic region and the N-terminal folded domain had reduced binding to GST-BRCA2 fragment (Figure 3E, Supplementary Figure S4D). We conclude that, in solution, the additional β-sheet predicted by AlphaFold is not present at the interface between DMC1 and PhePP. However, the folded domain contributes to binding, even if it is not directly in contact with the BRCA2 peptide. Our crystal structure suggests that a cation-π interaction exists between BRCA2 K2413 and DMC1 linker residue F89, which might be stabilized in the presence of the N-terminal domain (Figure 3A).
A- and P-motifs and sites
After experimentally establishing the mechanism of interaction between the BRCA2-PhePP motif and DMC1 recombinase, we explored whether these findings extend to other similar motifs and interactions (Figure 4, Supplementary Figures S5 and S6). Proline in the fourth position (P2409 in PhePP) is essential for the binding of the PhePP P-motif and never occurs in the BRC A-motifs. We designated the sites these motifs bind to on the recombinases as P- and A-sites (Figures 1 and 4A, B). We used AlphaFold to predict if other P-motifs could bind to the P-sites (Figure 4C, Supplementary Figure S5). For the C-terminal BRCA2 TR2, with a well-established role in RAD51 filament binding and stabilization, AlphaFold predicted binding to the P-sites of both RAD51 and DMC1 with medium ipTM scores. For the P-motif of RAD51AP1, the predictions again showed binding to the P-interfaces of the recombinases, despite this motif having tryptophan instead of phenylalanine in the first position. Finally, in FIGNL1, we noticed a previously undescribed P-motif close to its characterized BRC-like motif that binds RAD51 (49), and AlphaFold predicted that this motif binds to the P-interfaces of both recombinases.
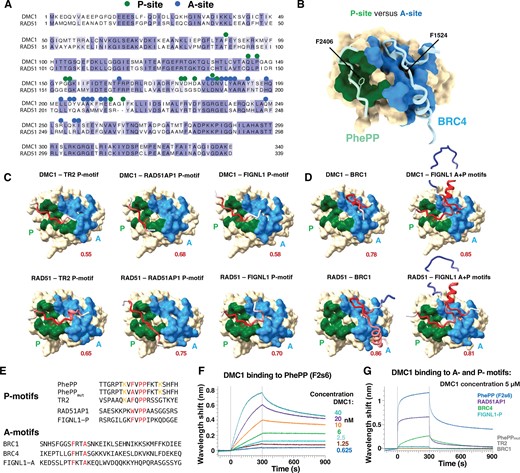
A- and P- recombinase-binding motifs and sites. (A) Alignment of the sequences of human RAD51 and DMC1 recombinases. Green and blue dots correspond to residues buried at >30% upon binding of DMC1 to PhePP (P-site) and RAD51 to BRC4 (A-site), respectively. (B) Representation of the surfaces binding to PhePP (P-site; green) and BRC4 (A-site; blue), as defined in (A), onto the 3D structure of RAD51 bound to BRC4 (PDB 1N0W). To display the PhePP peptide bound to the RAD51 P-site, the crystal structures of DMC1-F2s6 and RAD51-BRC4 were superimposed, and the structure of DMC1 was hidden. (C, D) AlphaFold models of the DMC1 and RAD51 complexes with P- and A-motifs from different proteins (see also Supplementary Figure S5). The ATPase domain surface of the recombinase is displayed, with the P- and A-sites colored in green and blue, respectively. Peptides containing P- or A-motifs are shown as cartoons, colored based on the AlphaFold IDDT score (red - accurate to white - poorly defined). The phenylalanine side chain of the motifs is shown as sticks. (E) Sequences of P- and A-motif peptides. (F) BioLayer Interferometry experiment performed using an immobilized biotinylated PhePP (F2s6) peptide and increasing concentrations of DMC1. The measured apparent Kd is about 1 nM, which is unexpectedly higher than the affinity measured in solution by ITC (Supplementary Figure S2E). (G) BioLayer Interferometry experiments revealing that P- and A-motifs bind with different affinities to DMC1 (see also Supplementary Figures S6A-B). Using a DMC1 concentration of 5 μM, an interaction was detected only in the case of PhePP, RAD51AP1, BRC4 and FIGNL1-P. Apparent affinities for RAD51AP1, BRC4 and FIGNL1 were about 1.6, 4.3 and 16.2 μM, respectively. No binding was detected for PhePPmut, TR2 and BRC1.
In parallel, we verified that the A-motif of BRC1, one of BRC repeats with the strongest affinity for both RAD51 (41) and DMC1 (16), is predicted to bind to the A-site of both RAD51 and DMC1 (Figure 4D, Supplementary Figure S5B). As the A- and P-motifs of FIGNL1 are only separated by 40 residues, we modelled how the conserved region containing both of them interacts with the recombinases and found that simultaneous binding of the two motifs to their corresponding sides was predicted with high (ipTM > 0.8) scores.
We then experimentally tested if the identified P-motifs of BRCA2 (PhePP and TR2), RAD51AP1 and FIGNL1, as well as A-motifs of BRCA2 BRC1 and BRC4, bound to DMC1 (Figures 4E–G, Supplementary Figure S6). Using BioLayer Interferometry (BLI), we found that PhePP (F2s6) is the strongest binder: it shows an affinity of about 1 nM for the recombinase (Figure 4F, Supplementary Figure S6A). This is much higher than the micromolar affinity measured by ITC in solution (Supplementary Figure S2E) and is likely an overestimate due to the very slow rate of dissociation of DMC1 from biotinylated F2s6 immobilized on the streptavidin-coated biosensor. Such a slow dissociation rate was not observed in the case of the other peptides: we detected micromolar interactions between immobilized RAD51AP1 P-motif and DMC1, as well as BRC4 A-motif and DMC1 (Figure 4G, Supplementary Figure S6B). Finally, a weak interaction was observed between FIGNL1 P-motif and DMC1. However, no interaction could be detected when we tested the P-motifs of the PhePP mutant F2406A and TR2, as well as the A-motif of BRC1. Taken together, AlphaFold predictions and BLI measurements support our generalization that different A- and P- motifs bind to their corresponding distinct sites on the recombinases, and demonstrate that binding energies can be distributed over a wide range, a subset of the interactions being characterized by micro- to nanomolar affinities.
PhePP binding stabilizes DMC1–DNA nucleoprotein filaments
Our crystal structure revealed that PhePP can bind to DMC1 oligomers and suggested that in addition to the main interface, the adjacent DMC1 protomer is also engaged (Figure 5A). The contact between PhePP and the adjacent protomer was only partially resolved in our structure as a PhePP-K2413∼DMC1-F89 cation–π interaction. If this contact contributes significantly to the binding energy, a non-oligomerizing variant of DMC1 is predicted to have a reduced affinity to PhePP, and a higher affinity to BRCs. This is supported by previous experiments performed with TR2, BRC4 and the non-oligomerizing F86E variant of RAD51 (30,31). We produced an analogous DMC1 F85A variant, and tested its binding to GST-fused peptides PhePP (F2s3), PhePP variant F2406A and TR2. The F85A substitution indeed substantially reduced DMC1 binding to PhePP, increased its binding to BRC4 (Figure 5B, Supplementary Figure S7A), and abolished its ability to polymerize on ssDNA as determined by EMSA (Figure 5E, lanes 1, 2, 8, Supplementary Figure S7B). Notably, monomeric DMC1 F85A co-precipitated more efficiently with the F2406A than with wild-type PhePP, suggesting that monomerization unmasks a DMC1 site that PhePP peptide can bind when its interaction with the P-site is disrupted by the F2406A substitution. The impact of F85A on TR2 binding was similar to its effect on PhePP binding, suggesting that both P-motifs preferentially bind oligomeric recombinases.
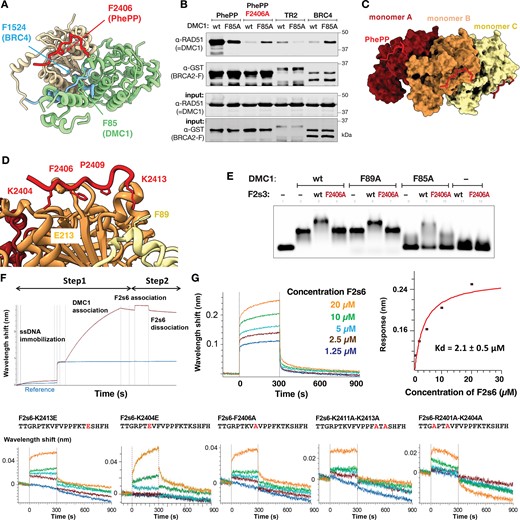
PhePP binds DMC1 oligomers. (A) Superposition of the crystal structure of a dimer of DMC1–PhePP onto the crystal structure of RAD51 ATPase domain bound to BRC4. The two DMC1 protomers are in wheat and green, as in Figure 2C. Only the best resolved PhePP (F2s6) peptide is shown (in red). RAD51 is not displayed and the BRC4 peptide is in blue. In DMC1 green protomer, the side chains of F85 at the interface with the wheat protomer and F89 at the interface with PhePP are shown in sticks. In PhePP, the side chains of two residues important for binding to DMC1, i.e. F2406 and K2413 (interacting with F89), are also represented. In BRC4, only the side chain of F1524, essential for binding RAD51, is displayed. (B) GST pull-down assay with GST-tagged BRCA2 peptides and either wild type DMC1 or its F85A variant (replicated in Supplementary Figure S7A). (C) Docking of three PhePP peptides onto the cryo-EM structure of the DMC1–ssDNA filament (PDB 7C9C). Each peptide was positioned by superimposing the DMC1 filament monomer (either A, B or C) onto the crystal structure of DMC1 (81–339) bound to PhePP, as shown in Supplementary Figure S8. DMC1 (81–339) structures were subsequently hidden. The ssDNA binds on the opposite side of the filament, as compared to PhePP, it is thus not visible in this view. (D) Zoom-in from (C). The side chains of PhePP important for binding to DMC1 are displayed in sticks. The side chains of the DMC1 residues in front of the two lysines are displayed. The same side chains were identified in front of the two lysines in the crystal structure of DMC1–PhePP. (E) EMSA analysis of the filaments formed under Ca-ATP conditions by wild-type or substitution variant (F85A, F89A) DMC1 on a 90 nt ssDNA in the absence or presence of F2s3 or its F2406A variant. (F, G) BLI curves corresponding to the interaction between PhePP (F2s6) or different mutated PhePP peptides F2s6-F2406A, F2s6-K2404E, F2s6-K2413E, F2s6-K2411A-K2413A, F2s6-R2401A-K2404A and ssDNA-DMC1 filaments. On panel F, a schematic representation of the BLI experiment is displayed: the biotinylated ssDNA is coupled to a streptavidin sensor, DMC1 is assembled onto ssDNA, and different concentrations of F2s6 peptides are added. On panel G, the binding curves show that F2s6 binds to the ssDNA-DMC1 filaments with a micromolar affinity (Kd value obtained in the steady-state mode), whereas for its variants, no significant binding was detected in our conditions. A replicate is shown in Supplementary Figure S7D.
Next, we tested if the observations made with the ring-shaped octameric DMC1–PhePP complex are valid for the helical nucleoprotein filament conformation. Fitting our PhePP–DMC1 crystal structure onto the available cryo-EM structure of DMC1 presynaptic filament on ssDNA (47,48) revealed no steric clashes between the peptide and the filament (Figure 5C, Supplementary Figure S8). The local environment of the PhePP residues is similar in the crystal structure and in the DMC1–PhePP-ssDNA model (Figure 5D). Consistent with this, addition of PhePP but not its F2406A variant to DMC1–ssDNA filaments reduced electrophoretic mobility of the DMC1–ssDNA complex, indicating that PhePP can bind DMC1 in its functional filamentous form (Figure 5E, lanes 1–4). An upshift resulting from F2s3 addition was also observed with DMC1 F85A monomeric variant (lanes 8–10), indicating that change in mobility under the experimental conditions reflects binding as well as or rather than polymerization. To study the PhePP–DMC1 filament interaction quantitatively, we measured the affinity of F2s6 for DMC1 assembled onto ssDNA using BLI, the biotinylated ssDNA being immobilized on the streptavidin-coated biosensor (Figure 5F). We found an affinity of 2.1 ± 0.5 μM, close to the affinity measured in solution by ITC between F2s6 and DMC1 (around 3 μM; Figure 5G, Supplementary Figures S2E, S7D). Replacing positively charged residues flanking the P-motif impaired binding in all cases we analyzed. This effect was consistent with the results of GST pull-downs, although it was difficult to measure Kd larger than ∼20 μM (10 × decrease in affinity) in our BLI setup, whereas lower affinity binding could probably be detected by GST-pulldown (Figure 3A, Supplementary Figure S4A, B).
We then asked if the dependence of PhePP binding efficiency on DMC1 oligomerization is reciprocal, i.e. whether PhePP binding can stabilize DMC1 nucleoprotein filament, as has been reported for TR2-RAD51 (30,31). In our filament model (Figure 5C,D, Supplementary Figure S8), the PhePP-K2413 is close to F89 from the linker domain of the adjacent DMC1 protomer, suggesting that it is still able to make a cation–π interaction. Based on this, we purified the F89A variant of DMC1 (Supplementary Figure S2A), established using EMSAs and ATPase activity assays that it can polymerize on ssDNA as wild-type (Figure 5E, lanes 1–2, 5, Supplementary Figure S7C) and then tested the effect of PhePP on these filaments. The electrophoretic mobility of both wild-type and F89A DMC1 filaments was reduced upon addition of PhePP but not of PhePP-F2406A (Figure 5E, lanes 3–4, 6–7). While this could indicate that filaments are stabilized by PhePP independently of F89, we note that the mobility of DMC1-F85A ssDNA complexes is also reduced upon addition of PhePP. Since DMC1 F85A does not form filaments, we conclude that reduced mobility represents peptide binding rather than filament stability. Therefore, we directly visualized DMC1 (wild-type and variants) filaments on tailed (3′-overhang) ds/ssDNA substrates using SFM and on ssDNA using TEM (Figure 6). We found that PhePP but not its F2406A variant significantly increased the average length of the filaments (Figure 6A–F). This stabilizing effect was not observed when the DMC1 F89A variant was used, supporting the role played by F89 in DMC1 polymerization upon addition of PhePP (Figure 6B). We also compared the stabilizing effects of the shorter (F2s6) and the longer (F2s3) peptides using TEM, and observed similar effects, although a higher concentration of the short peptide was required to achieve the same magnitude (e.g. 8 μM versus 2 μM in Figure 6C–F). Since ITC (Supplementary Figure S2E) and previous GST pull-downs (20) did not reveal a difference between F2s3 and F2s6, it is possible that the longer peptide forms additional interactions specifically with the DNA-bound DMC1. Alternatively, difference in peptide production (recombinant versus synthetic), concentration estimation, and assay condition could contribute. Finally, analysis of the F2s6 variants by EMSA performed under the same TEM conditions (Figure 6G) confirmed the importance of the positively charged positions flanking the core PhePP motif for F2s6 binding to the DMC1 ssDNA filaments. EMSA experiments also showed that PhePP binds DMC1 filaments formed on ss and dsDNA similarly. Taken together with our structural analysis and pull-downs, these results demonstrate that PhePP binding is not only compatible with the recombinase oligomerization into a nucleoprotein filament, but actually depends on it and stabilizes it.
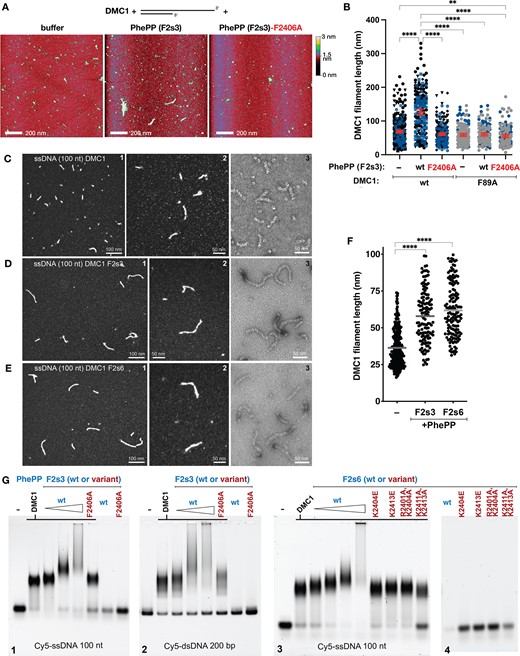
PhePP stabilizes DMC1 filaments. All experiments were performed under Ca-ATP conditions. (A) Representative images of the SFM scans of the complexes formed by DMC1 on a DNA molecule corresponding to resected DSB end, in the presence of the PhePP (F2s3) peptide, its F2406A variant or storage buffer as negative control. (B) Quantification of the wild-type and F89A DMC1 filament lengths in the presence of BRCA2 F2s3 (PhePP wild-type and F2406A variant), as analyzed by SFM. Symbol shapes and colors correspond to independent experiments (n = 2–4). Red lines indicate mean and 95% confidence intervals. Statistically significant differences (p< 0.05) are indicated, as determined by ANOVA with Kruskal–Wallis test. (C–E) TEM statistical analysis of DMC1 filaments. (C1–3) Representative views of DMC1–ssDNA (100 nt) filaments imaged using positive (C1-2) or negative staining (C3). (D1–3) Same as (C) but in presence of 2 μM F2s3. (E1–3) Same as (C) but in presence of 8 μM F2s6. (F) Statistical analysis of the filament lengths. (G) EMSA analysis of DMC1 filaments. Filaments were assembled in the same conditions as for TEM analysis in presence of various concentrations of F2s3 (1, 2 and 4 μM) and F2s3 F2406A mutant (4 μM) on the ssDNA (100 nt) (G1), or on the dsDNA (200 bp) (G2), in presence of various concentrations of F2s6 (2, 4, 8 and 15 μM) and F2s6 variants (8 μM) on the ssDNA (100 nt) (G3). The effect of peptides was also tested in absence of DMC1 on ssDNA (G4).
Discussion
The essential role of BRCA2 in somatic and meiotic HR stems from its interactions with the RAD51 and DMC1 recombinases mediated by short motifs in the central and C-terminal regions of BRCA2 (Figure 1). The core sequence of these motifs is four amino acids long with an invariant phenylalanine in the first position and a highly constrained or invariant residue in the fourth (alanine or proline). The recombinases are also very similar in sequence and interactions with BRCA2, and nearly identical structurally (44). Yet in meiosis, where both of them are present, they behave differently. To which extent these functional differences are intrinsic or are regulated by accessory proteins like BRCA2 was not clear. We recently described a mouse strain with a deletion in BRCA2 that resulted in crucial separation of function: DMC1 localization to meiotic DSBs in oocytes was abolished, but RAD51 was not. Here, we provide a molecular mechanistic explanation for that mouse phenotype. We determined the crystal structure of DMC1 in complex with the PhePP domain of the deleted region, analyzed the interaction computationally and biochemically and established its effect on DMC1 activity. The structure led to two key findings. The first is that despite the similarity in sequence, the phenylalanine-containing motifs bind to different sites. We classified the motifs and the corresponding interfaces into A- (FxxA) and P- (FxxP) and propose that our findings with PhePP–DMC1 extend to other P-motifs, as the recently identified P-motif in TR2. Second, we provide further evidence and molecular explanation to the opposite effects of the A- and P- motifs on the ability of the recombinase to oligomerize into filaments.
In our crystal structure, the P-site of the DMC1 ATPase domain contacts BRCA2 residues T2403-K2413 through a surface of about 500 Å2 (Figure 3A), which is half the size of the A-interface in the BRC4-RAD51 structure (35). Two BRCA2 residues, F2406 and P2409, contribute most to the P-interface (buried surface area per residue > 100 Å2), and their substitution to alanine impairs binding, as observed by pulldown and BLI (Figures 3C and 4G). The two lysines K2404 and K2413, as well as the F2410, are also buried in the structure of the complex (>70 Å2). The contribution of these lysines, as well as other conserved positively charged residues flanking the P-motif, to DMC1 binding, is supported by additional GST pull-down experiments, but the effect of their substitutions is smaller than for the P-motif residues and is masked at high concentrations (compare poorly expressed in bacteria, low concentration F2 substitution series in Supplementary Figure S4A to the stable, high concentration F2s3 series in Figure 3C). The absolute conservation of F2410 is arguably the most puzzling property of the PhePP motif, not explained by any of our data on PhePP–DMC1 interaction. This phenylalanine is unique to PhePP and not present in the other P-motifs (Figure 4E). Lastly, the PhePP peptide binds to the P-site of DMC1, which is accessible in the octameric form of DMC1 (Figures 3A, D). Substitutions of DMC1-G153 and G154 close to Phe2406 abolish binding. Moreover, mutating DMC1 F85 into alanine, which prevents DMC1 oligomerization, disrupts interaction of DMC1 with PhePP (Figures 3D and 5B).
From this structural analysis, we hypothesized that PhePP binding could further stabilize the DMC1 oligomers. Consistently, we experimentally found that F2s6 and F2s3 not only bind to the oligomeric form of DMC1, but also bind to and stabilize the DMC1 oligomers in the functional filament configuration (Figures 5E–G and 6A–G). In our model of the DMC1–ssDNA–PhePP complex, the PhePP domain interacts with the DMC1 ATPase domain through the same interface as observed in the DMC1–PhePP crystal structure. Experimentally, we showed that both F2406 and the positively charged residues flanking the P-motif significantly contribute to the binding of PhePP to the DMC1 ssDNA filaments, as observed by BLI and EMSA (Figures 5E-G, 6G). BRCA2 K2413 is an exception, as we saw its effect only on filament binding by BLI and EMSA, but not on octameric DMC1 binding by pulldown, even with a charge reversal F2413E substitution. Moreover, substitution of DMC1 D182 or D186, which are close to BRCA2 K2413 and could contribute to the main P-interface by salt bridge formation, have no effect on octameric DMC1 binding. On the opposite, substitution of DMC1 F89, also close to BRCA2 K2413, strongly affects DMC1 ssDNA filament stabilization, suggesting that the cation-π interaction between these two residues is critical for the function of the DMC1-BRCA2 interaction (Figure 6B).
While none of the PhePP–DMC1 contacts observed in the crystal structure involve the N-terminal DMC1 domain, PhePP poorly binds to DMC1 lacking first 81 amino acids. This could indicate that the protomer-spanning interface extends beyond the F89 position in the DMC1 linker domain and also involves the N-terminal domain (Figure 3E, Supplementary Figure S4D). Notably, previous crystallographic studies of octameric full-length DMC1 did not show electron density corresponding to the N-terminal domain of DMC1 (44,45), while we observed it in DMC1 chain A. This domain interacts with the ATPase domain of the adjacent (chain B) DMC1 monomer (Supplementary Figure S3B), which in turn interacts with a well-defined BRCA2 peptide. We did not find any crystal contact restraining the position of the observed N-terminal domain, suggesting that PhePP binding to the chain B ATPase domain stabilizes the (mainly electrostatic) interactions between this domain and the chain A N-terminal domain, as well as the position of the N-terminal domain. We conclude that the P-site extends beyond the DMC1 ATPase domain of one protomer to the linker of the adjacent protomer, with the N-terminal domain of the adjacent protomer being able to indirectly contribute to the binding interface. This favors the formation of the DMC1 nucleofilament and stabilizes it.
Our study benefited from the power of AlphaFold modeling. The prediction of a novel interaction mechanism for DMC1 and PhePP was unexpected, and we were unable to trace its origin to any known recombinase structure. It was fully corroborated experimentally by the crystal structure (Figure 2), mutagenesis (Figure 3) and functional assays (Figures 5 and 6). Therefore, based on additional AlphaFold modelling, sequence analysis and previously published data including our own, we hypothesized that other P-motifs will interact with recombinases via the same interface and with similar functional consequences. Among these, the P-motif of the TR2 domain is most notable. Our findings on PhePP–DMC1 are fully consistent with and suggest a molecular explanation to the seminal observations on RAD51-TR2 (30,31): preferential binding to oligomeric and reduced binding to monomeric form of recombinase and the stabilizing effect, which can counteract the disruptive effect of BRC. It is also supported by multiple studies that showed that the C-terminal domain of BRCA2 promotes RAD51 function and replication fork protection in cells. These biochemical and cell biological studies focused on phosphorylation of S3291, and the adjacent P-motif positioned next to it has only recently attracted attention(29). Thus, S3291 may exert its effect by modulating RAD51 interaction with the adjacent P-motif. The role of the conserved region of FIGNL1 L292-K346, containing both A-and P-motifs that might simultaneously bind to recombinases (as predicted by AlphaFold), is also intriguing. The phenylalanine of the A-motif is essential for binding RAD51, but the P-motif is also involved in this interaction (51). We consistently observed binding of the P-motif of FIGNL1 to DMC1. All this data suggests that the A- and P- motifs of FIGNL1 simultaneously bind to the contiguous A- and P-sites in RAD51 and DMC1, thus promoting the dissociation of the recombinases from nucleoprotein complexes at specific stages of HR.
The definition of two classes of recombinase-binding motifs, which bind to distinct but contiguous A- and P-sites, calls for re-evaluation of the previous data on recombinases and proteins that bind them and points to new experiments to explore them. The large sequence diversity of the A- and P-motifs, outside of their core FxxA and FxxP residues, suggests a variability in function (Figure 4E). The important arginine and lysine residues identified in PhePP are only partially conserved in TR2 and not conserved in the P-motifs of RAD51AP1 and FIGNL1, even if all these motifs are highly positively charged. How the distribution of these residues regulates binding of P-motifs to two adjacent recombinase protomers (π-cation interaction, involvement of the linker and the N-terminal domain) is still unclear. Thus, our work provides a molecular explanation to DMC1 disruption in Brca2Δ12–14 mouse and to RAD51- and replication fork-stabilizing effects described for the BRCA2 TR2/CTRD/CTRB and opens a new avenue of mechanistic questions on the disruptive versus stabilizing effect of A- and P-motifs outside of BRCA2. While our paper was under review, two studies have been published corroborating our key conclusions (66,67).
Data availability
The crystal structure was deposited at the PDB under the reference 8QQE.
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We thank Chloé Quignot for implementing the AlphaFold-multimer at Institute of Integrated Biology of the Cell (I2BC), Florine Dupeux and all the HTX Lab staff for the crystallogenesis and crystallography experiments, Vincent Kruit for help setting up the ATPase assay, and Magali Nicaise-Aumont on the Macromolecular Interaction Platform of I2BC for access and help when working on the BLI instrument. We acknowledge ESRF and SOLEIL for the provision of synchrotron radiation facilities.
Funding
S.M. and S.Z.J. were supported by funding from the French Infrastructure for Integrated Structural Biology [https://www.structuralbiology.eu/networks/frisbi; ANR-10-INSB-05-01]; European Community H2020 Program under the project iNEXT Discovery [871037]; S.M. also acknowledges funding by the Institut National de la Santé et de la Recherche Médicale (INSERM). Funding for open access charge: Erasmus Medical Center. This study was supported by the Oncode Institute, which is partly financed by the Dutch Cancer Society (KWF) and by Josephine Nefkens Cancer Program infrastructure support.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments