





Abstract
The ZFX transcriptional activator binds to CpG island promoters, with a major peak at ∼200–250 bp downstream from transcription start sites. Because ZFX binds within the transcribed region, we investigated whether it regulates transcriptional elongation. We used GRO-seq to show that loss or reduction of ZFX increased Pol2 pausing at ZFX-regulated promoters. To further investigate the mechanisms by which ZFX regulates transcription, we determined regions of the protein needed for transactivation and for recruitment to the chromatin. Interestingly, although ZFX has 13 grouped zinc fingers, deletion of the first 11 fingers produces a protein that can still bind to chromatin and activate transcription. We next used TurboID-MS to detect ZFX-interacting proteins, identifying ZNF593, as well as proteins that interact with the N-terminal transactivation domain (which included histone modifying proteins), and proteins that interact with ZFX when it is bound to the chromatin (which included TAFs and other histone modifying proteins). Our studies support a model in which ZFX enhances elongation at target promoters by recruiting H4 acetylation complexes and reducing pausing.
Introduction
The ZFX family is a small set of human C2H2 zinc finger proteins that belong to the class of multiple-adjacent zinc finger proteins (1,2). The ZFX family has 3 family members, ZFX, ZFY and ZNF711, that are conserved throughout evolution. ZFX is located on the X chromosome and contains 13 C2H2-type zinc fingers in its DNA binding domain. ZFY is encoded on the Y chromosome and thus there is only one copy in the diploid human genome (3). The two proteins have 96% overall similarity and 99% similarity in their zinc finger domains, which suggest they may have a similar function (4). In contrast, ZNF711 (which is also located on the X chromosome) has less overall similarity compared to ZFX and ZFY and has amino acid changes that eliminate the structure of the third and seventh finger (Supplementary Figure S1A). The N terminal domain of ZNF711 also is not as highly related to ZFX as is the N terminal domain of ZFY.
Of the three family members, only ZFX has been associated with tumorigenesis. In fact, the high expression of ZFX found in many types of cancers (including prostate cancer, breast cancer, colorectal cancer, glioma, renal carcinoma, gastric cancer, gallbladder adenocarcinoma, non-small cell lung carcinoma, and laryngeal squamous cell carcinoma) has been associated with poor patient survival (5–14). Accordingly, we have previously shown that knockout of all ZFX family member activity in 293T cells greatly impairs cell proliferation (4).
Approximately half of the annotated human C2H2 zinc-finger proteins contain a KRAB repressor domain that recruits histone methylase complexes to create silenced chromatin (15,16); therefore, many C2H2 proteins are repressors of transcription (17). In contrast, the ZFX family members do not have a KRAB domain and our previous studies suggest that ZFX activates, rather than represses, transcription (4). However, the mechanism by which this family of transcription factors (TFs) regulates transcription remains poorly understood.
RNA Polymerase 2 (Pol2)-mediated gene regulation begins with the recruitment of components of the general transcriptional machinery (composed of the multi-protein complexes TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH) to the core promoter, which is defined as a region ±50 bp from the transcription start site (TSS) of a gene (18,19). The majority of core promoters in the human genome are located in CpG islands, defined as genomic regions extending upstream and downstream from the TSS that have high GC content and a high density of CpG dinucleotides. We have previously reported that ZFX, ZNF711 and ZFY bind at the majority of active CpG island promoters in multiple cell lines (4,20).
Although core promoters display only modest activity, promoter activity can be increased when site-specific TFs bind proximal to (and generally upstream of) the core promoter or to distal enhancers. TFs can enhance promoter activity by stabilizing the recruitment of the general transcriptional machinery and/or can positively affect promoter activity by recruiting specific co-regulators (such as histone modifying complexes) to the core promoter either by direct protein-protein interactions (when bound in the proximal promoter region) or via long-range chromatin looping (when bound to distal regulatory elements). In addition to pre-initiation complex (PIC) assembly and recruitment of histone modifying complexes, release of promoter-proximal paused Pol2 to productive elongation is another key step in regulating gene expression. Transcription elongation is regulated by numerous factors including transcription factors and co-regulators, elongation factors, histone chaperones, and chromatin modifiers and remodelers (21). According to the current model, Pol2 transcribes ∼30–60 bp downstream of the TSS and then pauses in a complex containing negative elongation factors NELF and DSIF. Upon recruitment of P-TEFb (a heterodimer of cyclin T and CDK9) and the subsequent phosphorylation of NELF, DSIF and Ser 2 on the Pol2 CTD, Pol2 escapes the pause site and transcribes through the gene body (22).
Because ZFX, ZNF711 and ZFY bind ∼200–250 bp downstream from transcription start sites (4,20), it is possible that these TFs are involved in regulating the productive elongation of Pol2. In this study, we investigate the function of ZFX in regulating transcriptional elongation using GRO-seq to profile transcriptionally engaged Pol2 genome-wide after ZFX knockdown. Additionally, we use TurboID-MS to characterize protein interactions that require the ZFX N-terminal domain and those that require chromatin-bound ZFX. Our findings support a model in which ZFX stimulates H4 lysine acetylation and Pol2 pause release.
Materials and methods
Cell culture
Human female kidney 293T (ATCC #CRL-3216) and female glioblastoma LN-229 (ATCC #CRL-2611) cells were obtained from ATCC (https://www.atcc.org/). DKO cells are derived from 293T but, after CRISPR-mediated deletions, lack expression of all ZFX family members (4). 293T and LN-229 were grown in DMEM supplemented with 10% fetal bovine serum (Thermo Fisher #10437036) plus 1% penicillin and 1% streptomycin at 37°C with 5% CO2. Cell lines were authenticated via the STR method and validated to be mycoplasma free using a universal mycoplasma detection kit (ATCC #30-1012K).
siRNA knockdowns
LN-229 cells were transfected in triplicate with 100nM of siRNA oligonucleotides targeting human ZFX (Horizon #L006572000005), ZNF593 (Horizon #L017089020050) or control oligonucleotides (Horizon #D0018101005) using Lipofectamine RNAiMAX (Thermo Fisher #13778150) for 72 h. The knockdown efficiency for each target gene is shown in Supplementary Figure S4.
Transfection of DKO cells with wt and mutant ZFX expression vectors
Three plates of 15cm2 DKO cells were transfected with 30ug of plasmid DNA with Lipofectamine 3000 Transfection Reagent (Invitrogen #L3000008) for 48 h.
Chromatin immunoprecipitation sequencing (ChIP-seq)
Previously published ZFX ChIP-seq data from 293T (4) was used. ZFX, ZNF593, H3K4me3, H4K5ac, H4K8ac and H4K12ac ChIP-seq experiments were performed using two 15 cm2 plates of LN-229 cells transfected with siRNAs as described above. Cells were crosslinked in 1% formaldehyde for 10 min followed by quenching with 0.14 M glycine for 30 min at room temperature. Crosslinked cells were lysed in ChIP lysis buffer (1% SDS, 50 mM Tris–HCl pH 8, 20 mM EDTA, c0mplete Protease Inhibitor EDTA-free tablet (Roche #11836153001); lysis volumes were based on cell pellet volumes. Sonication was carried out with a Diagenode Bioruptor Pico using conditions to generate fragments 400–800 bp in length. After sonication, 50–100 μg soluble chromatin was diluted 1:10 in ChIP Dilution Buffer (16.7 mM Tris–HCl, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl), pre-cleared for 1 h at 4°C with 30 ul of Magnetic Protein A/G beads (Pierce, #88803), and incubated overnight with antibody (volume per IP: 5 μl anti-ZFX, 10μl anti-ZNF593, 10 μl anti-H3K4me3, 25 μl anti-H4K5ac, 8 μl anti-H4K8ac and 25 μl anti-H4K12ac) (Supplementary Table S1A). The next morning, magnetic Protein A/G beads (Pierce, #88803) were added for an additional 2 h of incubation. Bead-immunocomplexes were washed twice for 5 min with each of the following buffers in order: Wash Buffer A (50 mM HEPES pH 7.9, 0.1% SDS, 1% Triton X-100, 0.1% deoxycholate, 1 mM EDTA, 140 mM NaCl), Wash Buffer B (50 mM HEPES pH 7.9, 0.1% SDS, 1% Triton X-100, 0.1% deoxycholate, 1mM EDTA, 500 mM NaCl), LiCl buffer (20 mM Tris–HCl pH8, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA, 250 mM LiCl), and TE (50mM Tris–HCl pH 8, 1 mM EDTA). Elution was performed in 150 μl of elution buffer (50 mM Tris–HCl pH8, 1 mM EDTA, 1% SDS) then ChIP samples and inputs (10 μl of precleared chromatin lysis plus 140 μl elution buffer) were reverse crosslinked overnight at 65°C. Samples were RNase A treated for 1 h at 37°C and Proteinase K treated for 2 h at 56°C and DNA was extracted with phenol/chloroform and ethanol precipitated. DNA pellets were resuspended in 12 μl of TE.
For FLAG ChIP, three plates of 15 cm2 DKO cells were transfected as described as above. 100 μg soluble chromatin and 12.5 ul of FLAG antibody (Supplementary Table S1A) were used for each IP. The rest of the ChIP protocol was performed as previously described (4,20).
ChIP-seq libraries were prepared using the KAPA HyperPrep kit (Roche #KK8503) following the manufacturer's protocol. Samples were sequenced on a Novaseq 6000. All ChIP-seq data were aligned to hg38 using Bowtie2 software (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Only reads that aligned to a unique position in the genome with no more than two sequence mismatches were retained for further analysis. Duplicate reads that mapped to the same exact location in the genome were counted only once to reduce clonal amplification effects. Normalization was done across samples using an equal number of uniquely mapped reads. All ChIP-seq experiments were performed in duplicate and the duplicates were compared using IDR (23) for the transcription factors and histone H3K4me3. For the spreading histone marks (H4ac), duplicates were compared to the ENCODE datasets (Supplementary Figure S6). ChIP-seq peaks were called using MACS2 (https://github.com/taoliu/MACS), followed by identifying common peaks between duplicates using bedtools (https://bedtools.readthedocs.io/en/latest/). Average tag density plots were generated using CEAS software (http://liulab.dfci.harvard.edu/CEAS). A list of the ChIP-seq experiments is shown in Supplementary Table S1A and ZFX promoter-bound genes are provided in Supplementary Table S2.
Global run-on sequencing (GRO-seq)
For the knockdown experiments, LN-229 cells were transfected as described above. After 72 h, 5 × 106 cells per nuclear run-on reaction were harvested and incubated in swelling buffer (10 μM Tris–HCl, 2 mM MgCl2, 3 mM CaCl2). Nuclei were isolated with lysis buffer (10 μM Tris–HCl, 2 mM MgCl2, 3 mM CaCl2, 10% glycerol, 1% NP-40). Nuclear run-on was performed at 30°C for 5 min in 10 mM Tris–HCl pH 8, 5 mM MgCl2, 300 mM KCl, 1 mM DTT, 500 μM ATP, 500 μM GTP, 500 μM Br-UTP, 2 μM CTP, 200 U/ml Superase In RNase Inhibitor (Invitrogen #AM2696), and 1% sarkosyl. Nuclear RNA was isolated with Trizol (Invitrogen #15596026). RNA was purified with Micro Bio-Spin P-30 Gel Columns (Bio-Rad # 7326250), fragmented with RNA Fragmentation Kit (Invitrogen # AM8740), and treated with 15 units mRNA decapping enzyme (NEB # M0608S) and 30 units T4 PNK (NEB # M0201S). RNA immunoprecipitation was performed 3 times with anti-BrdU-conjugated agarose beads (Santa Cruz Biotechnologies # sc-32323 AC). Library preparation was performed with TruSeq Small RNA Library Preparation Kit (Illumina # RS-200-0012 following the manufacturer's protocol. Samples were sequenced on a Novaseq 6000). Reads were trimmed using Trimgalore and aligned to hg38 using Tophat. Gro-seq experiments were performed in duplicates and normalization was done across samples using an equal number of uniquely mapped reads. Pause index analysis was done with Bedtools multicov. A list of the GRO-seq experiments is shown in Supplementary Table S1C.
RNA-seq
RNA-seq experiments were performed in triplicate and LN-229 and DKO cells were transfected as described above Total RNA was extracted using DirectZol RNA MiniPrep kit (Zymo #R2052) following the manufacturer's protocol. RNA integrity was checked using RNA 6000 Nano kit (Agilent Technologies #50671511) on a 2100 Bioanalyzer (Agilent Technologies #G2939AA). The RNA-seq libraries were prepared and sequenced by Novogene. Samples were sequenced on a Novaseq 6000 with 150 bp pair-ended reads. RNA-seq results were aligned to the human genome GRCh38 using STAR and quantified using HTseq and Rsem. Differentially expressed genes with absolute log2 fold change >0.58 and q-value cut-off of 0.05 were determined using DEseq2. A list of the RNA-seq experiments is shown in Supplementary Table S1B and lists of differentially expressed genes after knockdown of ZFX, ZNF93 or both proteins are provided in Supplementary Table S3.
Plasmid construction
Construction of ZFX zinc finger deletion mutants
ZFX mutant expression constructs were generated by amplifying the ZFX-Myc-DDK expression vector (Origene #RC214045) using primers with 15 bp complementary overhangs flanking different ZFs to create constructs containing ZF11-13 (4) or ZF12-13. The resulting constructs were transformed into CopyCutter™ EPI400™ Chemically Competent E. coli (Lucigen #C400CH10) and induced to high copy number according to the manufacturer's protocol. Plasmids were purified using Qiagen miniprep kit (Qiagen #D4068) and the deletions were validated via Sanger sequencing. Primers used for cloning and sequencing are listed in Supplementary Table S1.
Construction of ZFX-Turbo plasmids
ZFXTurbo plasmids described in this study were generated using the Gibson assembly protocol from existing commercial plasmids. The parent mammalian expression vector is pCMV6-Entry Origene (PS100001). Primers were ordered from Integrated DNA Technologies, with desalting the sole purification method used. All amplification reactions were carried out using Platinum SuperFi II 2X master mix (Invitrogen #12361010). Following PCR, the presence of the expected size fragments was verified by agarose gel electrophoresis. Fragments were purified by magnetic beads (Ampure XP, BeckmanCoulter #A63881) and quantified by spectroscopy. Purified fragments were combined at a molar ratio of 5:1 insert:vector for the Gibson reaction, utilizing the Gibson Assembly Master Mix (New England Biolabs #E2611). Following incubation, a portion of the reaction was transformed into CopyCutter competent cells (Lucigen #C400CH10) using the manufacturer's recommended protocol. Final plasmid clones were Sanger sequenced to insure sequence fidelity through the construction procedure. The sequences of all primers using in cloning can be found in Supplementary Table S1E.
Wild-type ZFX-TurboID (Farnham lab clone #2427)
The TurboID open reading frame sequence was amplified from 3xHA-miniTurbo-NLS_pCDNA3 (Addgene Plasmid #107172) using oligonucleotides A and B. The plasmid backbone including the ZFX coding region was amplified from the expression vector ZFX (NM_003410) Human Tagged ORF Clone from Origene (cat. #RC214045) using primers C and D. DNA sequence coding for two copies of the peptide GGGS was introduced into the amplification products to provide a flexible linker between ZFX and the TurboID biotin ligase.
ZFX[1-8]-TurboID (Farnham lab clone #2425)
The wild-type ZFX-TurboID plasmid was used as a starting plasmid to create a construct in which the TurboID ligase was fused to ZFX truncated after the 8th (of 13) zinc fingers. Amplification was carried out using primers E and F; Gibson assembly of the resulting single amplified backbone generated an in-frame deletion of ZFX zinc fingers 9–13 in the expressed protein.
ZFX[NTD]-TurboID (Farnham lab clone #2426)
This plasmid contains only the amino terminal domain (NTD) of ZFX fused to the TurboID ligase. Amplification was carried out using primers G and E and the full length wild-type ZF-TurboID plasmid as the starting material. Gibson assembly of this single resulting amplified fragment generated an in-frame deletion of all thirteen ZFX zinc fingers.
ZFX[FO]-TurboID (Farnham lab clone #2427)
This construct contains only the 13 zinc fingers of ZFX (FO, Fingers Only) fused to the TurboID biotin ligase. Because removing the entire amino terminal domain of the ZFX protein removes the putative nuclear localization sequence (NLS) of ZFX, an exogenous NLS from the nucleoplasmin gene was included to help guide this protein containing the ZFX 13 zinc fingers to the nucleus. The first cloning step involved Gibson assembly combining an amplified nucleoplasmin NLS sequence (obtained from a dCas9 expression vector, using primers H and I) with an amplified backbone from the Origene ZFX expression plasmid using primers J and K (use of these primers removes the NTD of ZFX). The resulting plasmid construct, which fuses the nucleoplasmin NLS with the zinc finger region of ZFX, was then amplified with primers M and N and combined with an amplified plasmid backbone product from the original Origene wt ZFX plasmid (which was created using primers N and O and does not contain any ZFX sequence).
Transactivation assays
The biological activity of the ZFX-TurboID protein was assayed following plasmid construction. Also, N-terminal and finger region deletion ZFX constructs were for tested for transactivation activity. Briefly, DKO cells (4) were transfected with expression plasmids or the parent vector using Lipofectamine 3000 (ThermoFisher #L3000015) according to manufacturer's instructions. After 24 h, cells were lysed in TRI Reagent (Zymo #R2050-1-200) and RNA was recovered by precipitation. Total RNA was converted to cDNA using iScript (Bio-Rad #1708841BUN). RT-qPCR was carried out using SYBR on a BioRad CFX 1000. Data points represent results from triplicate wells and duplicate RT-qPCR readings. Primers used to monitor expression of endogenous genes are provided in Supplementary Table S1D.
TurboID-MS
At least three biological replicates were performed for each Turbo construct assayed in LN-229 cells. LN-229 cells were grown to 60–70% confluence in two 15 cm dishes in DMEM supplemented with 10% HyClone dialyzed FBS (Cytiva #SH30079.03). Cells were transfected with 40 μg wt or mutant ZFX-Turbo or HA-Turbo plasmids using Lipofectamine 3000 (Thermo Fisher #L3000075) for 48 h. Biotin was added to a final concentration of 0.5mM. Biotinylation was stopped after a 15 min pulse by placing cells on ice. Cells were washed 2× with cold PBS, and nuclei were isolated using cell lysis buffer (5 mM PIPES pH 8, 85 mM KCl, 1% NP-40) followed by lysis of nuclei with RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholate, 1 mM EDTA, c0mplete Protease Inhibitor EDTA-free tablet (Roche #11836153001). 100 μl of Dynabeads MyOne Streptavidin T1 beads (Invitrogen #65601) washed 3× in PBS were added to 1mg of nuclear lysate and incubated for 30 min at room temperature. Beads were washed 3× in high salt RIPA buffer (50mM Tris pH 7.4, 0.8M NaCl, 1% NP-40, 0.25% deoxycholate, 1 mM EDTA, c0mplete Protease Inhibitor EDTA-free tablet (Roche #11836153001). Beads were washed 3× in 50mM ammonium bicarbonate. On-bead trypsin digest was carried out with 0.5 ul sequencing grade modified Trypsin (Promega #V5111) overnight at room temperature.
Mass spectrometry
Mass spectrometry was performed and analyzed at the UC Davis Genome Center Proteomics Core Facility (https://proteomics.ucdavis.edu/). Samples were run in data-independent mode on the Orbitrap Fusion Lumos (Thermo Fisher) or the Orbitrap Exploris 380 (Thermo Fisher) mass spectrometers. Data was analyzed using Spectronaut software (https://biognosys.com/software/spectronaut). Filtered interaction candidates (Supplementary Table S4) were determined using q-value <0.05 and log2 fold change >1 followed by removal of cytoplasmic proteins and contaminating proteins found in the Contaminant Repository for Affinity Purification (CRAPome; https://reprint-apms.org/) with >20% frequency. All hits prior to filtering are also listed in Supplementary Table S4. EnrichR (https://maayanlab.cloud/Enrichr) was used for gene ontology analyses.
Results
ZFX binds to thousands of promoters and activates hundreds of genes in LN-229 cells
Our goal in this study is to investigate the mechanisms by which ZFX regulates transcription. As noted above, ZFX is a member of a family of three C2H2 zinc finger transcription factors (ZFX, ZFY and ZNF711) that have very similar DNA binding domains (Supplementary Figure S1A); in our previous studies, we have shown that ZFX, ZFY and ZNF711 all bind to very similar sets of promoters in a given cell type (4). Multiple members of the ZFX family are expressed in most human tissues, making it difficult to disentangle the function of individual family members. Therefore, to reduce any potential competition between ZFX and the other family members in our current study, we used the female glioblastoma cell line LN-229 which has robust ZFX expression, but no detectable ZFY or ZNF711 expression (Supplementary Figure S1B). Another contributing factor in our selection of LN-229 cells was the discovery of preferential ZFX expression in glioma stem cells (GSC) in glioblastoma cases and the previous finding that ZFX knockdown inhibits GSC tumor growth and increases survival in mice with GSC-derived xenografts (10).
To characterize ZFX binding in LN-229 cells, we first performed ZFX ChIP-seq. Similar to what we previously observed in 293T cells, ZFX binds primarily in promoter regions (defined as ± 2kb from TSSs) in LN-229 cells (Figure 1A). The ChIP-seq tag density plots are also consistent with our previous studies of ZFX binding in 293T cells (4,20); specifically, the averaged ZFX peak signal is at the unusual location of + 200 to + 250bp downstream of TSSs, with a shoulder upstream at −200–250 bp (Figure 1B). It is important to note that our previous studies demonstrated that ZFX-regulated genes predominantly have ZFX binding in the downstream location (4,20). The majority of ZFX peaks in LN-229 cells are at the same promoters as in 293T cells (Figure 1C); a list of all ZFX-bound genes in LN-229 cells is provided in Supplementary Table S2.
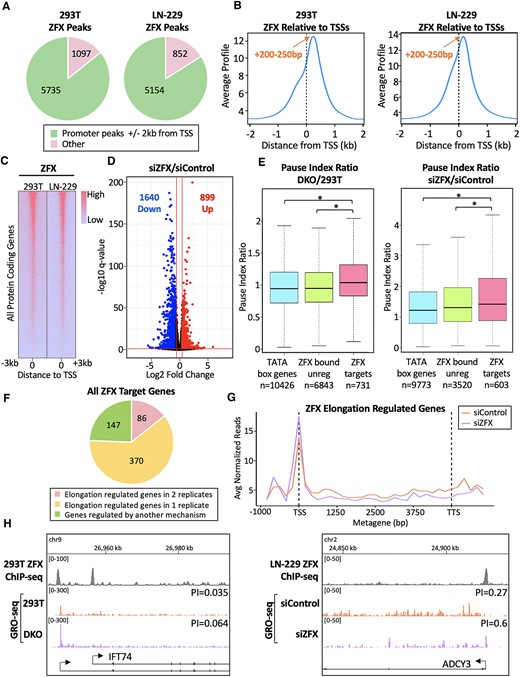
ZFX activates target genes by stimulating Pol2 pause release. (A) Shown are the numbers of ZFX peaks located inside or outside of promoter regions (defined as ± 2kb from TSSs) in 293T and LN-229 cells. (B) Average tag density profiles of ZFX ChIP-seq data centered at TSSs in 293T and LN-229 cells. (C) Heatmap of ZFX binding at all protein coding genes in 293T and LN-229 cells, ranked from high to low ZFX ChIP-seq signal in 293T cells. (D) Volcano plot showing RNA-seq data after siRNA-mediated knockdown of ZFX in LN-229 cells, compared to siControl; numbers of down- and up-regulated genes identified using Log2 FC >±0.58 and q-value <0.05 cut-offs are shown. (E) Left: Average pause index ratio (DKO/293T cells) at TATA box genes (blue), ZFX bound but not regulated genes (green), and ZFX target genes (pink); the numbers of genes in each category are shown. Right: Same as left, but plotting the pause index ratio of the indicated gene sets in siZFX- vs siControl-treated LN-229 cells. (F) Number of ZFX target genes regulated during elongation (PI ratio > 1.25). (G) GRO-seq metagene analysis of ZFX target genes regulated during elongation. (H) ZFX ChIP-seq and GRO-seq browser tracks at a ZFX target gene in 293T and DKO cells (left) and in LN-229 cells transfected with siC or siZFX (right).
We next tested the effects of ZFX on the regulation of the transcriptome in LN-229 cells. To do this, we performed RNA-seq after siRNA-mediated knockdown of ZFX. Changes in the transcriptome are reported using a volcano plot (Figure 1D). Differentially expressed genes with absolute log2 fold change >0.58 and q-value cut-off of 0.05 were determined using DEseq2; a list of all downregulated genes is provided in Supplementary Table S3. As expected, thousands of differentially regulated genes were identified after ZFX knockdown. The majority of these genes are downregulated, consistent with the classification of ZFX as a transcriptional activator (4,20). In this study, we refer to active, protein-coding genes that are both bound by ZFX and downregulated upon siZFX knockdown as ‘ZFX target genes’. In LN-229 cells, we identified 603 (of the 1640 down-regulated genes) as ZFX target genes; a list of these genes is provided in Supplementary Table S3. We note that ZFX binds to essentially the same set of promoters in all cells, but regulates a different subset of those promoters in different cell types (data not shown). Therefore, in the experiments described below we have compared ‘ZFX bound and regulated promoters’ (i.e. ZFX target genes) to ‘ZFX bound but not regulated’ promoters (see Figure 5C for an example of ZFX binding to both classes of these promoters).
Reduction of ZFX results in increased promoter-proximal Pol2 pausing at target genes
Because ZFX family members bind downstream of transcription start sites (4,20), it is possible that these TFs are involved in regulating the productive elongation of Pol2. We began by examining the effects of permanent loss of all ZFX family member expression upon transcription elongation. We have previously shown that eliminating expression of both ZFX and ZNF711 (which bind to the same promoters) in 293T cells causes larger gene expression changes and increased defects in cell proliferation than observed in the single ZFX or ZNF711 knockouts (4). Therefore, we used CRISPR-mediated ZFX and ZNF711 double knockout clones (referred to as DKO cells) to investigate ZFX family member-mediated transcription regulation. We performed GRO-seq to determine the amount of Pol2 pausing genome-wide. GRO-seq involves a nuclear run-on containing the detergent sarkosyl, which prevents new instances of Pol2 PIC assembly and re-initiation. The nuclear run-on reaction occurs in the presence of the UTP analog bromouridine-triphosphate (Br-UTP), which is incorporated into nascent RNAs by transcriptionally engaged Pol2. RNA immunoprecipitation then enriches for Br-UTP containing transcripts, which are sequenced to reveal the position, amount, and orientation of transcriptionally engaged Pol2 (24). We calculated the pausing index (PI) by dividing the number of GRO-seq reads in the promoter region (transcription start site to +200) by the number of GRO-seq reads in the gene body (+201 to the transcription termination site). All GRO-seq read counts and PIs are listed in Supplementary Table S7. We compared the PI ratio (DKO PI/293T PI) in 3 groups of active (FPKM > 0.5), protein-coding genes having >5 GRO-seq reads in the promoter region. Because ZFX binds to the majority of CpG island promoters and not TATA box promoters (20), we used TATA box-containing promoters as a control, along with ZFX-bound, but unregulated, genes. We found that, upon loss of ZFX family members, the average PI ratio at ZFX target genes is significantly higher when compared to both of these control groups (Figure 1E (left). Increased PIs at individual genes in DKO cells were also observed (Figure 1H (left) and Supplementary Figure S2B); higher PI indicates that permanent loss of ZFX and ZNF711 increases pausing at ZFX target promoters.
Because the permanent deletion of ZFX and ZNF711 may have resulted in severe, long-term effects on the cells, we also performed two biological replicates of GRO-seq after transient siRNA-mediated knockdown of ZFX in LN-229 cells (which do not express ZNF711 or ZFY). In both replicates, we found that the PI ratio (siZFX/siControl) at ZFX target genes is also significantly higher than in both control groups (Figure 1E (right) and Supplementary Figure S2C), indicating that even a short-term reduction in ZFX results in increased pausing at target promoters. Increased PIs after ZFX knockdown were also observed at individual genes (Figure 1H (right) and Supplementary Figure S2A). Out of 603 ZFX target genes, 370 were identified as being regulated during transcriptional elongation (PI ratio > 1.25) in either replicate, and 86 were identified in both replicates (Figure 1F). Additionally, a metagene analysis of the GRO-seq data in these 86 genes resulted in a clear increase in paused Pol2 and decreased elongation after siZFX (Figure 1G).The remaining 147 genes were not regulated during elongation in either replicate and are presumably regulated by another mechanism, likely during initiation. Taken together, these data suggest that ZFX regulates expression of target genes by stimulating paused Pol2 to release and continue elongation.
The first 11 C2H2 zinc fingers of ZFX are dispensable for DNA binding and transcriptional activity
Our previous studies showed that the last three zinc fingers (ZF11-13) of ZFX are sufficient for DNA binding and transactivation at a similar level as wt ZFX (4). These results are consistent with the current model that DNA binding by a zinc finger protein requires at least 3 adjacent zinc fingers, with each finger recognizing 3 or more bases in the major groove of the DNA helix. To test this concept and to extend our understanding of the mechanisms by which ZFX is recruited to chromatin, we created and assayed a ZFX deletion mutant having only two adjacent zinc fingers for DNA binding and activation activity. First, we constructed a FLAG-tagged ZFX expression vector containing the full length N-terminal domain and zinc fingers 12–13 (called ZFX[12–13]) (Figure 2A). FLAG ChIP-seq was performed following transfection of DKO cells with the ZFX[wt] and ZFX[12–13] constructs. Remarkably ZFX[12–13] demonstrated DNA binding, although impaired compared to wt ZFX (Figure 2B,C). Overall, ZFX[12–13] binds to the same promoters as wt ZFX (Figure 2C), at the same binding position +200–250 bp downstream of TSSs.
![ZFX zinc fingers 12–13 are sufficient for ZFX binding and transcriptional activation. (A) Diagrams of ZFX zinc finger mutants; the N-terminal domain and FLAG tag are represented by grey bars, zinc fingers are represented by grey arrows, and deleted zinc fingers are represented by grey dashes. (B) Tag density plot showing average wt ZFX and ZFX [12–13] ChIP-seq data near TSSs in transfected DKO cells. (C) Heatmap of wt ZFX and ZFX [12–13] binding at all protein coding genes, ranked from high to low signal in cells transfected with wt ZFX. (D) Volcano plots showing RNA-seq data from DKO cells transfected with wt ZFX and ZFX[12–13], compared to the empty vector control; numbers of up-regulated genes identified using Log2 FC>±0.58 and q-value <0.05 cut-offs are shown. (E) Venn diagram showing overlap of genes activated by wt ZFX and ZFX[12–13].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/52/12/10.1093_nar_gkae372/1/m_gkae372fig2.jpeg?Expires=1747862438&Signature=Orxg--0yhS4oPHWNltFb1iyPKjwKkXSPgJhxdzzzpwWdEVFWPYxKtwDZdi5aXTHqVH6G9yncbDFXMFeVqyYg50dsKNRu5m~PGg2RuGxFLX7IhwBg7fDVBwoDKrP-d7OQnFK4P4Arzr5Q5sxVl8mi58CS87IhiKINYXEMB5VZjcy8Q3vTsEIr~2P-2yIeeQ4tk10ngmf992-LAwkCGE~Sq0u1xKKuP-bWYpn2zOAoF1ZlbrnmsDCN4fCJ-uLx1~yIHyVRO-xMTWO-WkdYL~90Cv-0whrDegcr7jK~0J0nFttpGV29olmjMHcu6ZfEDFP1luxahs1yrc8ZxKR2RUE2-Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ZFX zinc fingers 12–13 are sufficient for ZFX binding and transcriptional activation. (A) Diagrams of ZFX zinc finger mutants; the N-terminal domain and FLAG tag are represented by grey bars, zinc fingers are represented by grey arrows, and deleted zinc fingers are represented by grey dashes. (B) Tag density plot showing average wt ZFX and ZFX [12–13] ChIP-seq data near TSSs in transfected DKO cells. (C) Heatmap of wt ZFX and ZFX [12–13] binding at all protein coding genes, ranked from high to low signal in cells transfected with wt ZFX. (D) Volcano plots showing RNA-seq data from DKO cells transfected with wt ZFX and ZFX[12–13], compared to the empty vector control; numbers of up-regulated genes identified using Log2 FC>±0.58 and q-value <0.05 cut-offs are shown. (E) Venn diagram showing overlap of genes activated by wt ZFX and ZFX[12–13].
We next asked if ZFX [12–13] is capable of activating gene expression in DKO cells. To answer this question, we performed RNA-seq after transfection of DKO cells with the ZFX [12–13] construct or the empty vector control. Surprisingly, ZFX[12–13] activated genes nearly the same number of genes as did as wt ZFX (1728 versus 2072) (Figure 2D), with 1137 genes being activated by both ZFX proteins. (Figure 2E).
ZNF593 is identified as a ZFX interacting partner (ZIP) by TurboID, and functions to recruit ZFX to chromatin
Regulation of gene expression by TFs requires the coordinated interactions of multiple proteins. Different combinations of TFs are known to bind cooperatively to specific proximal promoter sequences using their DNA binding domains and synergistically effect transcriptional regulation (25). Most eukaryotic TFs also have unstructured activation or repression domains that recruit co-activators or co-repressor proteins (26). These transcription co-factors include proteins which function to modify histones and/or other protein methylases or kinases that mediate processes such as chromatin remodeling (27). Co-activators and co-repressors lack the ability to bind to DNA on their own, relying on their interactions with site-specific DNA-binding TFs to regulate transcription (26,27). Thus, identifying proteins that interact with ZFX is crucial to understanding its mechanism of transcriptional regulation.
One commonly used method to identify protein-protein interactions is co-immunoprecipitation (co-IP), using an antibody specific to a ‘bait’ protein to also pull down a ‘prey’; the prey can then be identified using Mass Spectrometry (MS). However, this method can fail to precipitate binding partners that interact weakly or transiently to the bait protein, and its success depends on antibody binding to an epitope that may be occluded when the two proteins interact (28). Therefore, to obtain a comprehensive profile of ZFX binding partners, we instead used a sensitive proximal protein labeling technique called TurboID MS (29). The TurboID biotin ligase is more catalytically active than other enzymes used in similar assays, including BioID and BioID2, resulting in ∼3–6-fold more biotinylation within 30 min, within a radius of ∼35 nm (30,31).
To perform TurboID MS, we first constructed an expression vector encoding ZFX fused to the Turbo biotin ligase at its C-terminus and a vector solely expressing the HA-tagged Turbo biotin ligase as a control (Figure 3A). To test the functionality of the ZFX-Turbo expression construct, LN-229 cells were grown in dialyzed, biotin-free cell culture media and transfected with either the ZFX-Turbo or HA-tagged Turbo expression plasmids for 48 h. Biotin was then added to a final concentration of 0.5mM for a 15-min pulse of biotinylation. Expression of both fusion proteins in the LN-229 cells was validated via western blotting with an anti-BirA antibody, which detects the Turbo protein (Supplementary Figure S3A). Western blotting with streptavidin confirmed that the biotinylation activity by ZFX-Turbo was functional, as compared to mock transfected cells (Supplementary Figure S3B). To confirm that the transcriptional activity of ZFX was still intact when fused to the biotin ligase, we performed a transactivation assay using a known ZFX target promoter, LORNF2 (4). DKO cells were transfected with constructs expressing wt ZFX, an inactive ZFX mutant with the zinc fingers deleted (ZFX[NTD]; this ZFX mutant protein does not bind to DNA), or ZFX-Turbo. Importantly, our results show that ZFX-Turbo activates LORNF2 to an even greater extent than does wt ZFX, and that activation by ZFX[NTD] is equal to that of the empty vector control (Supplementary Figure S3C).

ZFX and ZNF593 cooperatively bind DNA. (A) Schematic of wt ZFX-TurboID and HA-TurboID control constructs. (B) Functional classification of ZIPs identified using wt ZFX-TurboID in LN-229 cells. (C) Enrichment of ZNF593 in ZFX-TurboID experiments in 293T, LN-229, U87 and C42B cells. (D) Heatmap of ZFX and ZNF593 ChIP-seq data at promoters of all protein coding genes, ranked by high to low ZFX binding. (E) Average ZNF593 tag density in siControl- (green) and siZFX-treated (orange) cells. (F) Average ZFX tag density in siControl- (pink) and siZNF593-treated (purple) cells. (G) Volcano plot showing down- and up-regulated genes after ZNF593 knockdown, compared to siControl; numbers of genes identified using Log2 FC >±0.58 and q-value <0.05 cut-offs are shown. (H) Same as (F), but with simultaneous ZFX and ZNF593 knockdown. (I) ZFX (pink) and ZNF593 (green) ChIP-seq signals after ZNF593 and ZFX knockdown, respectively; also shown are RNA-seq tracks after ZFX and ZNF593 knockdown (blue). (J) Heatmap of RNA-seq data after siRNA-mediated knockdown of ZFX or ZNF593, or simultaneous knockdown of both ZFX and ZNF593, as compared to treatment with control siRNAs. Genes are ranked from the most down-regulated (top, blue) to most upregulated (bottom, pink) in Log2 fold change in FPKM after knockdown. (K) Overlap of downregulated genes after siZFX, siZNF593 or simultaneous siZFX + siZNF593 knockdowns.
Having confirmed that ZFX-Turbo can activate a known target promoter, we performed TurboID in LN-229 cells. Following transfection and the biotinylation pulse, enrichment of biotinylated ZFX-proximal proteins from nuclear extracts was performed using streptavidin-coated beads. Proteins were subsequently trypsin-digested and identified using MS. Valid ZFX-interacting proteins were determined by comparison of proteins biotinylated in cells expressing ZFX-Turbo versus HA-Turbo, using ≥3 biological replicates. The identified proteins were filtered by eliminating contaminating cytoplasmic proteins and those found in the Contaminant Repository for Affinity Purification Proximity Dependent Biotinylation dataset (32) with ≥20% frequency. ZFX-proximal proteins were chosen based on q-value <0.05 and log2 fold change >1. For simplicity, we refer to ZFX-Turbo-enriched proteins as being ZIPs (ZFX-interacting proteins). However, we recognize that we cannot infer a direct interaction with ZFX from this proximal-dependent labelling technique. A list of all proteins identified as ZIPs in LN-229 cells as well as the filtered, high-confidence lists (see Methods), can be found in Supplementary Table S4). We found enrichment of proteins classified as TFs, including ZFX (presumably due to auto-biotinylation of ZFX-Turbo), transcription co-factors, and RNA-binding proteins (Figure 3B). Gene ontology analyses of the ZIPs resulted in enrichment of proteins involved in transcriptional processes (Supplementary Table S5).
We also performed ZFX-Turbo in 293T (human embryonic kidney), C42B (prostate cancer), and U87 (glioblastoma) cells, identifying proteins involved in transcriptional regulation in each cell type (data not shown). Strikingly, the only protein we identified as a ZIP in every one of the tested cell lines including LN-229 was the transcription factor ZNF593 (Figure 3C). Therefore, we were interested to characterize the relationship between ZNF593 and ZFX. ZNF593 is a small (∼16kd) protein that contains one C2H2 zinc finger flanked by about 40 disordered amino acid residues on each terminus (33). Very little is known about the function or activity of ZNF593. However, it has been shown to have a negative effect on binding of the OCT2 transcription factor using an in vitro gel mobility shift assay (34). As one C2H2 finger is unlikely to confer DNA-binding activity and there are no other known DNA binding domains in the protein, it is possible that ZNF593 may interfere with OCT2 binding via protein-protein interactions. No other studies of the role of ZNF593 in regulation of transcription have been reported. Therefore, we have investigated the genomic binding pattern of ZNF593 and whether it has a positive or negative effect on ZFX function.
We first performed ZNF593 ChIP-seq in LN-229 cells. Interestingly, the strongest ZNF593 peaks were found at the same promoters as the strongest ZFX peaks (Figure 3D). Next, we plotted the average ZNF593 ChIP-seq tag density relative to all TSSs. We found that ZNF593 has an almost identical binding pattern as ZFX, with an average location of binding +200–250 bp downstream of the TSS (Figure 3E, green line). This evidence for ZFX and ZNF593 promoter co-localization led us to ask if knockdown of ZFX has an effect on ZNF593 recruitment and vice versa. We performed ZNF593 ChIP-seq after ZFX knockdown and observed a decrease in ZNF593 binding compared to control (Figure 3E, I, and Supplementary Figure S4B). We then performed ZFX ChIP-seq after ZNF593 knockdown and observed an even greater decrease in ZFX binding compared to control (Figure 3F, I, and Supplementary Figure S4B). It is important to note that ZNF593 expression was not affected by ZFX knockdown, and ZNF593 expression was not affected by ZFX knockdown (Supplementary Figure S4A). These results demonstrate that the decreased DNA binding of each protein is because of defective recruitment, not downregulation of gene expression.
We also asked if ZFX and ZNF593 binding have additive effects on transcriptional activation. We performed RNA-seq after ZNF593 knockdown, and compared the results to a simultaneous knockdown of both ZFX and ZNF593. We found 992 downregulated genes after ZNF593 knockdown (Figure 3G), and 1717 downregulated genes after the ZFX and ZNF593 double knockdown (Figure 3H). When comparing the fold change in FPKM for all protein coding genes, the most downregulated genes after ZNF593 knockdown were also the most downregulated after ZFX knockdown (Figure 3J). At individual genes, decreased ZFX and ZNF593 binding correlated with downregulation of these genes, demonstrating that binding of both proteins have direct effects on activation of target genes (Figure 3I, Supplementary Figure S4B). Many of these genes were also highly downregulated after the double knockdown (Figure 3J). By comparing the downregulated genes in all 3 knockdown conditions, we identified 842 genes that require only ZFX, 486 genes that require only ZNF593, 129 genes that require both, and 638 genes that require either one for activation (Figure 3J, K), suggesting individual, cooperative, and redundant functions of these two factors.
ZFX interacts with co-regulatory complexes via its acidic N-terminal domain and components of the general transcription machinery through proximal DNA interactions
As noted above, not all ZIPs will be DNA binding partners or even directly interact with ZFX. Instead, it is likely that many of the identified proteins are part of large regulatory complexes involved in ZFX-mediated transcription. To begin to develop an overview of ZFX-mediated transcription, we first asked if any of the ZIPs we identified are known to interact with each other. We used esyN (http://www.esyn.org/) (35) to build a protein interaction network of all LN-229 ZIPS, based on publicly available interaction data. Interestingly, our analyses of the LN-229 ZIPs identified 49 proteins that have been previously demonstrated to have known interactions (out of the total of 66 identified ZIPs; see Supplementary Table S6). Interestingly, these ZIPs coalesce into three groups of known interactors (Figure 4C).
![Using TurboID, the ZFX NTD is required for identification of COMPASS family and NSL complexes, and chromatin binding is required for identification of TAFs and the TIP60 complex. (A) Diagrams of various ZFX constructs fused to the TurboID biotin ligase. The N-terminal domain is represented by grey bars and the zinc fingers present in each construct are represented by grey arrows. The regions of ZFX required to detect the different groups of interacting proteins shown in panel C are also indicated. (B) Heatmap showing the ZIPs identified with each ZFX mutant protein; protein groups identified in panel C are labeled next to the corresponding clusters. (C) Network of protein interactions among ZFX interacting proteins; blue circles indicate proteins identified with wt ZFX and ZFX[FO], green circles indicate proteins identified with wt ZFX, ZFX[1–8] and ZFX[NTD, and white circles indicate proteins that do not fall into either group.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/52/12/10.1093_nar_gkae372/1/m_gkae372fig4.jpeg?Expires=1747862438&Signature=N53iO32DTsTakNUkbTGdrc2EKubXHQxqABUcKFZ2EuNLx4ZksgeoqOPRPr8LgSd88d~az-NoUkDkZ8Uw8zSKAK391sJrbFH4ZyBYItJXbaAsoTIfUIchEDltk6z7wQXAsXhYKRxqodMKg9LHyzWjRrml1MleRyifBt-kPdT5-VnxeARCqSSK4E8z1jNCo1S9MiRvP16t6jfRGLvgliR7c2~rEbS6cYCbxfChLetW9bsezglrCGF2W3R781qpx~H6sUpZNabHea9dr5pmDJRyaf4oakGCX-hVEOEBwONM4~SjxpmJ61M~zQ94z0Jsy5ab~GekB81SG3b04Rog8OVy-Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Using TurboID, the ZFX NTD is required for identification of COMPASS family and NSL complexes, and chromatin binding is required for identification of TAFs and the TIP60 complex. (A) Diagrams of various ZFX constructs fused to the TurboID biotin ligase. The N-terminal domain is represented by grey bars and the zinc fingers present in each construct are represented by grey arrows. The regions of ZFX required to detect the different groups of interacting proteins shown in panel C are also indicated. (B) Heatmap showing the ZIPs identified with each ZFX mutant protein; protein groups identified in panel C are labeled next to the corresponding clusters. (C) Network of protein interactions among ZFX interacting proteins; blue circles indicate proteins identified with wt ZFX and ZFX[FO], green circles indicate proteins identified with wt ZFX, ZFX[1–8] and ZFX[NTD, and white circles indicate proteins that do not fall into either group.
Group 1 contains subunits of the COMPASS family of H3K4 methyltransferase complexes. These complexes mediate H3K4 mono-, di- and tri- methylation, the last of which is a mark of transcriptionally active promoters (36). Two core subunits belonging to all COMPASS and COMPASS-like complexes are in group 1, WDR5 and ASH2L (37–39). Importantly, we also identified SETD1A and KMT2A (also called MLL1), the catalytic subunits of two distinct COMPASS complexes, SETD1/COMPASS and MLL1/2 COMPASS-like, respectively (39–42). Other COMPASS complex-specific subunits were present in group 1 including CXXC1, which is specific to the SETD1/COMPASS complex, and the tumor suppressor protein MEN1, which is specific to the MLL1/2 COMPASS-like complex (43,44). The cell proliferation factor HCFC2, which is a subunit of both COMPASS complexes, was also found in group 1 (45). Further, the identification of the H3K4me3 reader PHF8, a H3K9me2/me1 demethylase, was consistent with the previously shown interaction of PHF8 interaction with the ZFX family member ZNF711 (46,47). Fittingly, the chromatin remodeler CHD8 that recruits H3K4 methyltransferases was also identified in group 1 (48). Components of the lysine acetyltransferase complex NSL were also found to interact within group 1, including KANSL1/3 and MCRS1, and WDR5 (49). Notably, the NSL complex is thought to work in tandem with COMPASS family of complexes to promote H3K4me2 (50). The two complexes share the subunits WDR5 (identified in Group 1), OGT and HCF1, suggesting that they may even form a larger complex together (49).
Group 2 includes components of the general transcriptional machinery, namely TAFs 1–9, which, along with TBP, constitute the DNA-binding general transcription factor TFIID (51). Using ZFX mutant proteins in the TurboID experiments, we demonstrated that ZFX interacts with the TAFs only when it is bound to DNA. This suggests that it is the co-localization of ZFX and the TAF complex on the promoter region that is necessary for biotinylation of TAFs by ZFX-Turbo. A previous study has shown that TAFs, unlike components of the other general TFs, can bind downstream of the start site (52), perhaps explaining how ZFX, which has the majority of its binding sites downstream of the TSS, can efficiently biotinylate the TAFs, but not other components of the general transcriptional machinery (which bind upstream of or at the TSS). It is also possible that the lack of detection of other general TFs or of other proteins implicated in pausing (such as NELF or DSIF) may be because the biotinylatable residues of these proteins are not within the appropriate distance (∼35 nm) to the Turbo fusion protein or are not accessible to the biotin ligase.
Group 3 was enriched in components of the histone acetyltransferase and chromatin remodeling TIP60 complex, including the catalytic subunit KAT5, EPC1/2, TRRAP, MBTD1, EP400 and BRD8 (53,54). The TIP60 complex is known to acetylate lysines on histones H2A and H4 (H4K5, H4K8, H4K12 and H4K16) (55). Interestingly, the NSL complex (identified in Group 1) also contains KAT5 and has been shown to be capable of acetylating H4K5, H4K8 and H4K16 (56). The ability to classify the ZIPs into known regulatory complexes demonstrate the validity of the TurboID data.
It is possible that ZFX is in proximity to certain ZIPs only when it is bound to DNA but in proximity to others before binding to the genome. ZIPs that require genomic co-localization may be lost if DNA-binding mutants of ZFX are used in the TurboID experiments, whereas ZIPs that require an intact transactivation domain may be lost if the N-terminus of ZFX is deleted. To delineate the regions of ZFX that interact with the identified ZIPs, we performed TurboID with a series of ZFX deletion TurboID constructs (Figure 4A). We have previously established that a ZFX protein lacking fingers 9–13 cannot bind to chromatin (4). Thus, we first created ZFX [1–8]-Turbo] which includes the full length NTD but lacks zinc fingers 9–13. We also created ZFX[NTD], which lacks the entire zinc finger domain and therefore should also lack DNA binding activity. As shown in Supplementary Figure S3C, ZFX[NTD] has no transactivation ability. We next created ZFX[FO]-Turbo, which encodes only the zinc fingers. Although the activation domain has not yet been fully mapped in the ZFX protein, the N-terminus has a high occurrence of acidic residues (ZFX amino acids 1–425 have a pI of 3.94), a common characteristic of activation domains (57). Therefore, we expect that ZFX[FO]-Turbo can bind to chromatin, but not activate transcription. We performed TurboID-MS experiments in triplicate for each mutant ZFX protein and determined which of the ZIPs required DNA binding or the NTD for identification in the biotinylation experiments.
First, we identified protein interactions that only occur when ZFX is bound to DNA (blue ZIPs) (Figure 4B,C). We found that proteins from group 2 (TAFs 3, 4, 5, 6 and 8) and group 3 (BRD8 and EPC2) require DNA binding activity of ZFX for identification in the TurboID experiments. In contrast, group 1 was enriched for proteins that require the ZFX NTD for identification in the TurboID experiments (green ZIPs) (Figure 4B,C). Specifically, 2 subunits of the SETD1/COMPASS complex (SETD1A and CXXC1), 2 subunits of the NSL complex (KANSL1 and KANSL3), and ASH2L, ASXL1, BAP1, CHD8 and KDM1B require the ZFX NTD for identification in the TurboID experiments.
Preferential acetylation of H4 lysines via ZFX co-activator recruitment drives transcriptional activation of target genes
Recruitment of co-activators, including histone-modifying complexes, by site-specific transcription factors is a crucial mechanism in facilitating transcriptional regulation. Because our TurboID experiments identified components of the COMPASS methyltransferase complex and the NSL and Tip60 acetyltransferase complexes, we reasoned that ZFX may bring these chromatin modifiers to target promoters, stimulating histone modifications.
As shown above, we identified components of the COMPASS methyltransferase complex as ZIPs. This complex mediates trimethylation of histone H3 at lysine 4 (H3K4me3), which is associated with open chromatin and is highly enriched at promoters of active genes (38). Therefore, we hypothesized that ZFX may function to regulate transcription via recruitment of COMPASS complexes, increasing the levels of H3K4me3. To test this, we performed H3K4me3 ChIP-seq in LN-229 cells before and after ZFX knockdown. We observed a decrease in H3K4me3 for the subset of highly ZFX bound target genes (n = 301) (Figure 5A, orange versus pink). However, ZFX knockdown also resulted in a decrease in the average H3K4me3 levels in the set of all active promoters, which suggests a small global decrease of H3K4me3 (Figure 5A, green versus purple). These results suggest that although ZFX may recruit COMPASS complex components to promoters, H3K4 methylation is not specifically correlated with ZFX-mediated gene regulation.
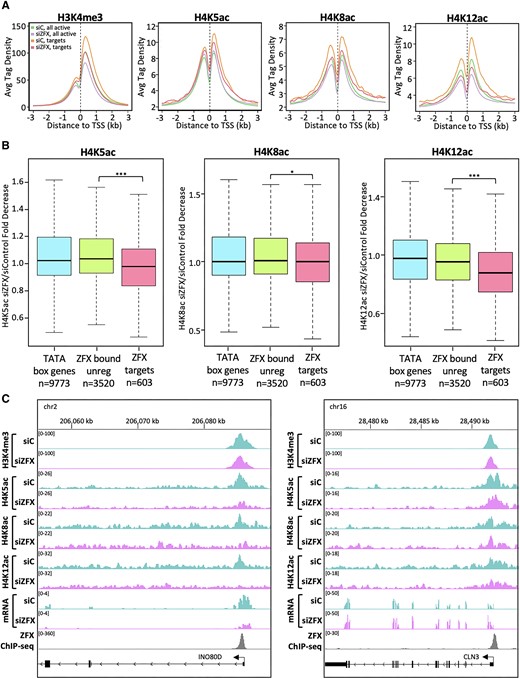
Reduction of ZFX results in decreased H4Kac at ZFX target genes. (A) Average H3K4me3 and H4K5/8/12ac tag density in siControl- and siZFX-treated cells at promoters of all active coding genes (siControl:green and siZFX:purple) and at promoters having robust binding of ZFX in control LN-229 cells (siControl:orange and siZFX:pink). (B) Boxplot showing fold decrease of H4K5ac, H4K8ac, and H4K12ac tags at TATA box promoters (blue), ZFX bound but not regulated promoters (green), and ZFX target genes (pink). (C) H3K4me3, H4K5ac, H4K8ac, H4K12ac and ZFX ChIP-seq and RNA-seq signals a ZFX target gene (left) and a ZFX bound, but unregulated gene (right).
We next focused on the histone acetylating complexes NSL and TIP60. We chose to assess the levels of NSL/TIP60-mediated H4K5ac and H4K8ac (56,58), and TIP60-mediated H4K12ac (58,59) before and after ZFX knockdown. The subset of highly ZFX-bound target promoters had greater H4K5/8/12ac compared to the average level of all active genes in the control (Figure 5A, orange versus green). This was not observed with the subset of target promoters weakly bound by ZFX, suggesting that levels of H4K5/8/12ac correlate with the degree of ZFX binding (Supplementary Figure S5A). After ZFX knockdown, average levels of H4K5/8/12ac at target promoters strongly bound by ZFX decreased to a larger extent than that observed at all active genes. (Figure 5, drop from orange to pink vs. drop from green to purple). These results demonstrate that these marks are preferentially written at highly ZFX bound target promoters and subsequently have a greater magnitude of decrease after ZFX knockdown.
While there are ∼600 ZFX target genes in LN-229 cells, there remain thousands of genes that are bound by, but not regulated by ZFX. Therefore, we asked if relatively greater decreases in H4K5/8/12ac are correlated with ZFX binding and regulation, rather than just ZFX binding alone. To test this, we compared the fold decrease in H4K5/8/12ac ChIP-seq tags at ZFX target promoters to TATA box promoters and promoters that are ZFX bound, but not regulated. For H4K5/8/12 but especially H4K5ac and H4K12ac, we found ZFX-bound target genes have significantly greater fold decreases than both control groups (Figure 5B). At individual ZFX bound genes, we observed that the decreases in H4K5/8/12ac correlated with an mRNA decrease whereas there weren’t significant changes in H3K4me3 regardless of whether gene expression is regulated by ZFX (Figure 5C). Overall, these data strongly suggest that the mechanism by which ZFX activates transcription involves acetylation of H4 lysines, especially K5 and K12, but not methylation of H3K4me3.
Discussion
ZFX is a potent transcriptional activator that binds at the unique location of +200–250 bp downstream of the start sites of active CpG island promoters (4,20). This binding location suggests that ZFX may regulate transcription at a step subsequent to initiation. Here, we used GRO-seq to investigate the function of ZFX in regulating transcriptional elongation. After eliminating ZFX family members in 293T cells and transiently decreasing ZFX levels in LN-229 cells, we calculated the average pausing index at ZFX target genes. In both cases, our data suggest that ZFX stimulates release of paused Pol2 at ZFX target genes.
In this study, we have expanded on our previous findings that the first 10 zinc fingers of ZFX are not required for DNA binding or transactivation (4). Surprisingly, we now show that a truncated ZFX protein having only the last two fingers (fingers 12 and 13) can also associate with chromatin and transactivate target genes. These findings defy the usual expectation that three adjacent zinc fingers are required for DNA binding (1,2). However our ChIP-seq data is consistent with published data from an in vitro filter binding assay showing that zinc fingers 12 and 13 from mouse ZFY, a highly similar ZFX family member, can bind DNA (60). Interestingly, our ZFX-TurboID experiments identified the single zinc finger-containing protein ZNF593 as a ZIP in multiple cell lines. Additionally, our data support a model in which ZFX and ZNF593 are co-localized on chromatin, cooperate to activate gene expression, and that ZNF593 helps (directly or indirectly) to bring ZFX to its binding site downstream of promoters. Because CpG island promoters are very GC rich, the other ZFX zinc fingers may also enhance stabilization of ZFX. This is a possible explanation why loss of ZFX zinc fingers 1–11 does not change the location of the binding, but simply makes binding weaker. This is consistent with our previous ChIP-exo data (4) which demonstrated an average of 2–3 ZFX ChIP-exo peaks per promoter, suggesting multiple alternate DNA contact points of ZFX with a promoter region.
Characterization of ZFX protein interactions is crucial for understanding its mechanism of promoting transcriptional elongation. Therefore, we used the proximity labeling technique, TurboID-MS, to identify ZIPs in LN-229 cells. We identified DNA binding proteins, components of chromatin-modifying complexes, and components of the general transcription machinery. Using published protein interaction databases, we found that the ZIPs from LN-229 cells can be classified into three interaction groups, plus additional site-specific DNA binding proteins not previously associated with any of the other ZIPs.
Our TurboID experiments with ZFX mutants determined that several histone-modifying complexes associate with ZFX through either the NTD (e.g. Tip60) or are proximal to ZFX when it is bound to DNA (e.g. COMPASS and NSL). We note that several histone acetylation marks have previously been shown to be involved in transcriptional elongation, such as H3K9ac (61) and H3K18/27ac (62,63). Our follow-up ChIP-seq experiments examining levels of H4K5/8ac (marks mediated by NSL and Tip60) and H4K12ac (a mark mediated by Tip60) revealed that reduction of ZFX results in a preferential decrease in H4K5/12ac levels at ZFX target promoters, as compared to the promoters of other active genes. Our results presented here are consistent with the demonstrated interaction between H4K5ac and the super elongation complex (SEC) via interaction with its AF9 and ENL (61). We also identified BRD8 (a subunit of the TIP60 complex) and DIDO1 in our ZFX-TurboID experiments. In yeast, the BRD8 and DIDO1 homologs (Brd8 and Bye-1, respectively) are known to interact and Bye1 is known to bind stimulate Pol2 pause release (64). Therefore, based upon our TurboID and ZFX knockdown experiments, we propose a connection between binding of ZFX, H4 acetylation, and productive transcriptional elongation. However, further studies would be needed to show the detailed mechanism of histone acetylation in ZFX-mediated gene activation.
Our TurboID experiments also identified components of the COMPASS complex as ZIPs; this complex is responsible for placing the H3K4me3 mark at active promoters. Although knockdown of ZFX caused a small global change in H3K4me3, the decrease was not larger at ZFX target promoters. Interestingly, a non-enzymatic domain of SETD1A (the primary catalytic component of COMPASS complexes), has been demonstrated to regulate paused Pol2 release when bound just downstream of TSSs, without altering levels of H3K4me3. SETD1A-mediated pause release is achieved by interacting with cyclin K, which recruits CDK9 (a component of P-TEFb) to the promoter region. (65). Therefore, it is possible that a ZFX-COMPASS interaction also contributes to release of paused Pol2 independent of effects on levels of H3K4me3.
In summary, we have investigated the mechanisms by which ZFX regulates transcription. We have defined which zinc fingers of ZFX are required for chromatin binding and have identified another zinc finger protein (ZNF593) that contributes to robust recruitment of ZFX to CpG island promoters. We have also identified histone-modifying proteins that are proximal to ZFX before (e.g. SETD1A) and after (e.g. EPC2) ZFX is bound at the promoter regions. In all, our studies suggest that one mechanism by which downstream binding of ZFX regulates transcription is to stimulate release of a paused Pol2 to stimulate productive elongation.
Data availability
All ChIP-seq and RNA-seq data generated in this study (summarized in Supplementary Table S1) have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/); accession GSE241284. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.proteomexchange.org) with the dataset identifier PXD044709.
Supplementary data
Supplementary Data are available at NAR Online.
Acknowledgements
We thank Novogene for sequencing RNA-seq and ChIP-seq samples, the USC Center for High Performance Computing (hpc.usc.edu), the UC Davis Proteomic Core Facility for mass spectrometry analyses, Azenta for assistance with plasmid sequencing, and the USC Office of Research. We thank S. Frietze, University of Vermont Medical Center, for the 3xHA-miniTurbo-NLS_pCDNA3, Addgene Plasmid #107172 and Z. Wu and S.K. Rhie, USC Keck School of Medicine, for assistance with RNA-seq analysis. Graphical abstract created using BioRender.com.
Author contributions: Conceived and designed the experiments: E.H. and P.J.F. Designed and performed experiments: E.H., K.H., Y.L., C.M.N. and S.S. Performed data analysis: E.H. and N.R.Z. Wrote and edited the manuscript: E.H. and P.J.F.
Funding
National Institutes of Health [1R01GM133450, P30CA014089, F32CA264890]; Margaret Kersten Ponty Endowed Postdoctoral Fellowship Award in Oncology Research at the Norris Comprehensive Cancer Center. Funding for open access charge: National Institutes of Health [1R01GM133450].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments