





Abstract
Family A DNA polymerases (PolAs) form an important and well-studied class of extant polymerases participating in DNA replication and repair. Nonetheless, despite the characterization of multiple subfamilies in independent, dedicated works, their comprehensive classification thus far is missing. We therefore re-examine all presently available PolA sequences, converting their pairwise similarities into positions in Euclidean space, separating them into 19 major clusters. While 11 of them correspond to known subfamilies, eight had not been characterized before. For every group, we compile their general characteristics, examine their phylogenetic relationships and perform conservation analysis in the essential sequence motifs. While most subfamilies are linked to a particular domain of life (including phages), one subfamily appears in Bacteria, Archaea and Eukaryota. We also show that two new bacterial subfamilies contain functional enzymes. We use AlphaFold2 to generate high-confidence prediction models for all clusters lacking an experimentally determined structure. We identify new, conserved features involving structural alterations, ordered insertions and an apparent structural incorporation of a uracil-DNA glycosylase (UDG) domain. Finally, genetic and structural analyses of a subset of T7-like phages indicate a splitting of the 3′–5′ exo and pol domains into two separate genes, observed in PolAs for the first time.
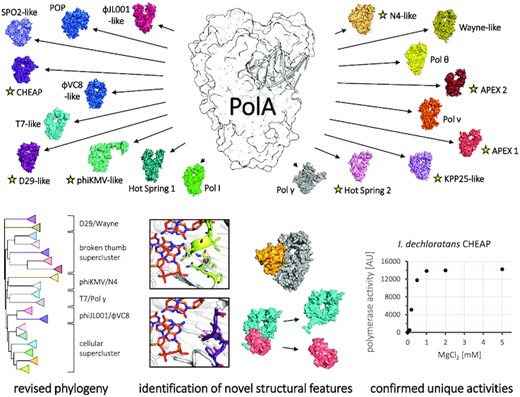
We distributed PolA sequences into 19 clusters, identifying eight novel subfamilies (yellow star). We present revised PolA phylogeny, describe novel structural idiosyncrasies and confirm functionality of two new bacterial subfamilies.
INTRODUCTION
In the course of evolution, nucleic acids emerged as the universal information carriers of life. The ‘central dogma’ of molecular biology describes how this information flows from DNA to RNA (and back), and from RNA to proteins (1). Despite having an auto-replicative potential that may have played a role during the very origins of life (2), contemporary nucleic acids are efficiently replicated by protein enzymes in a condensation reaction of nucleotide triphosphates, exploiting Watson–Crick base pairing with the templating strand as the fundamental mechanism for replication fidelity. DNA is the dominant support of genetic information found in all cellular organisms, as well as in an important fraction of the virus world, and its synthesis in cellulo relies on a variety of DNA polymerases (3).
There are eight distinct families of DNA polymerases (DNAPs or Pols): A, B, C, D, X, Y, PrimPol (AEP superfamily) and reverse transcriptases (4,5). While the first seven are DNA dependent, the last one uses RNA as a template; with the exception of PrimPol and several members of family B (6,7), all families need RNA or DNA primers to initiate replication. Some of the eight families are specialized in processive replication, whereas others are involved in re-priming this replication at blocked replication forks or in the repair of DNA damage (4,7). Family A polymerases (PolAs)—the first DNA polymerase family to be discovered and biochemically characterized (3)—essentially consist of a polypeptide chain folded into a polymerase (pol) domain and a proofreading 3′–5′ exonuclease (exo) domain (8); the additional 5′–3′ exonuclease domain is dispensable and does not interfere with the polymerase function and fidelity. While in cellular organisms PolA enzymes perform roles pertaining to whole-genome replication, recombination or repair (9–11), they also efficiently duplicate DNA of eukaryotic organelles (mitochondria and plastids), bacterial plasmids (12) and bacteriophages (e.g. T3/T7, SPO1 and SPO2) (13–15). Due to their relative simplicity, they are routinely used in diverse DNA amplification techniques (16,17), for instance in polymerase chain reaction (PCR)-based diagnostics against the severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2), which recently caused a major global health crisis (18). The archetypal member of the PolA family is Escherichia coli Pol I DNA polymerase and the derived so-called Klenow fragment, which lacks the 5′–3′ exonuclease domain.
Intriguingly, up to now PolAs represent the sole known family of DNA polymerases with representatives that moved away from the strict classical Watson–Crick base paring scheme. These polymerases (called DpoZ) operate in ZTGC-DNA phages, and have evolved a shifted specificity towards 2-aminoadenine (Z) instead of adenine at the templating thymine's Watson–Crick edge (19,20). Other phages, such as YerA41, developed PolA variants that are able to accept hypermodified thymine nucleotides in a templating strand during the replication of the phage DNA, which cannot be processed by commercially available polymerases (21). However, phages with similar modifications on 5-hydroxymethylcytosine, such as glucosylation (phages T-even: T2, T4 and T6) or arabinosylation (phage RB69), possess dedicated PolBs instead of PolA (22,23).
At the structural level, all PolAs share the Klenow fold, first identified in the X-ray structure of the Klenow fragment of E. coli DNA polymerase I (8). DNA polymerases B, Y, PrimPol and reverse transcriptases, as well as phage-like single-subunit RNA-dependent RNA polymerases, also possess the Klenow fold and are evolutionarily interconnected (24). On the other hand, families X and C share a different nucleotidyltransferase fold, the Polβ fold, while PolDs have yet another fold most commonly found in multisubunit RNA polymerases (the so-called double-psi β-barrel fold) (4).
The PolA family has been explored in the past using various phylogenetic analyses. Nevertheless, such classifications are either outdated (25), or focus only on a particular subfamily (26,27) or a particular group of biological entities (28–30). These methods rely heavily on multiple sequence alignments (MSAs), which are challenged by the divergent nature of very large datasets of sequences and vary in accuracy depending on the algorithm used, demanding specialized solutions for different cases (31,32). Lastly, the resulting phylogenetic trees assume a priori a fixed (and common) mutation rate—a synchronized molecular clock—for all PolA sequences; yet, a significant departure from this hypothesis is expected there, due to the divergent molecular specializations or horizontal transfers between carrier species having various life cycles, among other factors (33).
A non-hierarchical sequence clustering based on the Fruchterman–Reingold algorithm (34) avoids the above issues altogether. It aims not to provide exact phylogenetic relationships between the particulars, but rather to project and regroup them in Euclidean space based on their pairwise similarities. In short, the algorithm first randomly distributes the sequences in three-dimensional (3D) space, and then updates the position of each sequence by using pseudo-forces derived from the resemblance of each pair, until convergence is achieved. It was implemented in a computer program CLANS (35), which employs the BLAST algorithm (36) to assign the similarity scores to each sequence pair. CLANS has recently been used to investigate family B DNA polymerases (PolBs) and superfamily AEP, complementing the phylogenetic analyses and leading to the discovery of new subfamilies (37,38).
Inspired by these findings, we applied this clustering analysis to PolAs, using an up to date and fully comprehensive library of sequences. We confirmed its accuracy by correctly delineating known PolA subfamilies, unifying them in one global distribution that still captures their reported relationships. We distinguished five previously uncharacterized major groups, and three minor ones showing high similarity to other subfamilies. For each new PolA cluster, we determined its composition, occurrence and phylogenetic connections with other subfamilies. We used AlphaFold2 (39), the most recent and powerful protein structure prediction program (40), to investigate the architecture of the eight new and four still structurally undescribed PolA subfamilies. Consistent, high-confidence structural predictions revealed novel structural features, in the form of ordered insertions, domain assimilation or exo/pol gene splitting. Additionally, we determined that representatives of a known hot spring-associated subfamily consistently appear in archaea as well, demonstrating the presence of these PolAs across all domains of life. Finally, we tested the catalytic activity of the enzymes from two previously unexplored bacterial groups. Both act as a templated DNA polymerase, as expected, yet display distinct polymerase and exonuclease activity levels as well as different divalent metal ion dependencies.
MATERIALS AND METHODS
PolA protein sequence acquisition and clustering
All 60 975 sequences tagged as DNA polymerase A (HMMER-defined PFAM ID: PF00476 (41)) in the UniProt database (42) were downloaded in October 2021 and filtered, removing fragmentary (shorter than 350 amino acids) or incomplete (containing residue X) sequences. Using the BLAST algorithm (version 2.2.26) (36), the remaining sequences were compared against each other to assess their likeness; sequences with identity >70% were further removed from the dataset. The final selection (8109 sequences) was manually supplemented with several described PolAs of interest, sequences with a known 3D structure as well as all available DpoZ sequences (φVC8-like and Wayne-like) with pairwise identity <90%, due to their current under-representation.
The FASTA file containing 8136 final sequences was processed by the CLANS web-utility from the MPI Bioinformatics Toolkit (43). The clustering simulation was conducted using the Java version of CLANS (35) with default parameters. The sequences were randomly distributed in 3D space, converging to individual clusters after several hundred steps of the simulation. The simulation was let to run for a total of 6000 steps, during which no further modification of the positions appeared. The simulation was run without applying a P-value cut-off: additional simulations with cut-offs of 10−10 and 10−20 resulted, respectively, in an identical cluster distribution but with a reduction in size for most clusters, or further shrinking and fragmentation of the clusters. Clusters were determined with the network-based clustering tool, with a minimum of 40 sequences per cluster and active offset. Clusters φVC8-like and Wayne-like were selected manually, although they can be detected automatically using a lower threshold of at least 20 sequences per cluster. Several, independent simulations converged to almost identical cluster distributions, with no discrepancy regarding the critical details. Simulations not enriched with additional φVC8-like/Wayne-like DpoZ sequences resulted in equivalent distributions, barring the smaller size of clusters #18 and #19.
Similarities with PolAs of interest outside of the dataset were routinely assessed with BLAST searches at the NCBI (44).
Sequence analysis, structure prediction and phylogeny
Protein sequences making up each cluster were extracted with CLANS. For every subfamily, the sequences were aligned with Clustal Omega (45) applying default parameters. The alignments were used to generate sequence logos with WebLogo (46), or were displayed directly with ESPript3 (47).
The existing PolA structures were found through UniProt (42); completeness of the dataset was confirmed with Dali (48) queries. A local version of ColabFold (49) running 18 iterations of the AlphaFold2 algorithm (39) was used to predict PolA 3D models for all sequences selected for phylogenetic analysis (five per cluster) without a crystallographic structure. The highest ranking models of each run were superposed and analysed: they converged towards similar conformations—especially close within the clusters—and obtained high predicted local distance difference test (pLDDT) confidence scores, generally in the [70, 98] range. Known and predicted structures were visualized with Pymol (50).
Five representatives of each cluster were selected to create an MSA that comprises all clusters/subfamilies. Due to misalignments of additional, unrelated domains, all 95 sequences were truncated to their Klenow-like large fragments, i.e. the core PolA fold (3′–5′ exo and pol domains), based on PDB and AlphaFold2 structural models. The MSA was constructed on the truncated sequences in Clustal Omega with default parameters. The program also calculated a Neighbour–Joining tree without distance corrections, that was visualized with iTOL (51) on an unrooted dendrogram. The MSA was additionally used as an input for bootstrap analysis with MEGA X (52) (Maximum Likelihood method, 100 replicates, JTT model, uniform substitution rates).
Purification of Streptomyces sp. CT34 APEX and I. dechloratans CHEAP
The synthetic genes of Streptomyces CT34 (Actinobacterial Polymerases with a potentially Eclipsed eXonuclease or APEX subfamily) and Ideonella dechloratans (Cellular Highly Efficient Auxiliary Polymerases or CHEAP subfamily) were optimized for expression in E. coli (Supplementary Table S1) and synthesized using ThermoFisher's GeneArt service. The genes were cloned into a modified pRSF1-Duet expression vector with an N-terminal 14-histidine tag using New England Biolabs and Anza (Thermo Fisher Scientific) restriction enzymes. Escherichia coli BL21 Star (DE3) cells (Invitrogen) were transformed with the engineered plasmids. Bacteria were cultivated at 37°C in LB medium with kanamycin resistance selection and induced at an optical density (OD) = 0.6–1.0 with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After incubation overnight at 20°C, cells were harvested and homogenized in suspension buffer: 50 mM HEPES pH 8.0 (APEX) or 50 mM Tris pH 9.0 (CHEAP), 500 mM NaCl, 10 mM imidazole. After sonication and centrifugation of bacterial debris, corresponding lysate supernatants were supplemented with Benzonase (Sigma-Aldrich) and protease inhibitors (Thermo Fisher Scientific), 1 μl and one tablet per 50 ml, respectively. The proteins of interest were isolated by purification of the lysates on a HisTrap column (suspension buffer as washing buffer, 500 mM imidazole in elution buffer). Collected proteins were diluted to 75 mM NaCl and repurified on a HiTrap Heparin column with an elution at 1 M NaCl. Both purification columns were from Cytiva. Protein purity was assessed on a sodium dodecylsulphate–polyacrylamide gel electrophoresis (SDS–PAGE) 4–15% gel (BioRad) with a molecular weight ladder (Precision Plus Protein, Biorad) as control. The enzymes were concentrated to 3.4–3.9 mg/ml with Amicon Ultra 30k MWCO centrifugal filters (Merck), flash-frozen in liquid nitrogen and stored directly at −20°C, with no glycerol added.
Primer extension, exonuclease and thermostability assays
Polymerase activity tests were performed in 20 mM Tris–HCl pH 8.0, 20 mM NaCl, 5 mM MgCl2 and 1 mM MnCl2 (APEX only), unless specified otherwise. Reaction solutions contained 1 μM of templating oligo (dT10 and dN10 for APEX, dN10 for CHEAP; see Supplementary Table S2), 1 μM of FAM 5′-labelled DNA primer, 1 mM of dATP or a mix of four dNTPs and 1 μM of PolA. Solutions were incubated for 30 min at 37°C (APEX) or for 5 min at 20°C (CHEAP). The concentration of 3'→5' exo-Klenow polymerase used as a control was set at 1 U in 10 μl. Before adding the protein, DNA was hybridized by heating up to 90°C and gradually cooled to room temperature. Reactions were terminated by adding two volumes of a buffer containing 10 mM EDTA, 98% formamide and 1 mg/ml bromophenol blue, and stored at –20°C. Products were pre-heated at 95°C for 10 min, before being separated using PAGE and visualized by FAM fluorescence on a Typhoon FLA 9000 imager.
Exonuclease assays on the dN10 overhang template were conducted similarly for all enzymes (E. coli Pol I exo+, CT34 APEX and I. dechloratans CHEAP), except for the absence of four dNTPs in the reaction mixture. Solutions were incubated for 30 min at 37°C with no additional MnCl2, and for 2 h at 20°C with 5 mM MnCl2. Thermostability of I. dechloratans CHEAP was assessed by pre-incubating the enzyme for 10 min at room temperature, 70, 80 or 90°C before the primer extension test. All oligonucleotides were from Eurogentec, dNTPs from Fermentas (Thermo Fisher Scientific), chemicals from Sigma-Aldrich and 3'→5' exo-Klenow polymerase from New England Biolabs.
RESULTS
Clustering of available PolA sequences into 19 major subfamilies
In October 2021, we accessed the UniProt database (42) and extracted all non-fragmentary PolA protein sequences that share at most 70% sequence identity within the set. We expanded this dataset manually with 27 representative PolA entries, resulting in 8136 sequences on which we ran the clustering simulation using CLANS (35). After several hundred steps of the simulation, the sequences self-organized in 3D space in a stable manner, forming 19 distinguishable clusters (Figure 1): this distribution remained constant until the simulation's end at 6000 iterations.
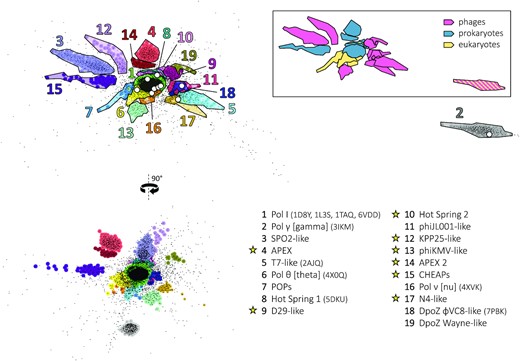
Non-hierarchical clustering of PolA sequences. A comprehensive dataset of all PolA sequences (black points) was obtained from UniProt in October 2021. The 3D distribution of the points was generated with CLANS (35) using the pairwise scores of sequence similarity (evolutionary distance) between individual polymerases. Two planar projections of this distribution are shown on the left (90° rotation). The 19 major clusters of PolA sequences, corresponding to PolA subfamilies, are coloured and numbered in decreasing order of size. Their names (known or proposed) and PDB codes (if applicable) are given in the key on the bottom right: yellow stars denote new subfamilies identified in this work. White dots in the top-left projection mark the position of known PolA X-ray structures. The same cluster projection was recoloured (framed insert on the top right), assigning one colour to each type of carrier species: phages (magenta), prokaryotes (blue) and eukaryotes (yellow). Double affiliation of clusters #2 and #8 is highlighted by coloured stripes.
The characteristics of the most prominent groups, numbered in decreasing order of size, are listed in Table 1. Seven of the eight largest clusters (#1, #2, #3, #5, #6, #7 and #8) determined in this work match the subfamilies annotated at NCBI’s Conserved Domain Database (CDD; superfamily cd06444) (53)—these are the most represented and explored PolA clades. Four other clusters (#11, #16, #18 and #19) represent lesser known subfamilies introduced recently in the literature (19,20,30,54), while the eight remaining clusters (#4, #9, #10, #12, #13, #14, #15 and #17) have not been described or recognized as separate subfamilies before. Importantly, reported phylogenetic relationships among the known subfamilies are consistent with the distribution of the clusters (see the following cluster descriptions).
Occurrence and main characteristics of the PolA subfamilies/clusters
| Cluster (colour) | No. of sequences (<70% id) | Occurrence | Subfamily (CDD ID) | Representative species (PDB structure) | Polymerase function | Conservation of motif DxE in the 3′–5′ exonuclease domain | Additional domains or binding partners |
|---|---|---|---|---|---|---|---|
| 1 (lime) | 4560 | ▪ Bacteria (all major phyla) | Canonical Pol I (cd08637) | ▪ E. coli (1D8Y) ▪ G. stearothermophilus (1L3S) ▪ T. aquaticus (1TAQ) ▪ M. smegmatis (6VDD) | ▪ Lagging strand synthesis ▪ Single-strand gap repair ▪ Removal of RNA primers ▪ Replication of plasmids | Partial (∼42% sequences with catalytic residues conserved) | ▪ 5′–3′ exonuclease (94% sequences, N-terminal fusion) |
| 2 (silver) | 313 | ▪ Mitochondria (opisthokonts) ▪ Several cyanophages (Caudovirales) | Pol γ (cd08641) | ▪ H. sapiens (3IKM) ▪ Phages A-HIS1, A-HIS2 | ▪ Replication of mitochondrial DNA ▪ Replication of phage DNA (putative) | Yes (all except some Glomeromycetes) | ▪ Interacts with PolγB dimer (H. sapiens), monomer (D. melanogaster) or functions as a single subunit (S. cerevisiae) |
| 3 (lavender blue) | 306 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Firmicutes | SPO2-like (cd08642) | ▪ Phage SPO2 | ▪ Replication of phage DNA | Yes (all) | ▪ No known partners |
| 4 (light red) | 290 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX (established in this study) | ▪ S. coelicolor ▪ Streptomyces sp. CT34 | ▪ UV sensitivity reduction ▪ Confirmed polymerase activity (this study) ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 5 (cyan) | 205 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | T7-like (cd08643) | ▪ Phage T7 (2AJQ; in a replisome complex: 5IKN) ▪ Phage S-SBP1 | ▪ Replication of phage DNA | Yes (all) | ▪ Interacts with host's thioredoxin through TBD insertion on thumb's tip (∼29% sequences with TBD ≥50 amino acids) ▪ Interacts with TOPRIM primase-helicase (shown for T7) ▪ Separate 3′–5′ exonuclease domain (S-SBP1-like) |
| 6 (yellow) | 143 | ▪ Eukaryotes | Pol θ (cd08638) | ▪ H. sapiens (4 × 0Q) | ▪ DNA repair (MMEJ) ▪ Template-dependent and -independent synthesis | No | ▪ Superfamily 2 helicase (HELQ, N-terminal fusion) |
| 7 (light blue) | 76 | ▪ Mitochondria and plastids (non-opisthokonts); replaced by Pol γ in opisthokonts | POPs (cd08640) | ▪ A. thaliana | ▪ Replication and repair of organellar DNA | Yes (∼89% sequences) | ▪ 5′–3′ exonuclease (∼8% sequences, N-terminal fusion) |
| 8 (dark green) | 75 | ▪ Diverse bacteria (mainly Aquificae and Cyanobacteria); replaces canonical Pol I in some Aquificae ▪ Euryarchaeota (Methanomicrobia) ▪ Apicomplexa apicoplasts | Hot Spring 1: Aquificae-like (cd08639) | ▪ A. aeolicus ▪ M. vulcani ▪ P. falciparum (5DKU) | ▪ Unknown role in prokaryotes ▪ Replication of apicoplast DNA ▪ Thermostable | Yes (∼93% sequences) | ▪ AEP primase-polymerase (∼7% of bacterial sequences, N-terminal fusion) ▪ Polyprotein in Apicomplexa: fused to TOPRIM primase and helicase (N-terminal) |
| 9 (purple red) | 74 | ▪ Actinomycetia phages (Caudovirales) ▪ Actinomycetia prophages | D29-like (established in this study) | ▪ Mycobacterium phage D29 | ▪ Replication of phage DNA (putative) | Yes (∼99% sequences) | ▪ No known partners |
| 10 (pale pink) | 68 | ▪ Diverse bacteria (mainly Acidobacteria and candidate division WWE3); does not replace canonical Pol I ▪ Diverse archaea (metagenomics-derived, putative) | Hot Spring 2 (shares a recent common ancestor with Aquificae-like) | ▪ Pyrinomonas methylaliphatogenes | ▪ Unknown role ▪ Thermostable (putative) | Yes (∼96% sequences) | ▪ No known partners |
| 11 (pink-red) | 59 | ▪ Phages (Caudovirales) | φJL001-like | ▪ Phage φJL001 | ▪ Replication of phage DNA (putative) | Yes (∼98% sequences) | ▪ No known partners |
| 12 (lilac) | 59 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | KPP25-like (established in this study) | ▪ Phage KPP25 | ▪ Untested ▪ Replication of phage DNA (putative) | Yes (all) | ▪ No known partners |
| 13 (aquamarine) | 59 | ▪ Phages of Proteobacteria (Caudovirales) ▪ Proteobacteria prophages | phiKMV-like (established in this study) | ▪ Phage phiKMV | ▪ Replication of phage DNA (putative) | Yes (∼95% sequences) | ▪ ∼100 amino acid insertion on thumb's tip, unrelated to TBD |
| 14 (brown-red) | 53 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX 2 (established in this study) | ▪ M. pelagius | ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 15 (dark violet) | 52 | ▪ Bacteria (mainly Proteobacteria); does not replace canonical Pol I ▪ Euryarchaeota (Methanomicrobia) ▪ Related to CCPols from staphylococcal MGEs | CHEAPs (established in this study) | ▪ I. dechloratans | ▪ Confirmed polymerase activity (this study) ▪ Highly efficient polymerase and exonuclease activities ▪ Replication-related (probable) | Yes (∼88% sequences) | ▪ No known partners ▪ CCPols lack the majority of 3′–5′ exonuclease domain and form a primase-helicase complex with MD, Cch2 |
| 16 (orange) | 52 | ▪ Metazoa | Pol ν | ▪ H. sapiens (4XVK) | ▪ DNA cross-linking rescue ▪ Germline meiotic homologous recombination | No | ▪ Interacts with Pol θ-like superfamily 2 helicase (HELQ) |
| 17 (light brown) | 45 | ▪ Phages (Caudovirales) ▪ Proteobacteria prophages | N4-like (established in this study) | ▪ Phage N4 ▪ Phage KPP21 | ▪ Replication of phage DNA (putative) | Yes (all) | ▪ N-terminal family 4 UDG domain (catalytic residues unconserved) |
| 18 (dark blue) | 21 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ φVC8-like | ▪ Phage φVC8 (7PBK) | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| 19 (dark gold) | 19 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ Wayne-like | ▪ Phage Wayne | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| Cluster (colour) | No. of sequences (<70% id) | Occurrence | Subfamily (CDD ID) | Representative species (PDB structure) | Polymerase function | Conservation of motif DxE in the 3′–5′ exonuclease domain | Additional domains or binding partners |
|---|---|---|---|---|---|---|---|
| 1 (lime) | 4560 | ▪ Bacteria (all major phyla) | Canonical Pol I (cd08637) | ▪ E. coli (1D8Y) ▪ G. stearothermophilus (1L3S) ▪ T. aquaticus (1TAQ) ▪ M. smegmatis (6VDD) | ▪ Lagging strand synthesis ▪ Single-strand gap repair ▪ Removal of RNA primers ▪ Replication of plasmids | Partial (∼42% sequences with catalytic residues conserved) | ▪ 5′–3′ exonuclease (94% sequences, N-terminal fusion) |
| 2 (silver) | 313 | ▪ Mitochondria (opisthokonts) ▪ Several cyanophages (Caudovirales) | Pol γ (cd08641) | ▪ H. sapiens (3IKM) ▪ Phages A-HIS1, A-HIS2 | ▪ Replication of mitochondrial DNA ▪ Replication of phage DNA (putative) | Yes (all except some Glomeromycetes) | ▪ Interacts with PolγB dimer (H. sapiens), monomer (D. melanogaster) or functions as a single subunit (S. cerevisiae) |
| 3 (lavender blue) | 306 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Firmicutes | SPO2-like (cd08642) | ▪ Phage SPO2 | ▪ Replication of phage DNA | Yes (all) | ▪ No known partners |
| 4 (light red) | 290 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX (established in this study) | ▪ S. coelicolor ▪ Streptomyces sp. CT34 | ▪ UV sensitivity reduction ▪ Confirmed polymerase activity (this study) ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 5 (cyan) | 205 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | T7-like (cd08643) | ▪ Phage T7 (2AJQ; in a replisome complex: 5IKN) ▪ Phage S-SBP1 | ▪ Replication of phage DNA | Yes (all) | ▪ Interacts with host's thioredoxin through TBD insertion on thumb's tip (∼29% sequences with TBD ≥50 amino acids) ▪ Interacts with TOPRIM primase-helicase (shown for T7) ▪ Separate 3′–5′ exonuclease domain (S-SBP1-like) |
| 6 (yellow) | 143 | ▪ Eukaryotes | Pol θ (cd08638) | ▪ H. sapiens (4 × 0Q) | ▪ DNA repair (MMEJ) ▪ Template-dependent and -independent synthesis | No | ▪ Superfamily 2 helicase (HELQ, N-terminal fusion) |
| 7 (light blue) | 76 | ▪ Mitochondria and plastids (non-opisthokonts); replaced by Pol γ in opisthokonts | POPs (cd08640) | ▪ A. thaliana | ▪ Replication and repair of organellar DNA | Yes (∼89% sequences) | ▪ 5′–3′ exonuclease (∼8% sequences, N-terminal fusion) |
| 8 (dark green) | 75 | ▪ Diverse bacteria (mainly Aquificae and Cyanobacteria); replaces canonical Pol I in some Aquificae ▪ Euryarchaeota (Methanomicrobia) ▪ Apicomplexa apicoplasts | Hot Spring 1: Aquificae-like (cd08639) | ▪ A. aeolicus ▪ M. vulcani ▪ P. falciparum (5DKU) | ▪ Unknown role in prokaryotes ▪ Replication of apicoplast DNA ▪ Thermostable | Yes (∼93% sequences) | ▪ AEP primase-polymerase (∼7% of bacterial sequences, N-terminal fusion) ▪ Polyprotein in Apicomplexa: fused to TOPRIM primase and helicase (N-terminal) |
| 9 (purple red) | 74 | ▪ Actinomycetia phages (Caudovirales) ▪ Actinomycetia prophages | D29-like (established in this study) | ▪ Mycobacterium phage D29 | ▪ Replication of phage DNA (putative) | Yes (∼99% sequences) | ▪ No known partners |
| 10 (pale pink) | 68 | ▪ Diverse bacteria (mainly Acidobacteria and candidate division WWE3); does not replace canonical Pol I ▪ Diverse archaea (metagenomics-derived, putative) | Hot Spring 2 (shares a recent common ancestor with Aquificae-like) | ▪ Pyrinomonas methylaliphatogenes | ▪ Unknown role ▪ Thermostable (putative) | Yes (∼96% sequences) | ▪ No known partners |
| 11 (pink-red) | 59 | ▪ Phages (Caudovirales) | φJL001-like | ▪ Phage φJL001 | ▪ Replication of phage DNA (putative) | Yes (∼98% sequences) | ▪ No known partners |
| 12 (lilac) | 59 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | KPP25-like (established in this study) | ▪ Phage KPP25 | ▪ Untested ▪ Replication of phage DNA (putative) | Yes (all) | ▪ No known partners |
| 13 (aquamarine) | 59 | ▪ Phages of Proteobacteria (Caudovirales) ▪ Proteobacteria prophages | phiKMV-like (established in this study) | ▪ Phage phiKMV | ▪ Replication of phage DNA (putative) | Yes (∼95% sequences) | ▪ ∼100 amino acid insertion on thumb's tip, unrelated to TBD |
| 14 (brown-red) | 53 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX 2 (established in this study) | ▪ M. pelagius | ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 15 (dark violet) | 52 | ▪ Bacteria (mainly Proteobacteria); does not replace canonical Pol I ▪ Euryarchaeota (Methanomicrobia) ▪ Related to CCPols from staphylococcal MGEs | CHEAPs (established in this study) | ▪ I. dechloratans | ▪ Confirmed polymerase activity (this study) ▪ Highly efficient polymerase and exonuclease activities ▪ Replication-related (probable) | Yes (∼88% sequences) | ▪ No known partners ▪ CCPols lack the majority of 3′–5′ exonuclease domain and form a primase-helicase complex with MD, Cch2 |
| 16 (orange) | 52 | ▪ Metazoa | Pol ν | ▪ H. sapiens (4XVK) | ▪ DNA cross-linking rescue ▪ Germline meiotic homologous recombination | No | ▪ Interacts with Pol θ-like superfamily 2 helicase (HELQ) |
| 17 (light brown) | 45 | ▪ Phages (Caudovirales) ▪ Proteobacteria prophages | N4-like (established in this study) | ▪ Phage N4 ▪ Phage KPP21 | ▪ Replication of phage DNA (putative) | Yes (all) | ▪ N-terminal family 4 UDG domain (catalytic residues unconserved) |
| 18 (dark blue) | 21 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ φVC8-like | ▪ Phage φVC8 (7PBK) | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| 19 (dark gold) | 19 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ Wayne-like | ▪ Phage Wayne | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
The clusters and their characteristics are listed in decreasing order of size. Only seven of the 19 clusters (#1, #2, #3, #5, #6, #7 and #8) correspond to PolA subfamilies with an assigned CDD identifier; a further four have been recognized in the literature (#11, #16, #18 and #19). The established or proposed names of the subfamilies are shown in column 4. Functional tests demonstrated that previously undescribed bacterial clusters #4 (APEX 1) and #15 (CHEAPs) comprise functional polymerases (Figures 7 and 8). Experimental 3D structures of subfamily representatives are available for seven clusters (#1, #2, #5, #6, #8, #16 and #18); their PDB code is provided in parentheses in column 5. In general, PolA subfamilies perform at least two different biological roles: DNA replication or repair. They frequently differ in the functionality of the 3′–5′ exonuclease domain and in the presence of additional domains (such as 5′–3′ exonuclease) or partners. Interestingly, all PolAs from phages seem to preserve their 3′–5′ exonuclease activity, regardless of subfamily.
Occurrence and main characteristics of the PolA subfamilies/clusters
| Cluster (colour) | No. of sequences (<70% id) | Occurrence | Subfamily (CDD ID) | Representative species (PDB structure) | Polymerase function | Conservation of motif DxE in the 3′–5′ exonuclease domain | Additional domains or binding partners |
|---|---|---|---|---|---|---|---|
| 1 (lime) | 4560 | ▪ Bacteria (all major phyla) | Canonical Pol I (cd08637) | ▪ E. coli (1D8Y) ▪ G. stearothermophilus (1L3S) ▪ T. aquaticus (1TAQ) ▪ M. smegmatis (6VDD) | ▪ Lagging strand synthesis ▪ Single-strand gap repair ▪ Removal of RNA primers ▪ Replication of plasmids | Partial (∼42% sequences with catalytic residues conserved) | ▪ 5′–3′ exonuclease (94% sequences, N-terminal fusion) |
| 2 (silver) | 313 | ▪ Mitochondria (opisthokonts) ▪ Several cyanophages (Caudovirales) | Pol γ (cd08641) | ▪ H. sapiens (3IKM) ▪ Phages A-HIS1, A-HIS2 | ▪ Replication of mitochondrial DNA ▪ Replication of phage DNA (putative) | Yes (all except some Glomeromycetes) | ▪ Interacts with PolγB dimer (H. sapiens), monomer (D. melanogaster) or functions as a single subunit (S. cerevisiae) |
| 3 (lavender blue) | 306 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Firmicutes | SPO2-like (cd08642) | ▪ Phage SPO2 | ▪ Replication of phage DNA | Yes (all) | ▪ No known partners |
| 4 (light red) | 290 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX (established in this study) | ▪ S. coelicolor ▪ Streptomyces sp. CT34 | ▪ UV sensitivity reduction ▪ Confirmed polymerase activity (this study) ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 5 (cyan) | 205 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | T7-like (cd08643) | ▪ Phage T7 (2AJQ; in a replisome complex: 5IKN) ▪ Phage S-SBP1 | ▪ Replication of phage DNA | Yes (all) | ▪ Interacts with host's thioredoxin through TBD insertion on thumb's tip (∼29% sequences with TBD ≥50 amino acids) ▪ Interacts with TOPRIM primase-helicase (shown for T7) ▪ Separate 3′–5′ exonuclease domain (S-SBP1-like) |
| 6 (yellow) | 143 | ▪ Eukaryotes | Pol θ (cd08638) | ▪ H. sapiens (4 × 0Q) | ▪ DNA repair (MMEJ) ▪ Template-dependent and -independent synthesis | No | ▪ Superfamily 2 helicase (HELQ, N-terminal fusion) |
| 7 (light blue) | 76 | ▪ Mitochondria and plastids (non-opisthokonts); replaced by Pol γ in opisthokonts | POPs (cd08640) | ▪ A. thaliana | ▪ Replication and repair of organellar DNA | Yes (∼89% sequences) | ▪ 5′–3′ exonuclease (∼8% sequences, N-terminal fusion) |
| 8 (dark green) | 75 | ▪ Diverse bacteria (mainly Aquificae and Cyanobacteria); replaces canonical Pol I in some Aquificae ▪ Euryarchaeota (Methanomicrobia) ▪ Apicomplexa apicoplasts | Hot Spring 1: Aquificae-like (cd08639) | ▪ A. aeolicus ▪ M. vulcani ▪ P. falciparum (5DKU) | ▪ Unknown role in prokaryotes ▪ Replication of apicoplast DNA ▪ Thermostable | Yes (∼93% sequences) | ▪ AEP primase-polymerase (∼7% of bacterial sequences, N-terminal fusion) ▪ Polyprotein in Apicomplexa: fused to TOPRIM primase and helicase (N-terminal) |
| 9 (purple red) | 74 | ▪ Actinomycetia phages (Caudovirales) ▪ Actinomycetia prophages | D29-like (established in this study) | ▪ Mycobacterium phage D29 | ▪ Replication of phage DNA (putative) | Yes (∼99% sequences) | ▪ No known partners |
| 10 (pale pink) | 68 | ▪ Diverse bacteria (mainly Acidobacteria and candidate division WWE3); does not replace canonical Pol I ▪ Diverse archaea (metagenomics-derived, putative) | Hot Spring 2 (shares a recent common ancestor with Aquificae-like) | ▪ Pyrinomonas methylaliphatogenes | ▪ Unknown role ▪ Thermostable (putative) | Yes (∼96% sequences) | ▪ No known partners |
| 11 (pink-red) | 59 | ▪ Phages (Caudovirales) | φJL001-like | ▪ Phage φJL001 | ▪ Replication of phage DNA (putative) | Yes (∼98% sequences) | ▪ No known partners |
| 12 (lilac) | 59 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | KPP25-like (established in this study) | ▪ Phage KPP25 | ▪ Untested ▪ Replication of phage DNA (putative) | Yes (all) | ▪ No known partners |
| 13 (aquamarine) | 59 | ▪ Phages of Proteobacteria (Caudovirales) ▪ Proteobacteria prophages | phiKMV-like (established in this study) | ▪ Phage phiKMV | ▪ Replication of phage DNA (putative) | Yes (∼95% sequences) | ▪ ∼100 amino acid insertion on thumb's tip, unrelated to TBD |
| 14 (brown-red) | 53 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX 2 (established in this study) | ▪ M. pelagius | ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 15 (dark violet) | 52 | ▪ Bacteria (mainly Proteobacteria); does not replace canonical Pol I ▪ Euryarchaeota (Methanomicrobia) ▪ Related to CCPols from staphylococcal MGEs | CHEAPs (established in this study) | ▪ I. dechloratans | ▪ Confirmed polymerase activity (this study) ▪ Highly efficient polymerase and exonuclease activities ▪ Replication-related (probable) | Yes (∼88% sequences) | ▪ No known partners ▪ CCPols lack the majority of 3′–5′ exonuclease domain and form a primase-helicase complex with MD, Cch2 |
| 16 (orange) | 52 | ▪ Metazoa | Pol ν | ▪ H. sapiens (4XVK) | ▪ DNA cross-linking rescue ▪ Germline meiotic homologous recombination | No | ▪ Interacts with Pol θ-like superfamily 2 helicase (HELQ) |
| 17 (light brown) | 45 | ▪ Phages (Caudovirales) ▪ Proteobacteria prophages | N4-like (established in this study) | ▪ Phage N4 ▪ Phage KPP21 | ▪ Replication of phage DNA (putative) | Yes (all) | ▪ N-terminal family 4 UDG domain (catalytic residues unconserved) |
| 18 (dark blue) | 21 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ φVC8-like | ▪ Phage φVC8 (7PBK) | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| 19 (dark gold) | 19 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ Wayne-like | ▪ Phage Wayne | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| Cluster (colour) | No. of sequences (<70% id) | Occurrence | Subfamily (CDD ID) | Representative species (PDB structure) | Polymerase function | Conservation of motif DxE in the 3′–5′ exonuclease domain | Additional domains or binding partners |
|---|---|---|---|---|---|---|---|
| 1 (lime) | 4560 | ▪ Bacteria (all major phyla) | Canonical Pol I (cd08637) | ▪ E. coli (1D8Y) ▪ G. stearothermophilus (1L3S) ▪ T. aquaticus (1TAQ) ▪ M. smegmatis (6VDD) | ▪ Lagging strand synthesis ▪ Single-strand gap repair ▪ Removal of RNA primers ▪ Replication of plasmids | Partial (∼42% sequences with catalytic residues conserved) | ▪ 5′–3′ exonuclease (94% sequences, N-terminal fusion) |
| 2 (silver) | 313 | ▪ Mitochondria (opisthokonts) ▪ Several cyanophages (Caudovirales) | Pol γ (cd08641) | ▪ H. sapiens (3IKM) ▪ Phages A-HIS1, A-HIS2 | ▪ Replication of mitochondrial DNA ▪ Replication of phage DNA (putative) | Yes (all except some Glomeromycetes) | ▪ Interacts with PolγB dimer (H. sapiens), monomer (D. melanogaster) or functions as a single subunit (S. cerevisiae) |
| 3 (lavender blue) | 306 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Firmicutes | SPO2-like (cd08642) | ▪ Phage SPO2 | ▪ Replication of phage DNA | Yes (all) | ▪ No known partners |
| 4 (light red) | 290 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX (established in this study) | ▪ S. coelicolor ▪ Streptomyces sp. CT34 | ▪ UV sensitivity reduction ▪ Confirmed polymerase activity (this study) ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 5 (cyan) | 205 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | T7-like (cd08643) | ▪ Phage T7 (2AJQ; in a replisome complex: 5IKN) ▪ Phage S-SBP1 | ▪ Replication of phage DNA | Yes (all) | ▪ Interacts with host's thioredoxin through TBD insertion on thumb's tip (∼29% sequences with TBD ≥50 amino acids) ▪ Interacts with TOPRIM primase-helicase (shown for T7) ▪ Separate 3′–5′ exonuclease domain (S-SBP1-like) |
| 6 (yellow) | 143 | ▪ Eukaryotes | Pol θ (cd08638) | ▪ H. sapiens (4 × 0Q) | ▪ DNA repair (MMEJ) ▪ Template-dependent and -independent synthesis | No | ▪ Superfamily 2 helicase (HELQ, N-terminal fusion) |
| 7 (light blue) | 76 | ▪ Mitochondria and plastids (non-opisthokonts); replaced by Pol γ in opisthokonts | POPs (cd08640) | ▪ A. thaliana | ▪ Replication and repair of organellar DNA | Yes (∼89% sequences) | ▪ 5′–3′ exonuclease (∼8% sequences, N-terminal fusion) |
| 8 (dark green) | 75 | ▪ Diverse bacteria (mainly Aquificae and Cyanobacteria); replaces canonical Pol I in some Aquificae ▪ Euryarchaeota (Methanomicrobia) ▪ Apicomplexa apicoplasts | Hot Spring 1: Aquificae-like (cd08639) | ▪ A. aeolicus ▪ M. vulcani ▪ P. falciparum (5DKU) | ▪ Unknown role in prokaryotes ▪ Replication of apicoplast DNA ▪ Thermostable | Yes (∼93% sequences) | ▪ AEP primase-polymerase (∼7% of bacterial sequences, N-terminal fusion) ▪ Polyprotein in Apicomplexa: fused to TOPRIM primase and helicase (N-terminal) |
| 9 (purple red) | 74 | ▪ Actinomycetia phages (Caudovirales) ▪ Actinomycetia prophages | D29-like (established in this study) | ▪ Mycobacterium phage D29 | ▪ Replication of phage DNA (putative) | Yes (∼99% sequences) | ▪ No known partners |
| 10 (pale pink) | 68 | ▪ Diverse bacteria (mainly Acidobacteria and candidate division WWE3); does not replace canonical Pol I ▪ Diverse archaea (metagenomics-derived, putative) | Hot Spring 2 (shares a recent common ancestor with Aquificae-like) | ▪ Pyrinomonas methylaliphatogenes | ▪ Unknown role ▪ Thermostable (putative) | Yes (∼96% sequences) | ▪ No known partners |
| 11 (pink-red) | 59 | ▪ Phages (Caudovirales) | φJL001-like | ▪ Phage φJL001 | ▪ Replication of phage DNA (putative) | Yes (∼98% sequences) | ▪ No known partners |
| 12 (lilac) | 59 | ▪ Phages (Caudovirales) ▪ Prophages mainly in Proteobacteria | KPP25-like (established in this study) | ▪ Phage KPP25 | ▪ Untested ▪ Replication of phage DNA (putative) | Yes (all) | ▪ No known partners |
| 13 (aquamarine) | 59 | ▪ Phages of Proteobacteria (Caudovirales) ▪ Proteobacteria prophages | phiKMV-like (established in this study) | ▪ Phage phiKMV | ▪ Replication of phage DNA (putative) | Yes (∼95% sequences) | ▪ ∼100 amino acid insertion on thumb's tip, unrelated to TBD |
| 14 (brown-red) | 53 | ▪ Bacteria (Actinobacteria); does not replace canonical Pol I | APEX 2 (established in this study) | ▪ M. pelagius | ▪ DNA damage repair (probable) | No; exo catalytic pocket is tightly sealed | ▪ No known partners |
| 15 (dark violet) | 52 | ▪ Bacteria (mainly Proteobacteria); does not replace canonical Pol I ▪ Euryarchaeota (Methanomicrobia) ▪ Related to CCPols from staphylococcal MGEs | CHEAPs (established in this study) | ▪ I. dechloratans | ▪ Confirmed polymerase activity (this study) ▪ Highly efficient polymerase and exonuclease activities ▪ Replication-related (probable) | Yes (∼88% sequences) | ▪ No known partners ▪ CCPols lack the majority of 3′–5′ exonuclease domain and form a primase-helicase complex with MD, Cch2 |
| 16 (orange) | 52 | ▪ Metazoa | Pol ν | ▪ H. sapiens (4XVK) | ▪ DNA cross-linking rescue ▪ Germline meiotic homologous recombination | No | ▪ Interacts with Pol θ-like superfamily 2 helicase (HELQ) |
| 17 (light brown) | 45 | ▪ Phages (Caudovirales) ▪ Proteobacteria prophages | N4-like (established in this study) | ▪ Phage N4 ▪ Phage KPP21 | ▪ Replication of phage DNA (putative) | Yes (all) | ▪ N-terminal family 4 UDG domain (catalytic residues unconserved) |
| 18 (dark blue) | 21 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ φVC8-like | ▪ Phage φVC8 (7PBK) | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
| 19 (dark gold) | 19 (<90% id) | ▪ ZTGC-DNA phages (Caudovirales) | DpoZ Wayne-like | ▪ Phage Wayne | ▪ Adenine-discriminative ▪ Replication of phage ZTGC-DNA | Yes (all) | No known partners |
The clusters and their characteristics are listed in decreasing order of size. Only seven of the 19 clusters (#1, #2, #3, #5, #6, #7 and #8) correspond to PolA subfamilies with an assigned CDD identifier; a further four have been recognized in the literature (#11, #16, #18 and #19). The established or proposed names of the subfamilies are shown in column 4. Functional tests demonstrated that previously undescribed bacterial clusters #4 (APEX 1) and #15 (CHEAPs) comprise functional polymerases (Figures 7 and 8). Experimental 3D structures of subfamily representatives are available for seven clusters (#1, #2, #5, #6, #8, #16 and #18); their PDB code is provided in parentheses in column 5. In general, PolA subfamilies perform at least two different biological roles: DNA replication or repair. They frequently differ in the functionality of the 3′–5′ exonuclease domain and in the presence of additional domains (such as 5′–3′ exonuclease) or partners. Interestingly, all PolAs from phages seem to preserve their 3′–5′ exonuclease activity, regardless of subfamily.
To further characterize the relationships between clusters, we performed a complementary phylogenetic analysis on representative cluster sequences. A Neighbour–Joining tree calculated in Clustal Omega (45) on PolA Klenow-like large fragments reflects the distribution of the clusters, revealing 5–6 superclusters/clades (Figure 2A). This is supported by a separate bootstrap analysis performed with the Maximum Likelihood method (Figure 2B) and corroborates previous phylogenetic studies (25,28). The most abundant supercluster consists of clusters #1, #6, #7, #8, #10 and #16 present in cellular organisms. The second one contains clusters #3, #4, #12, #14 and #15, all displaying disrupted helices in the thumb subdomain (see below). The remaining clusters form pairs, with either strong (#2 and #5; #11 and #18) or weak (#13 and #17; #9 and #19) bootstrap value support. The link between the #11–#18 pair and the first supercluster is also faint. As the connections between the superclusters are even weaker and ambiguous, their exact relationship and the origin of the tree cannot be unequivocally determined.
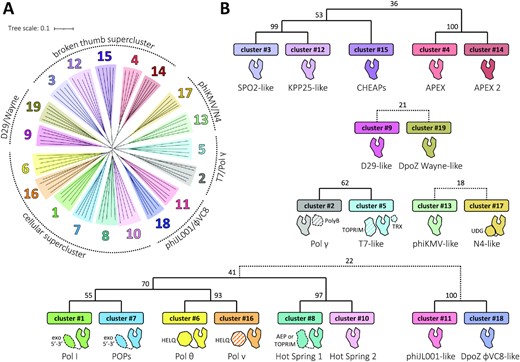
Phylogenetic relationships between the 19 PolA subfamilies. (A) Five representatives of each cluster were selected for the multiple sequence alignment of their corresponding Klenow-like large fragments, prepared with Clustal Omega (45) and visualized as an unrooted dendrogram (left). The branches were coloured to match the clusters’ colours as in Figure 1; they form five or six distinct groups (superclusters) of closely related subfamilies, presented schematically around the tree. (B) The superclusters with supporting bootstrap values between the clusters’ branches are presented on the annotated cladograms to the right and below the tree. The bootstrap values were generated with MEGA X (52), using the Maximum Likelihood method and taking 100 replicates. Dotted connections represent more distant relationships. A cartoon domain representation is shown below each cluster. Dashed contours represent domains/proteins present only for some members of a given subfamily; PolA-interacting proteins (separate polypeptide chains) are filled in stripes (see Table 1).
We also coloured the 3D map generated by CLANS as a function of the type of carrier species: prokaryotes, eukaryotes or viruses (phages) (Figure 1, framed insert). Whereas cellular—bacterial and eukaryotic—clusters tend to be the largest, phage clusters are abundant. This illustrates the general high diversification of the virosphere (55), drawing from frequent genetic transfers of replication-related genes between phages and their hosts (56). Two clusters belong to more than one type of carrier organisms (#2 and #8): for these cases, the horizontal transfer of polA genes between cellular hosts and phages has been evidenced in the literature (see cluster descriptions below).
For each cluster, we generated sequence logos of the key functional motifs in the polymerase domain (57,58) (Figure 3); a mapping of the motifs onto the E. coli Pol I structure is provided in Supplementary Figure S1. The strict conservation of crucial catalytic residues implies that every subfamily corresponds to functional polymerases. While this has been proved experimentally for 10 known clades, other clusters were lacking direct experimental data; here, we do provide such data for representatives of related bacterial clusters #4 and #15 (see below). The third conserved residue of motif B—that we refer to as position B3—participates in dideoxynucleotide discrimination (59) and modulates polymerase activity (60): it is the most variable conserved position, used to separate particular PolA clades in previous metagenomic studies (30). In the following, we pay special attention to this position.

Sequence motifs of the 19 PolA clusters. Each cluster is presented with a sequence logo, where the height of each residue type is proportional to its frequency. Motifs A, B, 6 and C, already recognized in the PolA family (57,58), are complemented with the TGR motif, also conserved in related Klenow-fold polymerases (DNA-dependent DNA and single-subunit RNA) (140,141). Positions of highly conserved residues are marked by orange triangles below. They are given a unique label corresponding to the motif they belong to and their order of appearance. Among them, position B3 shows the highest variability. Cluster #18-specific insertion in motif B is replaced with a blue ellipsis. See Supplementary Figure S1 for the structural context of the motifs, lining the DNA binding site and the polymerase catalytic site.
Below, we describe each cluster in turn, starting from #1, by far the most abundant cluster (56% of classified sequences), down to #19, the least abundant.
Cluster #1: Pol I
The largest cluster corresponds to the canonical bacterial Pol I (CDD cd08637), encompassing all known major phyla. This subfamily includes several well-described PolAs of species such as E. coli or Thermus aquaticus: the former was the first isolated and characterized DNA polymerase (3), while the latter is nowadays commonly used for in vitro DNA amplification (16). The vast majority of bacterial Pol I enzymes possess an additional N-terminal domain with 5′–3′ exonuclease activity (61). Independently of that fusion, their 3′–5′ exonuclease domain is often found to be deactivated with the mutation of one or several otherwise strictly conserved catalytic residues (62,63). Abundant in the cell (64), E. coli Pol I participates in the lagging strand synthesis, single-strand gap repair and in the removal of the RNA primer from Okazaki fragments through its 5′–3′ exonuclease activity; however, such functions can be partly compensated for by other DNA polymerases or nucleases (9,64–68). Cluster #1 PolAs may also be directly involved in the processive replication of plasmids (12).
Cluster #2: Pol γ
The second cluster contains nucleus-encoded DNA polymerases of mitochondria (CDD cd08641) in opisthokonts (animals and fungi), encoded in the cell nucleus. Known as subunit PolγA, they interact with the accessory subunit PolγB [homologous to class II aminoacyl-tRNA synthetases (69)] to form the functional Pol γ heterotrimer in humans (70) or the heterodimer in fruit flies (71); alternatively, they operate as a single subunit in yeast (72). Despite clear sequential and structural separation from other PolA subfamilies (73), it was suggested that Pol γ polymerases derive from T7-like PolAs; remarkably, mitochondria share with T7 phage not only their DNA polymerase but also their DNA primase and their RNA polymerase (25,74). This relationship is reproduced in our phylogenetic tree (Figure 2) and supports our clustering results, which place Pol γs as the most remote nebula of sequences, yet precisely behind the T7-like cluster #5 (Figure 1). Intriguingly, several cyanophages of the order Caudovirales have been found to contain Pol γ-like polymerases (26). These enzymes show the highest similarity with fungal Pol γ, although their functionality remains to be proven.
Cluster #3: SPO2-like
The third cluster represents a collection of bacteriophage PolAs (CDD cd08642), linked to the double-stranded DNA (dsDNA) order Caudovirales. Some of the carrier phages are found as prophages in bacterial genomes, mainly of the phylum Firmicutes. Like other PolAs of this subfamily, the polymerase of phage SPO2 (SP02) (15) carries a leucine in position B3 of motif B as a less common, non-aromatic variation (Figure 3). Cluster #3 is quite divergent in sequence from other explored PolAs and lacks a resolved representative 3D structure; however, polymerase models generated by AlphaFold2 for this and other clusters are convergent and show high confidence levels of prediction (see below).
Cluster #4: APEX 1
The fourth largest cluster determined in this work is new and corresponds to bacterial polymerases of the phylum Actinobacteria. The genetic context of these polymerases does not suggest a prophage origin, yet they co-exist alongside the canonical Pol I on the bacterial chromosome. A deletion of cluster #4 PolA in Streptomyces indicated its involvement in DNA repair (75), although this subfamily has not been previously tested for polymerase activity; however, below we confirm the functionality of its representative. These Actinobacterial polymerases carry a distinctive leucine in position B3 (Figure 3) and are indeed related to members of most other clusters sharing this characteristic, including cluster #3 (Figure 2). The 3′–5′ exonuclease of cluster #4 PolAs is expected to be non-functional, as it lacks a conserved DxE catalytic motif, or a conservative mutation thereof: in the following section, we also describe a distinctive reshaping in the exo catalytic pocket. In order to single out this abundant PolA subfamily and its closely related twin cluster (see below), we give them the name of Actinobacterial Polymerases with a potentially Eclipsed eXonuclease (APEX 1 and 2).
Cluster #5: T7-like
The fifth cluster concerns another, different set of Caudovirales PolAs (CDD cd08643), found in phage or prophage sequences predominantly in Proteobacteria. A provisional distribution of subclades constituting this cluster has been previously reported (76). Phage T7 replicative DNA polymerase, a cluster #5 PolA, binds the hosts’ thioredoxin for a truly processive polymerase activity (13); grafting the thioredoxin-binding domain (TBD) onto E. coli Pol I dramatically increases its processivity upon binding the cofactor (77). Nevertheless, the thioredoxin-binding motif does not consistently appear in all T7-related phages (78). The structure of the T7 replisome involving a hexameric TOPRIM primase-helicase has been recently determined (79,80). Cluster #5 PolAs contain a tyrosine in position B3 (Figure 3).
Surprisingly, several T7-like sequences include only the polymerase domain, entirely lacking the proofreading 3′–5′ exonuclease. One such polymerase has been modelled in silico, based on a metagenome fragment (29). Further below, we investigate their genomic context, revealing a recent domain splitting event.
Cluster #6: Pol θ
Cluster six encompasses Pol θ (CDD cd08638) present in many eukaryotes, with the exception of fungi. These DNA polymerases are both template dependent and independent. Due to their microhomology-mediated end joining activity, they are recognized as DNA repair enzymes (11,81). Nevertheless, much like cluster #1 PolAs, their physiological role seems partly redundant and pleiotropic, also extending towards replication control during cell division in animals and plants (82,83). PolA domains of Pol θ are fused on the N-termini to a large superfamily 2 (SF2) helicase domain, both having an experimentally determined structure in humans (84,85).
Cluster #7: POPs
The seventh PolA subfamily covers the polymerases of the DNA-containing organelles—plastids and mitochondria—in non-opisthokont eukaryotes (CDD cd08640). Originally detected in plants (86,87), they were dubbed POPs (Plant Organellar DNA Polymerases); due to their universality, they have been proposed to evolutionarily precede Pol γ in organelles (88). Despite also being encoded in the nucleus, phylogenetically these two groups are considerably distinct (87,88). POPs have a functional 3′–5′ exonuclease domain; a fusion with the domain of a 5′–3′ exonuclease was observed only in singular cases (28). Lastly, cluster #7 PolAs seem to share a relatively recent ancestor with several other cellular polymerases (clusters #1, #6, #8 and #16) (28); our phylogenetic tree captures such a relationship (Figure 2).
Cluster #8: Hot Spring 1 (Aquificae-like)
Cluster eight (CDD cd08639) consists of products of an adventurous (i.e. appearing in very disparate species) polA gene. The cluster's members were previously detected in apicoplasts of eukaryotic Apicomplexa (89) and diverse bacterial phyla (notably Aquificae) (90). In our dataset, we notice that they also appear in a group of archaeal Methanomicrobia from the phylum Euryarchaeota (e.g. Methanolobus vulcani, GenBank ID: WP_167879304): their genetic context is not indicative of a prophage sequence. This finding makes it the first PolA subfamily known to span across all three cellular domains of life. Frequent genetic transfers of these PolAs seem to be linked to a gene-sharing network specific to hot springs, populated by thermophilic viruses, their Aquificae hosts and archaea as well (27,91). PolA of Plasmodium falciparum shares with Aquificae polymerases not only sequence similarity, but also an unexpected high-temperature activity optimum (89); its X-ray structure has also been determined (92). Thus, a proposed model of their evolution involves horizontal gene transfer between phages, various bacterial phyla and apicoplasts, taking into account the loss of canonical Pol I in Aquificae (27). To underline this unique environmental context and the strong association of cluster #8 with its own twin cluster (#10), we will refer to these two clades as Hot Spring 1 and 2.
Similar polymerases are also found in thermophilic viruses/phages, such as Thermocrinis Great Boiling Spring virus (27,91); another viral metagenome-derived thermostable PolA called ‘3173 Pol’ has been adopted for reverse transcription–PCR applications, as it accepts RNA templates (17). These PolAs are distributed in the immediate proximity of clusters #8 and #10.
In apicoplasts, cluster #8 PolAs are fused on the N-terminus with TOPRIM primase and helicase domains, whose functionality as a polyprotein has been confirmed (89,93); in a subset of phages with a similar polymerase, a polyprotein fusion with a putative helicase domain has also been observed (27). Additionally, we observe that in several bacteria representing various phyla (i.e. Verrucomicrobia, Planctomycetota and Nitrospirae) the cluster #8 PolA is fused to an AEP primase-polymerase. In general, Hot Spring PolAs conserve the ‘canonical’ motif B3 phenylalanine (Figure 3), similarly to related Pol I and POPs (clusters #1 and #7).
Cluster #9: D29-like
The ninth largest cluster contains polymerases of Caudovirales bacteriophages (and their prophages) preying on Actinomycetia (phylum Actinomycetia). Although phage D29, the prototypical carrier phage of this PolA subfamily, was discovered almost 70 years ago (94), this is the first time that D29-like polymerases are recognized as a separate, highly diverged clade (Figure 2). Nevertheless, the crucial motifs of cluster #9 PolAs stay typical (Figure 3), including the well-conserved DxE catalytic motif in the 3′–5′ exonuclease domain; these polymerases are devoid of any significant insertions.
Cluster #10: Hot Spring 2
The 10th cluster, named Hot Spring 2, closely mimics cluster #8 of Aquificae-like PolAs (Hot Spring 1): the two groups share similar conservation profiles (Figure 3) and a recent common ancestor (Figure 2), although their separation is supported by both clustering and phylogenetic analyses. Organisms carrying cluster #10 polymerases involve bacterial- and metagenomics-derived putative archaeal species: they include predominantly distinct phyla (i.e. Acidobacteria, candidate division WWE, Nanoarchaeota and Thorarchaeota) but exclude Aquificae.
Cluster #11: φJL001-like
PolAs from cluster #11 belong to yet another fraction of Caudovirales phages. They are also found in multiple short sequence fragments annotated as bacteria, but such DNA portions directly correspond to the viral ones in length and composition. This subset of PolAs has been observed predominantly in marine virioplankton (29). It is so far the third cluster characterized by a leucine in position B3, despite an apparent interchangeability with phenylalanine (Figure 3) and lack of close homology with other leucine-bearing clusters, except for #18 (Figure 2). In this group resides the PolA of phage φJL001, whose genome has been described in detail (95). Nonetheless, cluster #11 still lacks an experimentally determined structural representative.
Cluster #12: KPP25-like
Next to cluster #3 one finds one more group of Caudovirales PolAs (Figure 1); their genes are often integrated as prophages into proteobacterial genomes as well. Up to now, this subfamily had remained completely undescribed, although a representative was identified in phage KPP25 through routine homology searches (96): we will therefore refer to cluster #12 PolAs as KPP25-like. In agreement with their evident relationship with SPO2-like PolAs (Figure 2), these DNA polymerases display the distinctive position B3 Leu variant (Figure 3). Their typical length is 600–650 amino acids, with all the activity-related residues conserved in both polymerase and 3′–5′ exonuclease domains.
Cluster #13: phiKMV-like
The 13th cluster comprises another set of PolAs found in phages of Proteobacteria, or in their prophages. This family is represented by the polymerase of phage phiKMV (97), previously described as a T7-like phage: nevertheless, phiKMV belongs to a distinct taxonomic subfamily, while the differences between T7-like and phiKMV-like PolAs are even more pronounced (98) (Figures 1 and 2). For example, cluster #13 polymerases have a phenylalanine in position B3, instead of a tyrosine specific to the T7 clade (Figure 3). Interestingly, they also possess an extensive (∼100 amino acid) insertion in the thumb subdomain, which could be reliably modelled for multiple representatives (see the following section). Its placement follows the helix H1, in contrast to the TBD of T7 PolA that precedes it (99). There seems to be no phylogenetic relationship between the two, although the peculiar, structured extension of phiKMV-like PolAs may also perform a role related to processivity.
Cluster #14: APEX 2
Similarly to cluster #10, cluster #14 acts as a twin cluster to a larger clade. Despite its strong resemblance to cluster #4 (APEX 1) in sequence and occurrence, the two subfamilies consistently split in a sufficiently large dataset (Figures 1 and 2). We note the exceptional motif conservation of the smaller cluster, named APEX 2, even on usually variable positions, suggesting its relatively recent separation. Cluster #14 represents the only PolA subfamily where an alanine replaces threonine in the TGR motif (Figure 3).
Cluster #15: CHEAPs
Cluster #15 is the last major cloud of bacterial PolA sequences. They are associated essentially with Proteobacteria, but, in a surprising parallel with the unrelated cluster #8 (Figure 2), also with some archaeal Methanomicrobia. Some of these PolAs are annotated as thermostable; however, a thorough search revealed that all such instances were inferred through homology and that no cluster #15 representative has been described before. Cluster #15 PolAs conserve all functional exonuclease and polymerase motifs, with a leucine found in position B3 (Figure 3), in agreement with their close homology to polymerases SPO2-like, KPP25-like and APEX (Figure 2). Much like APEX (clusters #4 and #14), these enzymes do not replace the canonical bacterial Pol I. An example of a reference carrier organism is I. dechloratans, a Betaproteobacteria (GenBank ID: WP_151124575): below, we confirm high templated polymerase and exonuclease activities, as well as the lack of thermostability of its PolA. We therefore name this subfamily Cellular Highly Efficient Auxiliary Polymerases (CHEAPs).
In the proximity of cluster #15, we observed a few truncated PolA sequences: although under-represented, they correspond to a unique class of PolAs (CCPols) with an incomplete exo domain (see below), which are associated with staphylococcal mobile genetic elements (MGEs) (100). CCPol of Stapylococcus aureus interacts with a small protein (MP) and a helicase (Cch2); importantly, the CCPol–MP complex displays priming activity (100). A supplementary phylogenetic evaluation confirms that CHEAP is the closest subfamily to CCPols.
Cluster #16: Pol ν
Cluster #16 represents polymerases ν, a young branch of PolAs that arose in animals (54). They display strong sequence similarity with Pol θ as well as with canonical bacterial Pol I (cluster #1), and were reported to match the indel profile of Pol θ (101). These tight evolutionary relationships (Figures 1 and 2) introduce some confusion as to the identity of protozoan Pol ν/Pol θ-like PolAs (102,103). Polymerases ν lack additional domains, although they do interact with a Pol θ-related superfamily 2 helicase (10). Despite being able to rescue DNA cross-linking in vitro, their physiological role is associated with meiotic homologous recombination in germline cells (54,10). The crystal structure of human Pol ν has been solved (104).
Cluster #17: N4-like
Cluster #17 is formed by yet another set of PolAs from Caudovirales phages of Proteobacteria, including their prophage form. They are comparatively long, usually comprising ∼800–900 amino acids: this results from the fusion on the N-terminus with a family 4 uracil-DNA glycosylase (UDG), an enzyme that typically removes uracil from DNA strands, leaving an abasic site (105). To this cluster belongs the polymerase of phage N4 (106). N4-like PolAs display distant homology to phiKMV-like PolAs (Figure 2), featuring a phenylalanine in position B3 as well (Figure 3).
There exist other well-known bacteriophages carrying a family A polymerase fused to a family 4 UDG domain: the most notable examples include Bacillus phages SPO1 (SP01) (14), SP-10 and SP-15 (107). Their polymerases do not cluster together with N4-like polymerases, although all UDG-PolAs are found in close proximity (Supplementary Figure S2). Importantly, all three phages contain modified uracil nucleotides in place of thymine in their genomes (108–110). The UDG domain of UDG-PolA was speculated to provide selectivity towards 5-hydroxymethyluracil (5hmU) (111), which can then be post-replicatively hypermodifed by glucosylation (112). N4-like phages are not known to modify their DNA, which is consistent with the observation that the catalytic residues in the UDG domain of cluster #17 PolAs have been replaced or deleted (see the following section).
Cluster #18: φVC8-like DpoZ
The penultimate and most recently described PolA clade concerns φVC8-like DpoZ enzymes found in some Caudovirales phages that replace their genomic adenine with 2-aminoadenine (Z), resulting in saturated interstrand hydrogen bonding in the phage DNA (19,113). As expected, these polymerases are Z-specific and substantially—although not completely—discriminate against adenine (19,20). Yet, the net incorporation of Z into the DNA of φVC8 and related phages is also modulated by a conserved dATPase (DatZ): importantly, the ZTGC-DNA cyanophage S-2L has DatZ, but lacks a Z-specific polymerase, demonstrating that DpoZ is in fact dispensable for a complete A-to-Z substitution (113–116). φVC8-like DpoZ show close similarity to φJL001-like PolAs from cluster #11 (Figures 1 and 2). We recently reported the experimental structure of the apo form of φVC8 DpoZ (20), providing a rationale for its specific sequence features (19), some of which were also found to be shared with φJL001 PolA.
Cluster #19: Wayne-like DpoZ
The last cluster contains Wayne-like DpoZ, the second group of PolA enzymes specific to 2-aminoadenine, also found in Caudovirales (19). Despite their equivalent functionality, φVC8-like DpoZ are clearly distinct from Wayne-like DpoZ enzymes (19,20) (Figures 1–4). Like the former, the latter show closer homology with an ATGC-DNA-related PolA subfamily (cluster #9), supporting the hypothesis of convergent DpoZ specialization (20), which stands in contradiction to a postulated congruent evolution with PurZ, a key enzyme in Z synthesis (19). Interestingly, both DpoZ clusters share a unique substitution, carrying phenylalanine in position B4 of motif B (Figure 3): it corresponds to the residue helping to discriminate between the dNTP and NTP substrates (117), in the vicinity of the steric gate with similar functionality (position A2) (118). Nevertheless, it is unlikely to influence the discrimination of adenine versus 2-aminoadenine (119).
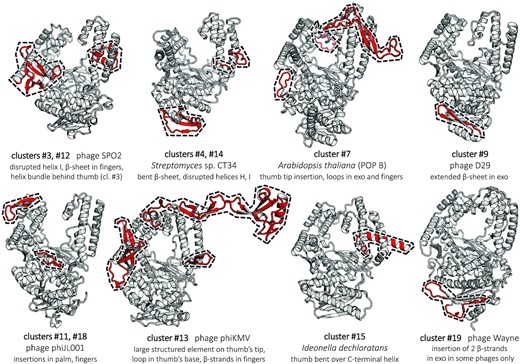
Ribbon representation of PolA structures predicted with AlphaFold2 (39), revealing idiosyncratic structural elements. Keys below the cluster representatives include the associated clusters, source organisms and the description of prominent features conserved within the clusters. These features are highlighted on the models in red and marked by a dashed contour; their amino acid sequence boundaries are specified in Supplementary Table S3. The remaining typical common structural elements are in grey. All structures are viewed from the same angle.
Predicted structural features of PolA subfamilies lacking a crystallographic structure
Structural differences among well-characterized PolA subfamilies are sometimes subtle (Supplementary Figure S3), yet they often prove to be functionally important. Therefore, we aimed to investigate structural features of all 12 clusters that lack an experimental structure (novel or not). We ran AlphaFold2 on five representative sequences in each subfamily. The resulting models, and in particular the observed new features, exhibited high confidence levels of prediction reflected in excellent pLDDT scores (39). These novel elements are characteristic of the individual PolA clusters and appear in all modelled representatives, greatly expanding the known diversity of the PolA fold (Figure 4). Three structured insertions stretch over ∼50 amino acids or more (Supplementary Table S3), joining the group of long PolA-specific insertions also found in T7-like polymerases (TBD) (77) and Pol γ (accessory-interacting determinant, AID) (73).
In cluster #3 PolAs (SPO1-like), we identified an expansion of a β-hairpin in the fingers subdomain by several novel β-strands (Figure 4). This structural element partly overlaps with structural elements in phage T7 PolA (cluster #5), found to be involved in template strand stabilization and in the conformational transition from the elongation to the editing modes (120). Additionally, one helix of the thumb subdomain is disrupted, although an additional stabilization is provided by multiple contacts with a bundle of small helices, whose position corresponds to a β-hairpin in Pol θ and Pol ν (Supplementary Figure S3). Cluster #12 polymerases (KPP25-like) show similar features, except for the presence of the helical bundle.
The ‘broken thumb’ subdomain reappears in related clusters #4, #14 and #15 (APEX 1, APEX 2 and CHEAPs), this time concerning both helices of the stem: in CHEAPs, the thumb bends backwards on a supporting C-terminal helix (Figure 4). We also observe a substantial reshaping of the 3′–5′ exo domain in both APEX subfamilies, resulting in a β-sheet being bent away from the catalytic pocket's side. In APEX 1, a flexible loop blocks the entrance to the inactive 3′–5′ exo site, although APEX 2 models show that this loop can assume a different conformation that unlocks the pocket (Supplementary Figure S4).
Cluster #7 POPs have two loop insertions located below and on the top of fingers, and an additional β-hairpin on the tip of the thumb subdomain. The latter is reminiscent of the TBD of T7 PolA (Figure 4; Supplementary Figure S3), despite their divergent topology.
PolAs D29-like and related Wayne-like DpoZ (clusters #9 and #19) have the typical Klenow fold that is only expanded by two β-strands in the exo domain. Likewise, PolA of phage φJL001 (cluster #11) has two insertions that are shared with φVC8 DpoZ (cluster #18) (20). Indeed, structural predictions of φJL001-like PolAs match closely the experimental structure of the latter (Figure 4; Supplementary Figure S3). In addition, despite quite diverging sequences, the shape of the mobile helices E1 and E2 engulfing the exonuclease's catalytic pocket (20) is conserved among the two subfamilies. Similar elements are also observed in phage T7 PolA, although in this case a corresponding phylogenetic connection is missing.
PhiKMV-like polymerases of cluster #13 are more heavily modified. They display a small insertion at the thumb's base, and a larger one involving multiple new β-strands on the interface between the exo domain and fingers subdomain, yet forming a different arrangement from that in SPO2-like/KPP25-like PolAs discussed above. Most importantly, phiKMV-like polymerases possess a long insertion at the thumb's tip that extends far away (60–70 Å) from the enzyme's core fold, through several β-strands. This insertion is structurally and phylogenetically unrelated to the TBD in T7-like PolAs, although it is equally well positioned for a potential interaction with nascent dsDNA (Supplementary Figure S5).
Unlike other N-terminal fusions, the uracil-DNA glycosylase domain of all UDG-PolAs—inside and outside of cluster #17—occupies a well-defined position in the structure, sandwiched between the edges of the 3′–5′ exonuclease and polymerase domains (Figure 5A). Interestingly, the catalytic sites of the three domains face each other and are almost equidistant (Figure 5B). We observe that an unfolded nascent DNA strand could possibly access either the exo or the UDG active site through relatively simple conformational transitions. In some UDG-PolAs, although not in N4-like enzymes, the family 4 UDG domain has retained all catalytic residues participating in the uracil base excision (121) (Figure 5C). In cluster #17 polymerases, however, the UDG domain lacks a short helix and an important β-hairpin carrying a histidine residue crucial for covalent binding of dU along the catalytic path. It is possible that the UDG domain confers a second editing mode in non-N4-like UDG-PolAs with an active UDG domain, which may be linked to the presence of 5hmU in the DNA of phage SPO1 or similar phages with UDG-PolA (108). Such a possibility is supported by the presence of genes related to nucleotide modification and dUTP processing directly upstream of the UDG-PolA gene in a prophage of Caldanaerobius polysaccharolyticus (NCBI ID: WP_026487659), where the UDG domain has all the catalytic residues conserved (Figure 5C). Alternatively, in the case of UDG inactivation, the domain could possibly increase polymerase processivity during replication.
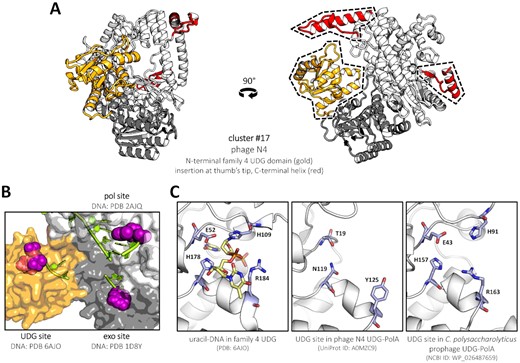
The predicted structure of phage N4 DNA polymerase, representing cluster #17, containing a supplementary domain matching family 4 uracil-DNA glycosylases (UDGs). (A) Ribbon representation, front and side view. The particular position of the UDG domain (gold) on the interface between the exo (grey) and pol (white) domains is conserved among the clade and other, unclassified UDG-PolAs. Other features specific to N4-like UDG-PolAs (red) are mapped and labelled as in Figure 4. (B) Surface representation, top-sideways angle. The three catalytic sites of N4 UDG-PolA are filled with DNA strands modelled from other structures (DNA strands shown as filled lime sticks, nucleotide substrates represented by purple spheres) and labelled above or below. All three pockets reside equidistantly from each other (30–35 Å). (C) Comparison of a functional family 4 UDG protein UdgX (left) with AlphaFold2 models of representative UDG-PolAs. While a deletion and several mutations in the catalytic site clearly inactivate the glycosylase domain of N4-like enzymes (middle), some of the unclassified UDG-PolAs preserve all structural elements and catalytic residues (right). Descriptions including PDB or GenBank IDs are shown below the panels.
PolAs missing the 3′–5′ exonuclease domain: exo/pol domain separation (cluster #5) or formation of a complex with MP (CCPols)
In a number of full-length T7-like phages sequenced recently, such as phage S-SBP1, a truncated polymerase gene is found to correspond only to the pol domain, in agreement with previous results based on metagenomic data (29). Parsing complete genomic sequences, we established that this gene (which we call pol) places itself next to a gene of T7-like 3′–5′ exonuclease, which we call exo. A sequence comparison of truncated and complete close relatives revealed that the ancestor of S-SBP1-like PolAs arose through a +1 frameshift leading to a new STOP codon 13 bp downstream, which terminates the translation of the exonuclease domain (Figure 6A). The simultaneous appearance of a start codon 21 bp upstream of the frameshift ensures the translation of the remaining domain in the original reading frame. This finding indicates that the domains were separated at the genetic level through gene fission, and dismisses the possibility of a constitutive translational frameshift that has been observed in other phages (122). Moreover, an AlphaFold2 structure prediction of the binary complex between the two separate S-SBP1 exo and pol gene products results in a perfect superposition with a model of a related, uninterrupted PolA of phage Syn5; the new protein termini of the split polymerase are simply exposed to the solvent (Figure 6B). This implies that the association between the exo and pol domains is most probably preserved.
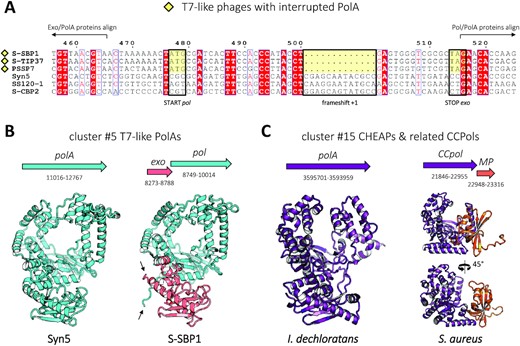
PolA enzymes missing the 3′–5′ exonuclease domain. (A) Genomic multialignment of T7-like PolAs (cluster #5) illustrating the genetic separation of the exo and pol domains. The disruption site of S-SBP1-like PolAs (yellow square) is compared with their close, but uninterrupted homologues. Phage names are given on the left. The start codon of pol, the stop codon of exo and the 1 nt deletion leading to a frameshift and termination of exo are conserved only among the interrupted PolAs (yellow boxes). Amino acid sequences of S-SBP1-like PolAs do not align with their uninterrupted homologues in the region of exo/pol overlap. (B) Predicted structure of a complete PolA of phage Syn5 (left, cyan) is essentially identical to the predicted complex of phage S-SBP1 exo and pol gene products (right, light red and cyan), except for the loose N- and C-termini introduced by the disruption (black arrows). Graphical representation of the corresponding genes along with their nucleotide boundaries is shown above the models, to scale. (C) Similar representation for exonuclease-truncated CCPol of S. aureus (dark violet, right, standard and 45°-rotated view) and its relative, I. dechloratans CHEAP (cluster #15, dark violet, left). CCPol keeps three helices of the exonuclease domain, through which it interacts with the MP protein (orange), according to the AlphaFold2 model.
Although phages with a split PolA are related to phage T7, it is not known whether their polymerases interact with the host's thioredoxin as well. These PolAs display a 32 amino acid deletion in the TBD region, shortening this structural element by half. An AlphaFold2 prediction failed to predict a complex of S-SBP1 PolA with either of the two thioredoxin proteins of the host, Synechococcus sp. WH7803 (123) (UniProt IDs: A5GN01, A5GM53).
In contrast to split T7-like PolAs, in the vicinity of the truncated CCPol gene we have not detected the complementary gene of the missing exo fragment (∼110 amino acids). Nonetheless, CCPols may retain the appropriate fold stability thanks to the three remaining exonuclease helices and unique interactions with MP and Cch2 (100). Indeed, AlphaFold2 predicts a stable MP–CCPol complex between the remainder of the exo domain and the MP protein, which forms a five-stranded β-barrel: their surface of interaction spans 796.4 Å2 (Figure 6C).
Catalytic activities of bacterial Streptomyces sp. CT34 APEX (cluster #4) and I. dechloratans CHEAP (cluster #15)
The conserved catalytic residues present in the pol domain of APEX (cluster #4 and #14 PolAs) indicate that the subfamilies should be functional. To confirm this experimentally, we cloned the gene of one such PolA found in the NCBI RefSeq database, present in the genome of Streptomyces sp. CT34 (GenBank WP_043265455). We overexpressed CT34 APEX in E. coli, purified its His-tagged version and subjected it to primer extension assays, using poly-thymine (dT10) or random nucleotide (dN10) overhanging sequence as templates (Figure 7A).
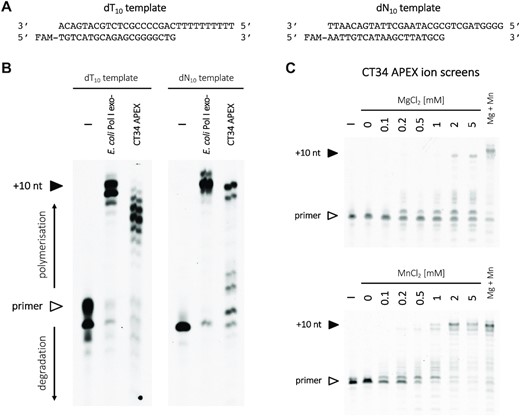
Polymerase activity of Streptomyces sp. CT34 APEX (cluster #4). (A) Two different substrates were used in the assay, with either dT10 or random dN10 template overhangs. (B) Enzymatic extension of the primer visualized on a polyacrylamide gel. Reaction mixtures were incubated at 37°C for 30 min. Lanes to the left represent a negative control without any polymerase, and a positive control with E. coli Pol I (Klenow fragment, 3′–5′ exo-). Bands corresponding to the primer are marked with a white arrow to the left, and fully extended products (+10 nt) with a black arrow. (C) Top: a screen for optimal MgCl2 concentration, between 0 and 5 mM, and no additional MnCl2. Bottom: a similar screen for MnCl2, with no MgCl2 added. The lanes to the left (–) correspond to a negative control, without polymerase; the lanes to the right (Mg + Mn) correspond to CT34 APEX in the presence of both 5 mM MgCl2 and 1 mM MnCl2.
We found the protein to be an active DNA polymerase, albeit not in a very processive way in our experimental conditions (Figure 7B). We screened the optimal Mg2+ concentration and found that the activity reaches a plateau at 5 mM; adding 1 mM Mn2+ increased it noticeably further (Figure 7C). Conversely, while the Mn2+ concentration screen levels off at 1 mM, we observed that adding 5 mM Mg2+ moderately improved the activity. We compared this behaviour with the optimal concentrations of divalent ions for different polymerases presented in a recent review (124), and conclude that CT34 APEX most closely resembles DNA polymerases performing DNA repair. This function would be consistent with its low processivity and exonuclease inactivation.
Using the same approach, we tested the activity of a reference enzyme from another newly defined bacterial PolA cluster. After purification of I. dechloratans CHEAP, we subjected it to a primer extension assay using the dN10 template. The protein is more active than E. coli Pol I, reaching its optimum at 1 mM Mg2+ with no additional Mn2+ needed (Figure 8A, B). Although automatic annotations of close homologues suggested possible thermostability of the enzyme, pre-incubation of I. dechloratans CHEAP at 70, 80 or 90°C for 10 min rendered the enzyme inactive (Supplementary Figure S6).
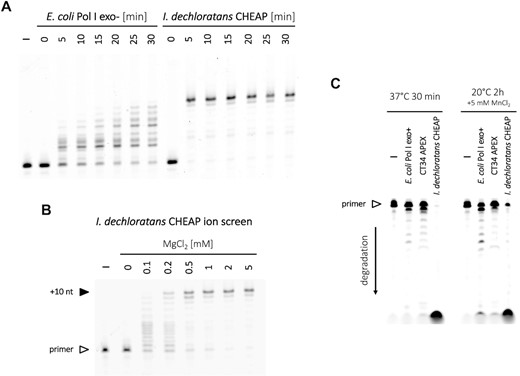
Primer extension activity of I. dechloratans CHEAP (cluster #15) and its strong exonuclease activity compared with CT34 APEX and E. coli Pol I. (A) Primer extension assay visualized on a polyacrylamide gel, with a dN10 overhang templating oligonucleotide (Figure 7A). E. coli Pol I (Klenow fragment, 3′–5′ exo-) and I. dechloratans CHEAP were incubated at 20°C for 0–30 min, as specified above the lanes. The lane to the left represents a negative control without any polymerase. (B) A corresponding MgCl2 concentration screen, between 0 and 5 mM, during 5 min incubation of the primed dN10 template with I. dechloratans CHEAP at 20°C. The negative control on the left had no polymerase added. (C) Exonuclease activity of E. coli Pol I (Klenow fragment), CT34 APEX and I. dechloratans CHEAP, shown for two different conditions specified above the gel images. Reaction mixtures included the dN10 overhang template strand and a labelled primer, with no added dNTPs.
Finally, we examined the exonuclease activities of CT34 APEX and I. dechloratans CHEAP (Figure 8C). Marginal degradation of the primer was observed for exonuclease-inactivated CT34 APEX: this trace activity probably arises from pyrophosphorolysis in the pol domain, which is inherently coupled to DNA and RNA polymerization as their reverse reaction (125,126). Contrastingly, I. dechloratans CHEAP shows pronounced exonuclease activity: this capacity for proofreading indicates that it acts as a genuine replicative DNA polymerase.
DISCUSSION
Being an ancient class of DNA replicators (24,127), family A of DNA polymerases displays the expected diversification of its extant progeny. The complete catalogue of major subfamilies, enriched by the newly determined clusters, allows for its holistic, up-to-date description. It lays the foundation for more sophisticated phylogenetic methods that could shed some light on the earliest evolutionary paths from which PolAs emerged. Nonetheless, our clustering does not include smaller subfamilies, such as mitochondrial PolAs of Trypanosoma-like euglenozoa and certain phages (128), phage T5-like PolAs (129), the aforementioned MGE-related CCPols (100) or other UDG-PolAs outside of the N4-like family: these already described polymerases do not form a proper cluster at the present time. Yet, the ever-growing number of deposited sequences promises that data available in the near future will be sufficient for more comprehensive analyses.
In our study, we could determine that the replacement of tyrosine or phenylalanine with leucine in position B3 is present in two separate superclusters: the ‘broken thumb’ supercluster encompassing APEX and CHEAPs, and the φJL001/φVC8 supercluster (Figure 2). Therefore, the position B3 as an evolutionary marker (30) should be used with caution and preferably in concert with full sequence data, in order to correctly infer common origins of given clades. To date, only one structure of a PolA carrying the leucine B3 variant has been experimentally determined—φVC8 DpoZ (PDB ID: 7PBK). The lack of a detectable relationship between the two DpoZ subfamilies—including their divergence in position B3—is a clear indication for functional convergence inside the PolA family concerning the incorporation of the base Z. New sequencing data could reveal whether the shift of specificity from A towards Z has also arisen in other DNA polymerase families, or other PolA subfamilies.
PolAs are known to structurally require the 3′–5′ exonuclease domain—even in an inactive form—for stability (130,131). The discovery of exo and pol domain separation in a number of T7-like phages (cluster #5) indicates that at least some PolA (S-SBP1-like) enzymes are split in vivo into two interacting components. Despite being an oddity among family A members, such a split is reminiscent of constitutive subunits DP1 (proofreading) and DP2 (elongation) of family D DNA polymerases (132), or multisubunit DNA-dependent RNA polymerases that also have their single-subunit counterparts (133). The separation could entail a differential regulation of the two functions at the gene level or might be necessary for a large conformational change and domain rearrangement during the catalytic cycle. In contrast, the unique example of the CCPol–MP complex predicted by AlphaFold2 demonstrates that a structural substitution of the 3′–5′ exonuclease domain is also possible.
The new structural features predicted by AlphaFold2 for 12 structurally unresolved clusters are all located outside of the catalytic sites; yet, they may contribute to the enzymes’ processivity, stability, their inherent essential dynamics or the binding of potential partners. Such appendices could indirectly influence the catalytic activity of a polymerase, for instance by modifying the conformational space spanned by helix O in the fingers domain, involved in mismatch detection (134). Globally, reliable 3D models can also inform deep phylogenetic searches, as the conservation of a structure takes precedence over that of a sequence (135).
The third domain with a possible UDG activity found in cluster #17 and other phage-related UDG-PolAs transcends a simple polypeptide chain fusion: in all predicted models, this family 4 UDG homologue maintains its firm position and proximity to both pol and exo sites without apparent clashes with the DNA reactants. In principle, PolA could smoothly integrate the UDG activity after polymerase backtracking and before proofreading (136,137), generating an abasic site before its removal by the exonuclease. It remains to be seen whether the putatively active domain found in some UDG-PolA sequences plays a role in the recognition of uracil, 5-hydroxymethyluracil or thymine, possibly participating in the maintenance of DNA modification in some phages. It is also conceivable that the UDG domain acts merely as a processivity module in UDG-PolAs with an inactive UDG, e.g. in N4-like enzymes.
Finally, we present the evidence that two prominent—so far unexplored—bacterial PolA subfamilies, referred to here as APEX and CHEAPs, consist of functional DNA polymerases: their respective activities may be structurally linked to specific substitutions in helix J, which participates in dsDNA binding and regulates the primer extension–proofreading equilibrium (138). Similar activity tests are still needed for phage-derived enzymes of the other new clusters.
Ultimately, detailed knowledge about the differences among the existing PolA subfamilies may inform the choice of specific polymerase candidates during goal-oriented mutagenesis or directed evolution. In a complementary approach, desirable PolA features—such as processivity factors or accessory domains—could be rationally selected and assembled in chimeric enzymes (139). In this way, engineered PolAs with desired traits would have the potential to meet new laboratory or biotechnological needs.
DATA AVAILABILITY
All data, including the final PolA dataset, all sequences from individual clusters, representative AlphaFold2 models and phylogenetic trees are available in the Supplementary Data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Sandrine Rosario for mass spectrometry experiments and Dr Sophia Missoury for helpful discussions and critical reading of the manuscript. We thank the Molecular Biophysics and Macromolecular Interactions Platform at Institut Pasteur for help in characterizing the purified proteins by mass spectrometry.
FUNDING
We thank ANR (Grant ANR 20 CE11 002603 BreakDance) for travelling funds allowing the completion of this project.
Conflict of interest statement. None declared.
Notes
Present address: Dariusz Czernecki, Medical Research Council Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, UK.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments