





Abstract
Schlafen11 (SLFN11) is one of the most studied Schlafen proteins that plays vital roles in cancer therapy and virus-host interactions. Herein, we determined the crystal structure of the Sus scrofa SLFN11 N-terminal domain (NTD) to 2.69 Å resolution. sSLFN11-NTD is a pincer-shaped molecule that shares an overall fold with other SLFN-NTDs but exhibits distinct biochemical characteristics. sSLFN11-NTD is a potent RNase cleaving type I and II tRNAs and rRNAs, and with preference to type II tRNAs. Consistent with the codon usage-based translation suppression activity of SLFN11, sSLFN11-NTD cleaves synonymous serine and leucine tRNAs with different efficiencies in vitro. Mutational analysis revealed key determinates of sSLFN11-NTD nucleolytic activity, including the Connection-loop, active site, and key residues essential for substrate recognition, among which E42 constrains sSLFN11-NTD RNase activity, and all nonconservative mutations of E42 stimulated RNase activities. sSLFN11 inhibited the translation of proteins with a low codon adaptation index in cells, which mainly dependent on the RNase activity of the NTD because E42A enhanced the inhibitory effect, but E209A abolished inhibition. Our findings provide structural characterization of an important SLFN11 protein and expand our understanding of the Schlafen family.
INTRODUCTION
Schlafen genes are a subset of interferon-stimulated response genes (ISGs) unique to mammals (1). The Schlafen (SLFN) protein family was originally discovered in mice in 1998 by Schwarz et al. (2). To date, ten SLFN members have been identified in mice (3,4) and have been categorized into three subgroups based on size and domain organization (Figure 1A). Short SLFN proteins (subgroup I) include mSLFN1, mSLFN1L and mSLFN2 with a molecular weight of 37−42 kDa. They harbor a highly conserved domain, also known as the Schlafen core domain or N-terminal domain (NTD) (1,3). Intermediate SLFN proteins (subgroup II) include mSLFN3 and mSLFN4 with a molecular weight of 58−68 kDa. They contain an additional C-terminal ‘SWADL’ domain, named after the conserved Ser-Trp-Ala-Asp-Leu segment within it (1,3). Long SLFN proteins (subgroup III) include mSLFN5, 8, 9, 10 and 14 with a molecular weight of 100−104 kDa. Long SLFNs possess a large C-terminal extension that belongs to Superfamily I helicases (3,5). Six SLFN proteins (SLFN5, 11, 12, 12L, 13 and 14) have been identified in humans. Except for two shorter members hSLFN12 and hSLFN12L belonging to subgroup II, all other human SLFNs belong to subgroup III (3,6). Phylogenic and sequence alignment analyses suggest that while SLFN5 and SLFN14 are direct orthologs between humans and mice, mSLFN3/4 are orthologs of hSLFN12/12L, and mSLFN8/9/10 are orthologs of hSLFN13 (4,7). By contrast, hSLNF11 lacks murine orthologs (7,8).
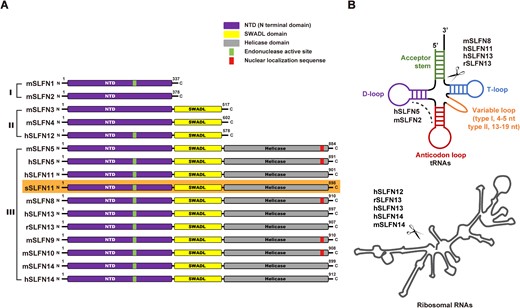
Domain organization and activity of SLFN protein family. (A) Domain architecture of the Schlafen protein family. SLFN proteins are divided into three subgroups: I (small proteins), II (medium proteins) and III (large proteins). Three conserved domains are found in SLFN proteins; the N terminal domain (NTD) is present in all SLFN proteins, the SWADL domain is present in subgroup II and III, and the helicase domain is present in subgroup III. Some SLFN proteins have a putative endonuclease active site (green bars) in their NTD, some SLFN harbors the nuclear localization signal (NLS, red bars). The NLS was predicted by using the cNLS Mapper (https://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi). This paper focuses on sSLFN11 which is highlighted with orange background. The length of SLFN diagrams is not scaled precisely to their size. (B) SLFN proteins exhibit nuclease activity on different RNA substrate, top, tRNAs, bottom rRNAs. SLFN proteins cleaving tRNAs and/or rRNAs are indicated. Some SLFN proteins do not have RNase activity. The letters h, m, r and s represent human, mouse, rat and sus scrofa, respectively.
SLFNs were initially found to be important for thymocyte maturation/activation and T cell quiescence in mice (2,4,7). Accumulating evidence demonstrates that SLFNs play vital roles in numerous biological processes, including T cell-mediated immunity, cell proliferation, cell differentiation, cancer biology, and restriction of viral infection (8). For instance, a recent paper reported that mSLFN2 protects tRNAs from oxidative stress-induced RNase cleavage and thereby counteracts translational suppression caused by T cell activation (9). mSLFN2 also alters the growth and differentiation of mouse hematopoietic cells and plays a vital role in T cell quiescence (9). hSLFN5 elicits regulatory effects on tumorigenesis depending on tumor type (10–12). hSLFN5 also exhibits antiviral activity against HSV-1 infection (13). hSLFN12 regulates differentiation of enterocytes and affects the degradation of several transcription factors (14). hSLFN14 inhibits influenza virus infection by blocking viral nucleoprotein expression and nuclear translocation (15). hSLFN14 also inhibits varicella zoster virus (VZV) replication by suppressing the translation of glycoprotein E and immediate-early protein 62, both required for VZV replication (15). Furthermore, SLFN14 was found to be associated with ribosomes, which results in ribosomal RNA degradation. Several heterozygous missense mutations in human slfn14 gene are responsible for defective megakaryocyte maturation and platelet dysfunction (16,17).
SLFN11 is one of the most studied Schlafen members and attracts significant attention in the areas of cancer treatment and virus restriction. SLFN11 is considered a promising biomarker for various cancer types and can enhance chemosensitivity through different mechanisms. For examples, SLFN11 is recruited to DNA damage sites through direct binding with replication protein A, where it inhibits checkpoint maintenance and homologous recombination (18). SLFN11 suppresses the expression of ATM and ATR involved in DNA damage responses through codon usage-based translation inhibition (19). SLFN11 interacts with damage-specific DNA binding protein 1 (DDB1)-CUL4 CDT2 E3 ubiquitin ligase, thereby promoting degradation of the replication licensing factor CDT1, which subsequently blocks replication irreversibly in response to replicative DNA damage and leads to cell death (20). SLFN11 remodels chromatin and stimulates chromatin accessibility, especially at active promoter regions during replication stress induced by DNA damaging agents (21). When replication is interfered with by DNA damage or improperly activated by cell cycle checkpoint inhibition, SLFN11 is recruited to stressed replication forks, which blocks replication by altering the chromatin structure (22).
Another important function of SLFN11 is virus restriction. SLFN11 expression is upregulated in CD4 + cells of human immunodeficiency (HIV) elite controllers and HIV patients, suggesting its role in suppressing HIV replication (23,24). Mechanisms underlying SLFN11-mediated HIV restriction include codon usage-based inhibition of viral protein expression, which is a similar mechanism adopted by SLFN11 in translational control of host proteins ATR and ATM (25,26). In addition to HIV, SLFN11 was also found to restrict replication of other viruses, including flavivirus (27).
Although human SLFN11 lacks murine orthologs, SLFN11 orthologs with high amino acid sequence similarity are present in many other mammals, suggesting that mammalian SLFN11 orthologs share similar functions. Stabell et al. compared HIV inhibition activity of different SLFN11 orthologs encoded by human and non-human primates and found that the HIV inhibitory activity of hSLFN11 is weaker than that of the other primate orthologs (28). They also demonstrated that SLFN11 orthologs target translation of mRNA transcripts with suboptimal codon usage regardless of virus or host origins (28). In addition to primate SLFN11 orthologs, other mammalian SLFN11 were also found to possess anti-retrovirus activity. For example, equine SLFN11 was found to restrict the replication of equine infectious anemia virus (belonging to the same family as HIV) in a codon usage-dependent manner (29). Cattle SLFN11 and African green monkey SLFN11 were also found to suppress the production of prototype foamy virus (PFV, a unique subfamily of retrovirus) via similar codon preference discrimination (30). These findings suggest that SLFN11 may share highly conserved functions across mammals.
Structural characterization of SLFNs has been proven challenging, which hinders elucidation of the molecular mechanism underlying this unique protein family. Previous studies showed that SLFN11, SLFN12, SLFN13 and SLFN14 harbor RNase activity (Figure 1B), albeit with differences in their RNA substrate specificities, especially for cleaving tRNA (15,16,19,31). Rat SLFN11 (rSLNF13) only cleaves type II tRNAs. hSLFN11 cleaves both type I and II tRNAs, although it cleaves type II tRNA with higher efficiency. Transfer RNAs can be categorized into type I and II classes depending on the size of their variable loop. Type II tRNAs contain the long variable loop (13–19 nt), whereas type I tRNAs contain the short variable loop (4–5 nt, Figure 1B) (32). Escherichia coli has three type II tRNAs (Ser-tRNA, Leu-tRNA and Tyr-tRNA). Of note, human has only two type II tRNAs because human Tyr-tRNA has a short variable loop (5 nt), thus makes it a type I tRNA. SLFN proteins cleave tRNA substrates at specific sites. Estimation from migration rate of the cleavage products in electrophoresis or direct sequencing the cleavage products have revealed that the primary cleavage site of rSLFN13 and hSLFN11 is between the acceptor stem and the T-loop of tRNA substrates (19,31).
By contrast, hSLFN5 and mSLFN2 only bind tRNAs but show no detectable nucleolytic activity (9,12). The crystal structure of rSLFN13 N-terminal domain (rSLFN13-NTD) has been determined, providing the first glimpse of the structure of an SLFN protein. rSLFN13-NTD is a U pillow-shaped pseudo-dimer that exhibits nucleolytic activity targeting type II tRNAs and ribosomal RNAs (19). A recent cryo-electron microscopy (EM) structure of the human PDE3A-SLFN12 complex revealed that hSLFN12-NTD shares a similar fold as rSLFN13-NTD which also harbors RNase activity (33). Intriguingly, a recent structural characterization of hSLFN5 demonstrates that hSLFN5-NTD shares a similar fold with rSLFN13-NTD and hSLFN12-NTD; however, hSLFN5-NTD lacks RNase activity despite binding various nucleic acid substrates, including tRNAs. The cryo-EM structure of full-length hSLFN5 revealed a tripartite domain organization comprising an NTD, a linker domain (SWADL domain), and a C-terminal SF1 helicase domain (12). Nevertheless, molecular determinants governing the nucleolytic activity of SLFN-NTD remain to be identified. Because the SLFN-RNA complex has not been structurally characterized, the RNA binding mode of SLFN, the RNase-active site in NTD, and the regulatory mechanism of the nucleolytic activity remain elusive.
It worth noting, that tRNAs are extensively modified in cells to ensure their stability, structure and translation efficiency. There are at least 93 known tRNA modifications (34), some of which are important to the correct folding of tRNAs. The absence of certain modifications can affect tRNA folding, thus making them susceptible to ribonucleases (35). Whether tRNA modification has impact on SLFN protein-mediated tRNA binding or cleavage demands further investigation.
Here, we determined the crystal structure of Sus scrofa SLFN11-NTD. Although sSLFN11-NTD shares a pincer-like overall fold with rSLFN13-NTD, hSLFN12-NTD, and hSLFN5-NTD, it exhibited distinct RNase activity. sSLFN11-NTD bound to tRNA with high affinity (KD = 1.9 μM) and exhibited broader substrate specificity; it cleaves type I and type II tRNAs, and rRNAs. sSLFN11 autoinhibited its own expression and that of HIV Gag and p24 in cells, based on codon usage frequency. sSLFN11-NTD cleaved synonymous-codon tRNAs with different efficiencies, providing evidence for its codon usage-based protein translation inhibition. Mutagenesis studies identified a panel of charged residues of sSLFN11-NTD located between the two jaws of the pincer-like molecule, among which E42 is a key regulator that controls the RNase activity of sSLFN11 and its translation inhibition function in cells. Furthermore, we identified three key residues of sSLFN11 (R139, K211 and K251) governing the RNase activity of sSLFN11-NTD.
MATERIALS AND METHODS
Protein expression and purification
The genes encoding Sus scrofa Schlafen family member 11 protein (sSLFN11, Gene ID: 100625890) and Rattus norvegicus Schlafen family member 13 (rSLFN13, Gene ID: 303378) were synthesized by Genscript and amplified by polymerase chain reaction (PCR). The DNA encoding residues 1–352 of sSLFN11 (sSLFN11-NTD) and 1–353 of rSLFN13 (rSLFN13-NTD) were individually cloned into a modified pET-28a vector between BamH I and Xho I restriction sites with a N-terminal 6× His-SUMO tag followed by a ULP1 (ubiquitin-like-specific protease 1) cleavage site. Plasmids encoding the mutant lacking residue 166–180 (Connection-loop) (termed as sSLFN11-NTD-Δ) and other sSLFN11-NTD mutants were generated by using site-directed mutagenesis (QuikChangeTM) and verified by DNA sequencing. To express rSLFN13, sSLFN11-NTD or its mutants, the plasmids were transformed into E. coli BL21 (DE3) competent cells. The bacteria were cultured at 37°C in Luria-Bertani broth (LB) to OD600 = 1.0. Then induced the bacteria with IPTG (isopropylthio-β-galactoside) to 0.5 mM and continued culturing at 18°C overnight. The bacteria cells were harvested by centrifugation at 5422 × g for 30 min. The cell pellets were resuspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole) and disrupted by ultrasonication. The lysate was clarified by centrifugation at 47 850 × g for 60 min and the supernatant was applied to Ni-NTA (QIAGEN) resin pre-equilibrated with lysis buffer. After washing the unbound proteins with lysis buffer, the resin with the bound proteins was incubated with ULP1 at 4°C overnight to remove the 6× His-SUMO tag. The untagged proteins (sSLFN11-NTD or its mutants, rSLFN13) were collected in flowthrough from the column, and were individually loaded to a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with gel filtration buffer (10 mM Tris–HCl, pH 8.0, 100 mM NaCl). Selenomethionine (Se-Met) derivatized sSLFN11-NTD-Δ was expressed in E. coli B834 (DE3) competent cells and grown in LeMASTER medium containing l-selenomethionine following the protocols suggested by the manufactures (Molecular Dimensions). The purification protocol was the same as for the native sSLFN11-NTD-Δ. The purified sSLFN11-NTD or its mutants was concentrated to 10 mg ml−1 and stored at −80°C for further experiments.
The gene encoding full-length of sSLFN11 (1–898 aa) (sSLFN11-FL) was amplified by PCR and inserted into a modified pFastbac-1 baculovirus transfer vector containing an N-terminal 6 × His-SUMO tag. sSLFN11-FL was overexpressed in sf21 insect cells using Bac-to-Bac Baculovirus Expression System (Invitrogen). One liter cell (2.0 × 106 cells ml−1) were infected with 30 ml baculovirus at 28°C for 48 h, cells were subsequently harvest by centrifugation at 5422 × g for 20 min. The cell pellets were re-suspended in the lysis buffer containing 25 mM Tris–HCl, pH 8.0, 300 mM NaCl, 2 mM MgCl2, 20 mM imidazole and lysed by ultrasonification (work 3.0 s, stop 5.0 s, power 200 W, time 20 min). The lysate was clarified by centrifugation at 47 850 × g for 60 min and the supernatant was incubated with Ni-NTA resin, the bound proteins were digested with Pre-Scission protease to remove the N-terminal 6 × His-SUMO tag. The untagged sSLFN11-FL was collected in flowthrough and then loaded to a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with gel filtration buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl). Purified sSLFN11-FL was concentrated to 1 mg ml−1 and stored at –80°C for further experiments.
Crystallization and structure determination
SeMet derivatized sSLFN11-NTD-Δ was concentrated to 10 mg ml−1 and crystallized by mixing 1 μl sample with 1 μl reservoir buffer containing 0.1 M HEPES pH 7.5, 22% PEG3350, 0.15 M Li2SO4 in a hanging drop vapor diffusion system at 18°C. The crystals of SeMet derivatized sSLFN11-NTD-Δ appeared after 3 days, the shape of the crystals is prismatic. Single crystals were transferred to reservoir buffer containing 10% glycerol before flash-freezing in liquid nitrogen. Highly redundant X-ray diffraction data of a SeMet sSLFN11-NTD-Δ crystal was collected at the Shanghai Synchrotron Radiation Facility (SSRF) at beamline BL19U1. The wavelength of the X-ray used for diffraction experiments was set at the peak absorption edge (0.979Å), which is the peak adoption edge of the Se atoms. The data was processed using XDS package (36). The crystal belonged to the space group of P6122, diffracted the X-ray 2.69 Å and contained 1 molecule per asymmetric unit. Because sSLFN11-NTD-Δ contained 10 SeMet, 10 Se atoms were available per asymmetric unit. In the diffraction data, Anomal Corr (percentage of correlation between random half-sets of anomalous intensity differences) with correlation significant at the 0.1% level was observed up to 3.05 Å resolution limit. The crystal structure was then solved by single wavelength anomalous diffraction (SAD) using software SHELXC/D/E, which yielded an interpretable initial electron density map. Manual model building was conducted using software Coot and the structure was finally refined using software PHENIX (37). The statistics of data collection and structure refinement are summarized in Table 1. Structural presentations were prepared using software Pymol.
| tRNA | GtRNAdb Gene Symbol | Sequence (5’ -> 3’) |
|---|---|---|
| tRNA-Leu TAA | tRNA-Leu-TAA-1–1 | ACCAGGATGGCCGAGTGGTTAAGGCGTTGGACTTAAGATCCAATGGACATATGTCCGCGTGGGTTCGAACCCCACTCCTGGTACCA |
| tRNA-Leu AAG | tRNA-Leu-AAG-1–1 | GGTAGCGTGGCCGAGCGGTCTAAGGCGCTGGATTAAGGCTCCAGTCTCTTCGGAGGCGTGGGTTCGAATCCCACCGCTGCCA |
| tRNA-Leu CAA | tRNA-Leu-CAA-2–1 | GTCAGGATGGCCGAGTGGTCTAAGGCGCCAGACTCAAGTTCTGGTCTCCGTATGGAGGCGTGGGTTCGAATCCCACTTCTGACACCA |
| tRNA-Leu CAG | tRNA-Leu-CAG-1–1 | GTCAGGATGGCCGAGCGGTCTAAGGCGCTGCGTTCAGGTCGCAGTCTCCCCTGGAGGCGTGGGTTCGAATCCCACTCCTGACACCA |
| tRNA-Ser AGA | tRNA-Ser-AGA-1–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTAGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-Ser CGA | tRNA-Ser-CGA-1–1 | GCTGTGATGGCCGAGTGGTTAAGGCGTTGGACTCGAAATCCAATGGGGTCTCCCCGCGCAGGTTCGAATCCTGCTCACAGCGCCA |
| tRNA-Ser GCT | tRNA-Ser-GCT-1–1 | GACGAGGTGGCCGAGTGGTTAAGGCGATGGACTGCTAATCCATTGTGCTCTGCACGCGTGGGTTCGAATCCCACCCTCGTCGCCA |
| tRNA-Ser TGA | tRNA-Ser-TGA-2–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTTGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-His GTG | tRNA-His-GTG-1–7 | GGCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGTGGCCGCAGCAACCTCGGTTCGAATCCGAGTCACGGCACCA |
| tRNA-Gly CCC | tRNA-Gly-CCC-1–1 | GCGCCGCTGGTGTAGTGGTATCATGCAAGATTCCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGCACCA |
| tRNA-Cys GCA | tRNA-Cys-GCA-2–2 | GGGGGTATAGCTCAGGGGTAGAGCATTTGACTGCAGATCAAGAGGTCCCTGGTTCAAATCCAGGTGCCCCCTCCA |
| tRNA-Thr TGT | tRNA-Thr-TGT-1–1 | GGCTCCATAGCTCAGGGGTTAGAGCACTGGTCTTGTAAACCAGGGGTCGCGAGTTCAAATCTCGCTGGGGCCTCCA |
| tRNA-Sec TCA | tRNA-Sec-TCA-1–1 | GCCCGGATGATCCTCAGTGGTCTGGGGTGCAGGCTTCAAACCTGTAGCTGTCTAGCGACAGAGTGGTTCAATTCCACCTTTCGGGCGCCA |
| tRNA | GtRNAdb Gene Symbol | Sequence (5’ -> 3’) |
|---|---|---|
| tRNA-Leu TAA | tRNA-Leu-TAA-1–1 | ACCAGGATGGCCGAGTGGTTAAGGCGTTGGACTTAAGATCCAATGGACATATGTCCGCGTGGGTTCGAACCCCACTCCTGGTACCA |
| tRNA-Leu AAG | tRNA-Leu-AAG-1–1 | GGTAGCGTGGCCGAGCGGTCTAAGGCGCTGGATTAAGGCTCCAGTCTCTTCGGAGGCGTGGGTTCGAATCCCACCGCTGCCA |
| tRNA-Leu CAA | tRNA-Leu-CAA-2–1 | GTCAGGATGGCCGAGTGGTCTAAGGCGCCAGACTCAAGTTCTGGTCTCCGTATGGAGGCGTGGGTTCGAATCCCACTTCTGACACCA |
| tRNA-Leu CAG | tRNA-Leu-CAG-1–1 | GTCAGGATGGCCGAGCGGTCTAAGGCGCTGCGTTCAGGTCGCAGTCTCCCCTGGAGGCGTGGGTTCGAATCCCACTCCTGACACCA |
| tRNA-Ser AGA | tRNA-Ser-AGA-1–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTAGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-Ser CGA | tRNA-Ser-CGA-1–1 | GCTGTGATGGCCGAGTGGTTAAGGCGTTGGACTCGAAATCCAATGGGGTCTCCCCGCGCAGGTTCGAATCCTGCTCACAGCGCCA |
| tRNA-Ser GCT | tRNA-Ser-GCT-1–1 | GACGAGGTGGCCGAGTGGTTAAGGCGATGGACTGCTAATCCATTGTGCTCTGCACGCGTGGGTTCGAATCCCACCCTCGTCGCCA |
| tRNA-Ser TGA | tRNA-Ser-TGA-2–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTTGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-His GTG | tRNA-His-GTG-1–7 | GGCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGTGGCCGCAGCAACCTCGGTTCGAATCCGAGTCACGGCACCA |
| tRNA-Gly CCC | tRNA-Gly-CCC-1–1 | GCGCCGCTGGTGTAGTGGTATCATGCAAGATTCCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGCACCA |
| tRNA-Cys GCA | tRNA-Cys-GCA-2–2 | GGGGGTATAGCTCAGGGGTAGAGCATTTGACTGCAGATCAAGAGGTCCCTGGTTCAAATCCAGGTGCCCCCTCCA |
| tRNA-Thr TGT | tRNA-Thr-TGT-1–1 | GGCTCCATAGCTCAGGGGTTAGAGCACTGGTCTTGTAAACCAGGGGTCGCGAGTTCAAATCTCGCTGGGGCCTCCA |
| tRNA-Sec TCA | tRNA-Sec-TCA-1–1 | GCCCGGATGATCCTCAGTGGTCTGGGGTGCAGGCTTCAAACCTGTAGCTGTCTAGCGACAGAGTGGTTCAATTCCACCTTTCGGGCGCCA |
| tRNA | GtRNAdb Gene Symbol | Sequence (5’ -> 3’) |
|---|---|---|
| tRNA-Leu TAA | tRNA-Leu-TAA-1–1 | ACCAGGATGGCCGAGTGGTTAAGGCGTTGGACTTAAGATCCAATGGACATATGTCCGCGTGGGTTCGAACCCCACTCCTGGTACCA |
| tRNA-Leu AAG | tRNA-Leu-AAG-1–1 | GGTAGCGTGGCCGAGCGGTCTAAGGCGCTGGATTAAGGCTCCAGTCTCTTCGGAGGCGTGGGTTCGAATCCCACCGCTGCCA |
| tRNA-Leu CAA | tRNA-Leu-CAA-2–1 | GTCAGGATGGCCGAGTGGTCTAAGGCGCCAGACTCAAGTTCTGGTCTCCGTATGGAGGCGTGGGTTCGAATCCCACTTCTGACACCA |
| tRNA-Leu CAG | tRNA-Leu-CAG-1–1 | GTCAGGATGGCCGAGCGGTCTAAGGCGCTGCGTTCAGGTCGCAGTCTCCCCTGGAGGCGTGGGTTCGAATCCCACTCCTGACACCA |
| tRNA-Ser AGA | tRNA-Ser-AGA-1–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTAGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-Ser CGA | tRNA-Ser-CGA-1–1 | GCTGTGATGGCCGAGTGGTTAAGGCGTTGGACTCGAAATCCAATGGGGTCTCCCCGCGCAGGTTCGAATCCTGCTCACAGCGCCA |
| tRNA-Ser GCT | tRNA-Ser-GCT-1–1 | GACGAGGTGGCCGAGTGGTTAAGGCGATGGACTGCTAATCCATTGTGCTCTGCACGCGTGGGTTCGAATCCCACCCTCGTCGCCA |
| tRNA-Ser TGA | tRNA-Ser-TGA-2–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTTGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-His GTG | tRNA-His-GTG-1–7 | GGCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGTGGCCGCAGCAACCTCGGTTCGAATCCGAGTCACGGCACCA |
| tRNA-Gly CCC | tRNA-Gly-CCC-1–1 | GCGCCGCTGGTGTAGTGGTATCATGCAAGATTCCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGCACCA |
| tRNA-Cys GCA | tRNA-Cys-GCA-2–2 | GGGGGTATAGCTCAGGGGTAGAGCATTTGACTGCAGATCAAGAGGTCCCTGGTTCAAATCCAGGTGCCCCCTCCA |
| tRNA-Thr TGT | tRNA-Thr-TGT-1–1 | GGCTCCATAGCTCAGGGGTTAGAGCACTGGTCTTGTAAACCAGGGGTCGCGAGTTCAAATCTCGCTGGGGCCTCCA |
| tRNA-Sec TCA | tRNA-Sec-TCA-1–1 | GCCCGGATGATCCTCAGTGGTCTGGGGTGCAGGCTTCAAACCTGTAGCTGTCTAGCGACAGAGTGGTTCAATTCCACCTTTCGGGCGCCA |
| tRNA | GtRNAdb Gene Symbol | Sequence (5’ -> 3’) |
|---|---|---|
| tRNA-Leu TAA | tRNA-Leu-TAA-1–1 | ACCAGGATGGCCGAGTGGTTAAGGCGTTGGACTTAAGATCCAATGGACATATGTCCGCGTGGGTTCGAACCCCACTCCTGGTACCA |
| tRNA-Leu AAG | tRNA-Leu-AAG-1–1 | GGTAGCGTGGCCGAGCGGTCTAAGGCGCTGGATTAAGGCTCCAGTCTCTTCGGAGGCGTGGGTTCGAATCCCACCGCTGCCA |
| tRNA-Leu CAA | tRNA-Leu-CAA-2–1 | GTCAGGATGGCCGAGTGGTCTAAGGCGCCAGACTCAAGTTCTGGTCTCCGTATGGAGGCGTGGGTTCGAATCCCACTTCTGACACCA |
| tRNA-Leu CAG | tRNA-Leu-CAG-1–1 | GTCAGGATGGCCGAGCGGTCTAAGGCGCTGCGTTCAGGTCGCAGTCTCCCCTGGAGGCGTGGGTTCGAATCCCACTCCTGACACCA |
| tRNA-Ser AGA | tRNA-Ser-AGA-1–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTAGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-Ser CGA | tRNA-Ser-CGA-1–1 | GCTGTGATGGCCGAGTGGTTAAGGCGTTGGACTCGAAATCCAATGGGGTCTCCCCGCGCAGGTTCGAATCCTGCTCACAGCGCCA |
| tRNA-Ser GCT | tRNA-Ser-GCT-1–1 | GACGAGGTGGCCGAGTGGTTAAGGCGATGGACTGCTAATCCATTGTGCTCTGCACGCGTGGGTTCGAATCCCACCCTCGTCGCCA |
| tRNA-Ser TGA | tRNA-Ser-TGA-2–1 | GTAGTCGTGGCCGAGTGGTTAAGGCGATGGACTTGAAATCCATTGGGGTTTCCCCGCGCAGGTTCGAATCCTGCCGACTACGCCA |
| tRNA-His GTG | tRNA-His-GTG-1–7 | GGCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGTGGCCGCAGCAACCTCGGTTCGAATCCGAGTCACGGCACCA |
| tRNA-Gly CCC | tRNA-Gly-CCC-1–1 | GCGCCGCTGGTGTAGTGGTATCATGCAAGATTCCCATTCTTGCGACCCGGGTTCGATTCCCGGGCGGCGCACCA |
| tRNA-Cys GCA | tRNA-Cys-GCA-2–2 | GGGGGTATAGCTCAGGGGTAGAGCATTTGACTGCAGATCAAGAGGTCCCTGGTTCAAATCCAGGTGCCCCCTCCA |
| tRNA-Thr TGT | tRNA-Thr-TGT-1–1 | GGCTCCATAGCTCAGGGGTTAGAGCACTGGTCTTGTAAACCAGGGGTCGCGAGTTCAAATCTCGCTGGGGCCTCCA |
| tRNA-Sec TCA | tRNA-Sec-TCA-1–1 | GCCCGGATGATCCTCAGTGGTCTGGGGTGCAGGCTTCAAACCTGTAGCTGTCTAGCGACAGAGTGGTTCAATTCCACCTTTCGGGCGCCA |
Transfection and whole cell lysis
Original or codon optimized genes of sSLFN11-FL or its mutants were cloned into the pcDNA6-V5-HisB vector between restriction sites BamH I and Xho I, and a C-terminal V5-tag was fused to the proteins. Gag-WT contains wild-type HIV-Gag gene sequence from pGag-EGFP (NIH AIDS Research & Reference Reagent Program, no. 11468), in which only the inhibitory RNA sequence was eliminated by introducing silent mutations (38), otherwise retaining the original viral sequence. Gag-LS contains codon optimized gene, in which all serine or leucine were encoded by the high-frequent codon, (Ser) AGC and (Leu) CUG, respectively. Gag-opt contains overall codon-optimized Gag gene. All optimized genes were synthesized by Genscript and were cloned to pcDNA6-V5-His B vector between restriction sites BamH I and Xho I; a C-terminal V5-tag was fused to the constructs. The genes encoding C-terminal V5-tagged EGFP, human SLFN11 and its mutants were synthesized and inserted into pcDNA6-V5-His B vector. The pNL4-3.Luc.R-E- plasmid, encodes an Env-defective, luciferase reporter expressing HIV-1 genome plasmid, was kindly provided by Dr Yuxian He.
HEK-293T cells were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U ml−1 penicillin (Solarbio) and 100 μg ml−1 streptomycin (Solarbio) at 37°C, in the presence 5% CO2. HEK-293T cells were transfected with the plasmids harboring original sSLFN11 gene (or its mutants E42A and E209A) alone or co-transfected with pNL4-3.Luc. R-E- plasmid. The plasmid harboring codon-optimized sSLFN11 gene, human Schlafen11 gene or indicated SLFN11 mutants was co-transfected with the V5-tagged Gag-WT, Gag-LS, Gag-opt, EGFP or pNL4-3.Luc.R-E- plasmid into HEK-293T cells using Lipofectamine 2000 (Invitrogen). At 48 h post-transfection, cells were harvested and lysed by pre-cooled RIPA lysis buffer (Solarbio) supplemented with proteinase inhibitor cocktail (final concentration 1 mM) (Roche) and 1 mM PMSF (phenylmethylsulfonyl fluoride) (final concentration 1 mM) at 4°C for 1 h. The lysate was centrifuged at 12 000 × g for 10 min in 4°C, and the supernatants were stored at −80°C used for further western blot assays.
Western blot
The supernatant of whole cell lysate was separated by 10% SDS-PAGE at 80 V for 30 min then 120 V for 1.5 h at room temperature, and transferred to a 0.22 μM PVDF (polyvinylidine difluoride membrane) membrane (Millipore) using a Criterion blotter (Bio-Rad) at 4°C, 300 mA for 2 h. The PVDF membrane was blocked for 1 h in the blocking buffer containing 5% skimmed milk dissolved in 1× TBST (Solarbio) at room temperature, washed the membranes with 1× TBST and then incubated the membranes with target-specific primary antibodies at 4°C overnight. The membranes were washed 10 min for 3 times with 1× TBST before incubating with corresponding secondary antibodies 1 h at room temperature. Washed the membranes 10 min for 3 times with 1× TBST again, then analyzed and visualized with Odyssey (LI-COR). Following antibodies used in this paper for Western Blot assays are list as below: Mouse anti-V5 antibody (Abclonal, 1:500, Cat#: AE017), Mouse anti-GAPDH monoclonal antibody (Abclonal, 1: 50000, Cat#: AC033), Rabbit polyclonal EGFP antibody (Wanleibio, 1:500, Cat#: WL03189), Rabbit monoclonal p24 antibody (Sino Biological, 1: 100, Cat#: 11695-RP02); Goat anti-Mouse secondary antibody IRDye@800CW (LI-COR, 1:10 000, Cat#: 926-32210) and Goat anti-Rabbit secondary antibody IRDye@800CW (LI-COR, 1:10000, Cat#: 926-32211).
In vitro transcription of tRNA substrate
To prepare unlabeled human tRNAs, we purchase the synthesized plasmids containing the DNA fragments corresponding to the T7 promoter plus human tRNA from Synbio-tech, the gene were ligated into the pUC57 vector using the restriction sites BamH I and Xho I. We obtained the transcription templates by PCR amplification of ligated DNA fragments and all the PCR products were extracted by DNA Gel Extraction kit (Axygen). The in vitro transcriptions were carried out by using the T7 in vitro Transcription kit (Biomisc) following the protocol provided, the tRNA transcripts were labeled with the Cy3 following the protocol which was provided by Label IT® Nucleic Acid Labeling kit (Mirus). Cy3 fluorescence signal could be detected and analyzed by using the Typhoon Trio Variable Mode Imager (GE healthcare). The tRNA gene sequences used in this study were chosen based on the predictions from GtRNAdb (http://gtrnadb.ucsc.edu/GtRNAdb2/index.html) and summarized in table above.
In vitro cleavage assay
For substrates screening assays, 1 μM sSLFN11-NTD was individually incubated with 50 nM Cy3-labeled ssRNA, ssDNA, dsRNA, dsDNA, stem–loop RNA or tRNA-Ser in each cleavage buffer (10 μl) containing 40 mM Tris–HCl, pH 8.0, 20 mM KCl, 4 mM MgCl2 and 2 mM DTT at 37°C for 30 min, the reaction was terminated with 2 × RNA loading Dye (95% formamide, 0.02% SDS, 0.02% bromophenol blue, 0.01% xylene cyanol, 1 mM EDTA) (NEB). The cleavage products were then analyzed by electrophoresis at 150 V for 40 min with 15% denaturing 8M urea-polyacrylamide gel (urea–PAGE) at room temperature, and the running buffer is 1× TBE. The gel was scanned and analysis with Typhoon Trio Variable Mode Imager. All the Cy3-labbled RNA and DNA strands used in this experiment are synthesized in Sangon Biotech (Shang Hai). The sequences of nucleic acids are summarized and listed in below table.
| Nucleic acids | Sequences (5’ -> 3’) | Modification |
|---|---|---|
| ssRNA | GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| ssDNA | GGCTTTTGACCTTTATGC | 5’-Cy3 |
| dsRNA | Sense:GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| Anti-sense:GCAUAAAGGUCAAAAGCC | ||
| dsDNA | Sense:GGCTTTTGACCTTTATGC | 5’-Cy3 |
| Anti-sense:GCATAAAGGTCAAAAGCC | ||
| stem-loop RNA | AUAAAGGUCAUUCGCAAGAGUGGCCUUUAU | 5’-Cy3 |
| Nucleic acids | Sequences (5’ -> 3’) | Modification |
|---|---|---|
| ssRNA | GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| ssDNA | GGCTTTTGACCTTTATGC | 5’-Cy3 |
| dsRNA | Sense:GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| Anti-sense:GCAUAAAGGUCAAAAGCC | ||
| dsDNA | Sense:GGCTTTTGACCTTTATGC | 5’-Cy3 |
| Anti-sense:GCATAAAGGTCAAAAGCC | ||
| stem-loop RNA | AUAAAGGUCAUUCGCAAGAGUGGCCUUUAU | 5’-Cy3 |
| Nucleic acids | Sequences (5’ -> 3’) | Modification |
|---|---|---|
| ssRNA | GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| ssDNA | GGCTTTTGACCTTTATGC | 5’-Cy3 |
| dsRNA | Sense:GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| Anti-sense:GCAUAAAGGUCAAAAGCC | ||
| dsDNA | Sense:GGCTTTTGACCTTTATGC | 5’-Cy3 |
| Anti-sense:GCATAAAGGTCAAAAGCC | ||
| stem-loop RNA | AUAAAGGUCAUUCGCAAGAGUGGCCUUUAU | 5’-Cy3 |
| Nucleic acids | Sequences (5’ -> 3’) | Modification |
|---|---|---|
| ssRNA | GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| ssDNA | GGCTTTTGACCTTTATGC | 5’-Cy3 |
| dsRNA | Sense:GGCUUUUGACCUUUAUGC | 5’-Cy3 |
| Anti-sense:GCAUAAAGGUCAAAAGCC | ||
| dsDNA | Sense:GGCTTTTGACCTTTATGC | 5’-Cy3 |
| Anti-sense:GCATAAAGGTCAAAAGCC | ||
| stem-loop RNA | AUAAAGGUCAUUCGCAAGAGUGGCCUUUAU | 5’-Cy3 |
For cleavage assays on in vitro transcript Cy3-labeled tRNAs, protein of increasing concentration or a certain concentration (1, 4 or 8 μM) was incubated with 50 nM Cy3-labeled tRNA in the cleavage buffer (10 μl) at 37°C for different time points as indicated, and the reaction was terminated by adding the 2× RNA loading dye. Where indicated, MgCl2 (4 mM), MnCl2 (4 mM), CaCl2 (4 mM), ZnCl2 (4 mM) or EDTA (4 mM) was added. The cleavage products were resolved in the 15% denaturing 8 M urea–PAGE with 1× TBE running buffer at room temperature at 150 V for 1.5 h. All the gels were scanned with Typhoon Trio Variable Mode Imager (GE healthcare).
For cleavage assays on native rRNAs and tRNAs, total RNA was isolated with TRIzol™ Regent (Invitrogen) from HEK293T cells following the manufacturer's protocol, including rRNAs and tRNAs and subsequently used in the cleavage assays. Protein with indicated concentration was incubated with 2 μg total RNA in cleavage buffer (10 μl) at 37°C for 30 min, an equal volume of 2× RNA loading Dye containing SYBR Gold (Invitrogen) was added to the reaction mixture to stop reaction. Cleavage products were analyzed by electrophoresis with 1% native agarose gel for the analysis of 28S and 18S rRNA at 130 V for 30 min in the running buffer (1 × TAE), after electrophoresis, the gel was visualized by UV light. Cleavage products were also resolved to 10% denaturing 8 M urea™PAGE at 120 V for 1 h 15 min for the analysis of small RNAs or tRNA, the gel was stained with SYBR Gold and then visualized by UV light.
Biolayer interferometry
We conducted our experiment at 25°C in the buffer containing PBS and 0.005% surfactant P20 by the Octet RED96e instrument (Fortebio). The volume for the solutions were 200 μl, and the assay was performed in black, solid, flat bottom 96-well plates with a shaking speed of 1000 rpm. The streptavidin (SA) biosensors were activated in double distilled water for at least 10 min. A time of 60 s was performed for sensor check before loading the biotin labeled tRNA-Ser UGA (200 nM), which took 300 s. A time of 50 s was taken for protein association in the following step. The dissociation was the last step and performed for 100 s. The reference baseline was recorded for a sensor loaded with ligand but no analyte which used to be subtracted to correct any systematic baseline drift. The tRNA labeled with 5’- biotin labeled was synthesized by Synbio-tech. 200 nM of biotinylated tRNAs were immobilized onto the surface of SA biosensors. To compare the binding ability between different mutant proteins with tRNA-Ser UGA, 12.5 μM sSLFN11-NTD or its mutants were used to associate with 200 nM tRNA-Ser UGA coated SA biosensors. All data were processed by Data Analysis 11.
Northern blot analysis of tRNAs
Total RNA was purified from HEK293T cells using TRIzol Regent and separated by 10% TBE-Urea gels, subsequently transferred onto Amersham Hybond™-N+ Nylon membranes (GE Healthcare) at 350 mA for 1 h in 0.5× TBE buffer. After transfer, crosslinked transferred RNA to Membrane at 120 mJ/cm2 using the UV-light crosslinking instrument (CL-1000 Ultraviolet Crosslinker). The membrane was pre-hybridized in the Dig easy hyb (Roche) at 42°C for 30 min, following hybridized the membrane with digoxigenin-labeled DNA oligo probes (synthesized in Tsingke) at 42°C for at least 6 h or overnight, respectively. Rinsing and washing the hybridized membrane with 1× SSC with containing 0.1% SDS 2 times for 20 min each. Later, the membrane was blocked with 1× Blocking solution (Roche) for at least 30 min, and then incubated the membrane with Anti-Digoxigenin AP (Roche, 1:10000) for 30 min at room temperature. Membrane was rinsed and washed 2 times again in wash-buffer (0.1 M maleic acid, 0.15 M NaCl pH 7.5, 0.3% Tween 20) for 15 min each, then analyzed and visualized with Tanon 5200. The DNA probe sequences used in this assay were chosen based on predictions from GtRNAdb. The sequences of the DNA probes are summarized and listed in table on next page.
| tRNA | Probe sequence (5’ -> 3’) |
|---|---|
| tRNA-Leu TAA | CCATTGGATCTTAAGTCCAACGCCTTAACCACTC |
| tRNA-Leu AAG | AGCCTTAATCCAGCGCCTTAGACCGCTCGGCCACGCTACC |
| tRNA-Leu CAA | GGAGACCAGAACTTGAGTCTGGCGCCTTAGACCA |
| tRNA-Leu CAG | CACGCCTCCAGGGGAGACTGCGACCTGAACGCAGCGCCTT |
| tRNA-Ser AGA | GATTTCTAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-Ser CGA | CCCCATTGGATTTCGAGTCCAACGCCTTAACCACTCGGCCA |
| tRNA-Ser GCT | GGATTAGCAGTCCATCGCCTTAACCACTCGGCCACCTCGTC |
| tRNA-Ser TGA | TGGATTTCAAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-His GTG | CGAGGTTGCTGCGGCCACAACGCAGAGTACTAA |
| tRNA-Thr TGT | AGGCCCCAGCGAGGATCGAACTCGCGACCCCTGG |
| tRNA-Sec TCA | CACTCTGTCGCTAGACAGCTACAGGTTTGAAGCCTGCACCCCAGAC |
| tRNA | Probe sequence (5’ -> 3’) |
|---|---|
| tRNA-Leu TAA | CCATTGGATCTTAAGTCCAACGCCTTAACCACTC |
| tRNA-Leu AAG | AGCCTTAATCCAGCGCCTTAGACCGCTCGGCCACGCTACC |
| tRNA-Leu CAA | GGAGACCAGAACTTGAGTCTGGCGCCTTAGACCA |
| tRNA-Leu CAG | CACGCCTCCAGGGGAGACTGCGACCTGAACGCAGCGCCTT |
| tRNA-Ser AGA | GATTTCTAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-Ser CGA | CCCCATTGGATTTCGAGTCCAACGCCTTAACCACTCGGCCA |
| tRNA-Ser GCT | GGATTAGCAGTCCATCGCCTTAACCACTCGGCCACCTCGTC |
| tRNA-Ser TGA | TGGATTTCAAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-His GTG | CGAGGTTGCTGCGGCCACAACGCAGAGTACTAA |
| tRNA-Thr TGT | AGGCCCCAGCGAGGATCGAACTCGCGACCCCTGG |
| tRNA-Sec TCA | CACTCTGTCGCTAGACAGCTACAGGTTTGAAGCCTGCACCCCAGAC |
| tRNA | Probe sequence (5’ -> 3’) |
|---|---|
| tRNA-Leu TAA | CCATTGGATCTTAAGTCCAACGCCTTAACCACTC |
| tRNA-Leu AAG | AGCCTTAATCCAGCGCCTTAGACCGCTCGGCCACGCTACC |
| tRNA-Leu CAA | GGAGACCAGAACTTGAGTCTGGCGCCTTAGACCA |
| tRNA-Leu CAG | CACGCCTCCAGGGGAGACTGCGACCTGAACGCAGCGCCTT |
| tRNA-Ser AGA | GATTTCTAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-Ser CGA | CCCCATTGGATTTCGAGTCCAACGCCTTAACCACTCGGCCA |
| tRNA-Ser GCT | GGATTAGCAGTCCATCGCCTTAACCACTCGGCCACCTCGTC |
| tRNA-Ser TGA | TGGATTTCAAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-His GTG | CGAGGTTGCTGCGGCCACAACGCAGAGTACTAA |
| tRNA-Thr TGT | AGGCCCCAGCGAGGATCGAACTCGCGACCCCTGG |
| tRNA-Sec TCA | CACTCTGTCGCTAGACAGCTACAGGTTTGAAGCCTGCACCCCAGAC |
| tRNA | Probe sequence (5’ -> 3’) |
|---|---|
| tRNA-Leu TAA | CCATTGGATCTTAAGTCCAACGCCTTAACCACTC |
| tRNA-Leu AAG | AGCCTTAATCCAGCGCCTTAGACCGCTCGGCCACGCTACC |
| tRNA-Leu CAA | GGAGACCAGAACTTGAGTCTGGCGCCTTAGACCA |
| tRNA-Leu CAG | CACGCCTCCAGGGGAGACTGCGACCTGAACGCAGCGCCTT |
| tRNA-Ser AGA | GATTTCTAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-Ser CGA | CCCCATTGGATTTCGAGTCCAACGCCTTAACCACTCGGCCA |
| tRNA-Ser GCT | GGATTAGCAGTCCATCGCCTTAACCACTCGGCCACCTCGTC |
| tRNA-Ser TGA | TGGATTTCAAGTCCATCGCCTTAACCACTCGGCCACGACTAC |
| tRNA-His GTG | CGAGGTTGCTGCGGCCACAACGCAGAGTACTAA |
| tRNA-Thr TGT | AGGCCCCAGCGAGGATCGAACTCGCGACCCCTGG |
| tRNA-Sec TCA | CACTCTGTCGCTAGACAGCTACAGGTTTGAAGCCTGCACCCCAGAC |
RESULTS AND DISCUSSION
sSLFN11-NTD cleaves tRNA and differentiates isoacceptors
SLFN family members share a conserved NTD. Despite similarity in amino acid sequence, some SLFN proteins (e.g. rSLFN13) harbor RNase activities attributing to their NTDs, while others (e.g. hSLFN5 and mSLFN2) do not cleave RNAs. To investigate the enzymatic activity of SLFN11, we attempted to express numerous mammalian SLFN11 orthologs, but we only managed to successfully express S. scrofa SLFN11 (sSLFN11-FL; Figure 2A, top). We observed a predominant peak corresponding to sSLFN11-FL monomers, and higher oligomeric species were also visible; therefore, sSLFN11-FL might have multiple oligomeric states in solution. Because the production of sSLFN11-FL from insect cells had very limited yield, we expressed a fragment of sSLFN11 (residues 1−352) containing its NTD (denoted sSLFN11-NTD) in E. coli (Figure 2A, bottom). sSLFN11-NTD was highly stable and thus suited to subsequent structural and biochemical characterization.
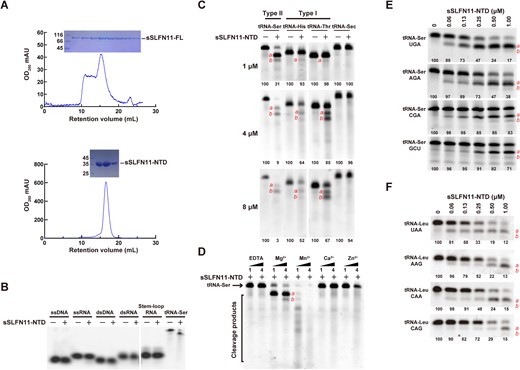
Expression and RNase activity of sSLFN11-NTD. (A) Size-exclusion chromatography of sSLFN11-FL (top) expressed in sf-21 cells and sSLFN11-NTD (bottom) expressed in E. coli. SDS-PAGE analyses of fractions corresponding to the peak in chromatograms are shown on top. (B) sSLFN11-NTD selectively cleaves tRNA. Purified sSLFN11-NTD (1 μM) was incubated with single-stranded DNA (ssDNA), single-stranded RNA (ssRNA), double-stranded DNA (dsDNA), double-stranded RNA (dsRNA), stem–loop RNA and tRNA-Ser, respectively. The reaction mixtures were incubated at 37°C for 30 min, and were resolved using denaturing Urea™PAGE. (C) sSLFN11-NTD cleaves different tRNAs. sSLFN11-NTD was incubated with tRNA-Ser, tRNA-His, tRNA-Thr and tRNA-Sec, respectively. The rection mixtures were analyzed by denaturing urea™PAGE. At low sSLFN11-NTD concentration (1 μM), type II tRNA was cleaved, but type I tRNAs were resistant to cleavage; at higher concentration (4 and 8 μM), both type I and II tRNAs were cleaved. tRNA-Sec was not cleaved by sSLFN11-NTD even at the highest concentration. Primary cleavage product is indicated with ‘a’ and secondary cleavage product is indicated with ‘b’. (D) Nucleolytic activity of sSLFN11-NTD requires divalent ions. sSLFN11-NTD catalyzed tRNA-Ser cleavage was carried out in presence of different divalent ions, magnesium (Mg2+), manganese (Mn2+), calcium (Ca2+) and zinc (Zn2+). Concentration of divalent ions (1 and 4 μM) are indicated on top of the gel. Same amount of EDTA was added to reaction as ion-free controls. (E) sSLFN11-NTD cleaved four tRNA-Ser isoacceptors with distinct efficiencies. sSLFN11-NTD exhibited higher nucleolytic activity on tRNA-Ser UGA than other isoacceptors. (F) sSLFN11-NTD cleaved different tRNA-Leu isoacceptors with distinct efficiencies. sSLFN11-NTD exhibited the highest nucleolytic activity on tRNA-Leu UAA and tRNA-Leu CAG was the most resistant isoacceptor. Numbers below gels in panel (C), (E) and (F) indicate the percentage of uncleaved RNA substrates in each reaction.
To characterize the RNase activity of sSLFN11-NTD, we tested its ability to cleave various nucleic acid substrates (Figure 2B). sSLFN11-NTD cleaved a serine tRNA (human tRNA-Ser UGA) but it was ineffective against other substrates. Furthermore, we examined the activity of sSLFN11-NTD in cleaving different in vitro transcribed tRNA types (Figure 2C). Whereas sSLFN11-NTD cleaved both type I and II tRNAs at high enzyme concentration (4−8 μM), it only efficiently cleaved type II tRNA (tRNA-Ser) at a lower enzyme concentration (1 μM). While the majority of tRNA-Ser was cleaved by sSLFN11-NTD, cleavage products from tRNA-His and tRNA-Thr were barely visible (Figure 2C, top). By contrast, sSLFN11-NTD could not cleave the rare selenocysteine tRNA (tRNA-Sec), even at the highest enzyme concentration used in our assays. Because many characterized ribonucleases (e.g. RNase E and rSLFN13) require divalent ions for nucleolytic activities, we compared the nucleolytic activity of sSLFN11-NTD in the presence of Mg2+, Mn2+, Ca2+ and Zn2+ (Figure 2D). Mg2+ and Mn2+ were essential for the tRNA cleavage activity of sSLFN11-NTD, and the enzyme was inactive in the absence of any metal ions (EDTA controls). Mn2+ stimulated the nucleolytic activity of sSLFN11-NTD to a greater extent than Mg2+, and neither Ca2+ nor Zn2+ could stimulate nucleolytic activity.
To further investigate its substrate specificity, we compared the nucleolytic activity of sSLFN11-NTD against four different tRNA-Ser isoacceptors (tRNA-Ser UGA, tRNA-Ser AGA, tRNA-Ser CGA, and tRNA-Ser GCU). These isoaccepting tRNAs recognize four serine codons (UCA, UCU, UCG, and AGC) with distinct frequencies in humans (per thousand) of 12.2, 15.2, 4.4 and 19.5 (http://www.kazusa.or.jp/codon/). tRNA-Ser GCU was the most resistant substrate to sSLFN11-NTD cleavage, and tRNA-Ser UGA was the most favored substrate (Figure 2E). Our results showed that the most frequently used tRNA-Ser GCU being the most resistant isoacceptor to sSLFN11-NTD cleavage, which is consistent with the function of SLFN11 in suppressing protein translation based on codon usage (25,39). The most vulnerable tRNA-Ser UGA to sSLFN11-NTD cleavage was still the second most frequently used Ser tRNA for translation, which explains why sSLFN11 limits the expression of host proteins.
Next, we investigated the selective activity of sSLFN11-NTD in cleaving another type-II tRNA isoacceptors, tRNA-Leu UAA, tRNA-Leu AAG, tRNA-Leu CAA, and tRNA-Leu CAG (Figure 2F). They recognize Leu codon UUA, CUU, UUG and CUG with frequencies of 7.7, 13.2, 12.9 and 39.6. Similar to the selectivity for tRNA-Ser isoacceptors, sSLFN11-NTD was the most effective in cleaving tRNA-Leu UAA that recognizes the least frequent codon UUA. By contrast, tRNA-Leu CAG recognizing the most frequent codon CUG was the most resistant isoacceptor to sSLFN11 cleavage.
Structural characterization of sSLFN11-NTD
To explore the structural basis underlying the RNase activity of sSLFN11-NTD, we carried out crystallographic investigations. sSLFN11-NTD failed to crystallize despite numerous screening attempts, which implied the presence of a flexible region in this fragment. By aligning the amino acid sequences of multiple SLFN proteins, we identified a highly variable region (residues 158−184) in sSLFN11-NTD (Supplementary Figure S1). In the structures of hSLFN5 (PDB: 7PPJ, 6RR9), hSLFN12 (PDB: 7LRE, 7LRD) and rSLFN13 (PDB: 5YD0), the topologically equivalent regions are largely disordered or missing, underscoring its flexible nature. Therefore, we engineered a mutant lacking residue 166−180 (denoted sSLFN11-NTD-Δ) for crystallization purposes. We then prepared selenomethionine-derivatized sSLFN11-NTD-Δ crystals for phasing given that there are ten methionines in this fragment. Crystals of sSLFN11-NTD-Δ diffracted X-rays to 2.69 Å resolution and contained one molecule per asymmetric unit. We solved the structure using single-wavelength anomalous dispersion and refined the structure using PHENIX (37). Statistics for data collection, structure refinement, and validation are summarized in Table 1.
Data collection and refinement statistics
| sSLFN11-NTD (1–352, Δ 166–180) (SeMet derivative, PDB: 7FEX) | |
|---|---|
| Data collection | |
| Space group | P6122 |
| Cell dimensions | |
| a, b, c (Å) | 64.99, 64.99, 344.02 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| X ray source | |
| Wavelength (Å) | 0.98 |
| Resolution range (Å) | 43.56–2.69 |
| Reflections unique | 22498a |
| Rsymb (last shell) | 0.14 (1.15) |
| I/σI (last shell) | 26.33 (3.22) |
| Completeness (%) (last shell) | 99.9 (99.8) |
| Redundancy (last shell) | 34.4 (34.8) |
| CC1/2 (last shell) | 99.90 (88.60) |
| Refinement | |
| Resolution range (Å) | 43.56–2.69 |
| No. reflections, % reflections in cross validation | 22498a, 5.42 |
| Rworkc/Rfreed (last shell) | 0.22/0.26 (0.33/0.39) |
| Atoms | |
| Non-hydrogen protein atoms | 2549 |
| Protein | 2549 |
| Solvent | 10 |
| B-factors average (Å2) | 66.87 |
| Protein (Å2) | 66.90 |
| Ligands (Å2) | 0 |
| Solvent (Å2) | 59.20 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.630 |
| % residues in favored regions, allowed regions, outliers in Ramachandran plot | 96.2, 3.8, 0 |
| sSLFN11-NTD (1–352, Δ 166–180) (SeMet derivative, PDB: 7FEX) | |
|---|---|
| Data collection | |
| Space group | P6122 |
| Cell dimensions | |
| a, b, c (Å) | 64.99, 64.99, 344.02 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| X ray source | |
| Wavelength (Å) | 0.98 |
| Resolution range (Å) | 43.56–2.69 |
| Reflections unique | 22498a |
| Rsymb (last shell) | 0.14 (1.15) |
| I/σI (last shell) | 26.33 (3.22) |
| Completeness (%) (last shell) | 99.9 (99.8) |
| Redundancy (last shell) | 34.4 (34.8) |
| CC1/2 (last shell) | 99.90 (88.60) |
| Refinement | |
| Resolution range (Å) | 43.56–2.69 |
| No. reflections, % reflections in cross validation | 22498a, 5.42 |
| Rworkc/Rfreed (last shell) | 0.22/0.26 (0.33/0.39) |
| Atoms | |
| Non-hydrogen protein atoms | 2549 |
| Protein | 2549 |
| Solvent | 10 |
| B-factors average (Å2) | 66.87 |
| Protein (Å2) | 66.90 |
| Ligands (Å2) | 0 |
| Solvent (Å2) | 59.20 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.630 |
| % residues in favored regions, allowed regions, outliers in Ramachandran plot | 96.2, 3.8, 0 |
a Friedel pairs are treated as different reflections.
bRsym = ∑hkl∑j |Ihkl,j - Ihkl|/∑hkl∑jIhkl,j, where Ihkl is the average of symmetry-related observations of a unique reflection.
cRwork = ∑hkl ||Fobs(hkl)|-|Fcalc(hkl)||/∑hkl|Fobs(hkl)|.
dRfree= the cross-validation R factor for 5% of reflections against which the model was not refined.
Data collection and refinement statistics
| sSLFN11-NTD (1–352, Δ 166–180) (SeMet derivative, PDB: 7FEX) | |
|---|---|
| Data collection | |
| Space group | P6122 |
| Cell dimensions | |
| a, b, c (Å) | 64.99, 64.99, 344.02 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| X ray source | |
| Wavelength (Å) | 0.98 |
| Resolution range (Å) | 43.56–2.69 |
| Reflections unique | 22498a |
| Rsymb (last shell) | 0.14 (1.15) |
| I/σI (last shell) | 26.33 (3.22) |
| Completeness (%) (last shell) | 99.9 (99.8) |
| Redundancy (last shell) | 34.4 (34.8) |
| CC1/2 (last shell) | 99.90 (88.60) |
| Refinement | |
| Resolution range (Å) | 43.56–2.69 |
| No. reflections, % reflections in cross validation | 22498a, 5.42 |
| Rworkc/Rfreed (last shell) | 0.22/0.26 (0.33/0.39) |
| Atoms | |
| Non-hydrogen protein atoms | 2549 |
| Protein | 2549 |
| Solvent | 10 |
| B-factors average (Å2) | 66.87 |
| Protein (Å2) | 66.90 |
| Ligands (Å2) | 0 |
| Solvent (Å2) | 59.20 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.630 |
| % residues in favored regions, allowed regions, outliers in Ramachandran plot | 96.2, 3.8, 0 |
| sSLFN11-NTD (1–352, Δ 166–180) (SeMet derivative, PDB: 7FEX) | |
|---|---|
| Data collection | |
| Space group | P6122 |
| Cell dimensions | |
| a, b, c (Å) | 64.99, 64.99, 344.02 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| X ray source | |
| Wavelength (Å) | 0.98 |
| Resolution range (Å) | 43.56–2.69 |
| Reflections unique | 22498a |
| Rsymb (last shell) | 0.14 (1.15) |
| I/σI (last shell) | 26.33 (3.22) |
| Completeness (%) (last shell) | 99.9 (99.8) |
| Redundancy (last shell) | 34.4 (34.8) |
| CC1/2 (last shell) | 99.90 (88.60) |
| Refinement | |
| Resolution range (Å) | 43.56–2.69 |
| No. reflections, % reflections in cross validation | 22498a, 5.42 |
| Rworkc/Rfreed (last shell) | 0.22/0.26 (0.33/0.39) |
| Atoms | |
| Non-hydrogen protein atoms | 2549 |
| Protein | 2549 |
| Solvent | 10 |
| B-factors average (Å2) | 66.87 |
| Protein (Å2) | 66.90 |
| Ligands (Å2) | 0 |
| Solvent (Å2) | 59.20 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.630 |
| % residues in favored regions, allowed regions, outliers in Ramachandran plot | 96.2, 3.8, 0 |
a Friedel pairs are treated as different reflections.
bRsym = ∑hkl∑j |Ihkl,j - Ihkl|/∑hkl∑jIhkl,j, where Ihkl is the average of symmetry-related observations of a unique reflection.
cRwork = ∑hkl ||Fobs(hkl)|-|Fcalc(hkl)||/∑hkl|Fobs(hkl)|.
dRfree= the cross-validation R factor for 5% of reflections against which the model was not refined.
sSLFN11-NTD-Δ is a pincer-shaped pseudo-dimer comprising two topologically similar subdomains (Figure 3A, B). The N-terminal subdomain of sSLFN11-NTD spans residues 1−165 (denoted N-domain), and its C-terminal subdomain spans residues 181−352 (denoted C-domain). The N- and C-domains are related to each another by pseudo 2-fold rotational symmetry (Figure 3B). The region (residues 166−180) we deleted from sSLFN11-NTD is located between the N- and C-domains, hence it was denoted the Connection loop. To investigate whether removal of the Connection loop altered the overall fold of sSLFN11-NTD, we predicted the full-length sSLFN-11 (sSLFN11-FL) structure using software AlphaFold2 (40) and superimposed our crystal structure of sSLFN11-NTD-Δ onto the resulting model (Figure 3C, top). Structural comparison gave a root mean square deviation (RMSD) of 1.61 Å, implying that truncation of the Connection-loop had minimal impact on the overall structure of sSLFN11-NTD. Of note, the predicted model of sSLFN11 showed that the Connection loop is surface-exposed and lacks regular secondary structures (Figure 3C), which explains why this region interfered with crystallization. The Connection loop had a low predicted LDDT (local distance difference test) score (pLDDT < 70), suggesting structure prediction for this region was unreliable. We superimposed structure of sSLFN11-NTD-Δ onto a recently published cryo-EM structure of human Schlafen11 (hSLFN11, PDB: 7ZEP) (31), which aligned 321 residues and gave an RMSD of 2.70 Å. The topologically equivalent Connection loop region in the hSLFN11 structure is also disordered (Figure 3C, bottom). The function of the Connection loop requires further investigation.
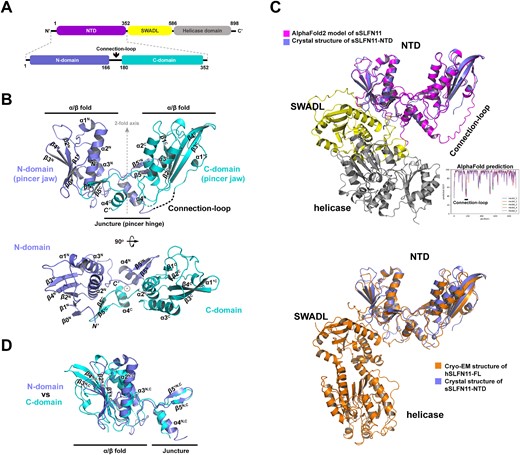
Crystal structure of sSLFN11-NTD. (A) 2D-diagram of sSLFN11 domain organization. N-terminal domain (NTD) is colored magenta, SWADL domain is colored yellow and C-terminal helicase domain is colored gray. sSLFN11-NTD comprises N-domain, Connection-loop and C-domain. (B) Overall structure of sSLFN11-NTD shown in ribbon model; secondary structures are labeled. sSLFN11-NTD exhibits a ‘pincer’ shape, comprising two topologically similar domain: N-domain (colored blue) and C-domain (colored cyan). Each domain comprises an α/β fold resembles pincer jaw, and a small domain that mediates interaction of N- and C-domains, resembling pincer hinge. The N- and C-domains are related by a pseudo two-fold axis. A region connecting N- and C-domains (166–180aa) is disordered in the crystal structure, which is indicated by a dashed line. This region was denoted ‘Connection-loop’. (C) Top, crystal structure of sSLFN11-NTD (blue) is superimposed with an AlphaFold2 model of sSLFN11-FL (rmsd, 1.6 Å); the model is colored by domains with the same color scheme as in (A). A long loop connecting N- and C-domain of NTD was predicted as an unstructured loop. Lower right insert, pLDDT scoring of structure prediction of sSLFN11 by AlphaFold2. pLDDT >90 indicates high model confidence, 90–70 for normal confidence, 70–50 for low confidence and <50 for very low confidence. Bottom, sSLFN11-NTD structure (blue) is superimposed with the cryo-EM structure of hSLFN11-FL (orange, PDB: 7ZEP), rmsd, 2.7Å. (D) Superimposition of the N- and C-domain of NTD, demonstrating that two domains share overall folding and most secondary structural elements.
Each N- and C-domain of sSLFN11-NTD comprises an α/β domain followed by a small domain. The α/β domains form two jaws of a pincer, while the smaller domains constitute the hinge of the pincer mediating interaction between the N- and C-domains. Each α/β domain comprises an antiparallel β-plane (the N-domain contains five-stranded β-sheets, and the C-domain contains four-stranded β-sheets) sandwiched by connecting α-helices and loops, a topology reminiscent of the AAA protein family. Especially, the C-domain of sSLFN11-NTD exhibited some similarity with the NTD of a putative DNA helicase from Nitrosomonas europaea (PDB: 2KYY; Dali Z-score = 8.0, RMSD = 2.6 Å), which belongs to the AAA_4 family. Superimposition of the N- and C-domains of sSLFN11-NTD showed that the two subdomains share a common fold (RMSD = 2.5 Å; Figure 3D).
Previous studies identified that SLFN-NTDs contain a highly conserved CCCH-type zinc finger in and revealed that the zinc finger is essential for protein stability (12,19,41). In the structures of rSLFN13-NTD, hSLFN5-NTD, and hSLFN12-NTD, a zinc ion is coordinated by a histidine and a cysteine (first and second zinc-coordinating residues) between the α3C helix and β3C strand, and by two consecutive cysteines (third and fourth zinc-coordinating residues) downstream of the β4C strand (Supplementary Figure S2A, B). Surprisingly, we found that the zinc-finger was missing in sSLFN11-NTD-Δ structure. sSLFN11 naturally lacks the fourth zinc-coordinating residue (it contains a G318 instead of the cysteines present in other Schlafen proteins at this position; Supplementary Figure S2B). Indeed, in the crystal structure of sSLFN11-NTD-Δ, the loop between α3C and β3C was largely disordered, and C283 (the second zinc-coordinating residue) could not be traced in the final model. Although H281 and C317 (the first and third zinc-coordinating residues) were located in sSLFN11-NTD, they adopted conformations distinct from their counterparts in zinc fingers of other SLFN-NTDs, which suggest that neither residue contributed to zinc coordination.
We searched the sSLFN11-NTD-Δ structure against all structures in the Protein Data Bank using the Dali server (http://ekhidna2.biocenter.helsinki.fi/dali/). Top hits included rSLFN13, hSLFN12, and hSLFN5, which gave the Dali Z-scores of 30.3−32.5 and RMSD values of 2.0−2.5Å (Supplementary Figure S2C, D). Structural similarity shared by different SLFN-NTDs suggests that they might be derived from a common evolutionary origin. However, to understand their distinct RNase activities requires in-depth investigation of their active sites.
Available Schlafen protein structures reveal that their NTDs share a similar fold; topologically similar N- and C-domains form a pincer-like pseudo-dimer, and each domain resembles a pincer jaw. However, little is known about the region joining the N- and C-domains (spanning α4N and α1’C, Supplementary Figure S1). The Connection-loop covers one of the least conserved regions among SLFN members and is largely disordered in available SLFN structures, underscoring its intrinsic flexibility. We omitted the Connection-loop to produce a crystallizable fragment (sSLFN11-NTD-Δ). Compared with sSLFN11-NTD, sSLFN11-NTD-Δ remained active cleaving RNA substrates but to a lesser extent (Supplementary Figure S3A−H), indicating that the Connection-loop contributes to RNase activity, despite being located on the outer side of the pincer while the catalytically important residues are on the inner sides (Figure 3B, C). Thus, it is possible that the Connection-loop performs a regulatory function in nucleolytic cleavage rather than a direct role in catalysis. It is also possible that other proteins regulate the RNase activity of sSLFN11-NTD through binding to the Connection-loop, possibly by modulating the opening between the two pincer jaws through tightening or loosening of the Connection-loop. Nevertheless, to fully understand the functions of the Connection loop, the structure of full-length SLFN11 complexed with a tRNA substrate is necessary.
Key residues for the nucleolytic activity of sSLFN11-NTD
sSLFN11-NTD exhibited different RNase activity from other SLFN members despite sharing similar primary and tertiary structures (Supplementary Figure S1, Supplementary Figure S2C). Whereas sSLFN11-NTD efficiently cleaved tRNA-Ser (type II tRNA) substrates at low concentration (∼0.06 μM), nucleolytic activity of rSLFN13-NTD only manifested at ∼8-fold higher concentration (0.50 μM; Figure 4A, top two gels). sSLFN11-NTD also cleaved tRNA-Gly (type I tRNA), albeit with lower efficiency, whereas rSLFN13-NTD could not cleave tRNA-Gly even at the highest enzyme concentration tested (4 μM; Figure 4A, bottom two gels).
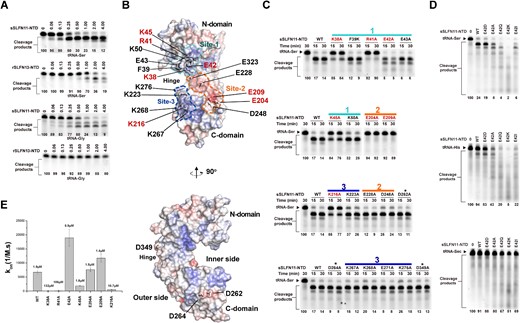
Molecular determinants for sSLFN11-NTD nucleolytic activity. (A) Comparison of tRNAs cleavage by sSLFN11-NTD and rSLFN13-NTD. Type I and II tRNA, tRNA-Gly and tRNA-Ser were incubated with purified sSLFN11-NTD and rSLFN13-NTD, and the reactions were resolved using denaturing urea−PAGE. Enzyme concentration are indicated on top of the gels. (B) Surface electrostatic potential plot of sSLFN11-NTD. Top, front view of the pincer shaped sSLFN11-NTD, in which inner side of the pincer faces toward the reader. Three charge sites, Site-1 (positive charge, colored cyan), Site-2 (negative charge, colored orange) and Site-3 (positive charge, colored blue), are delineated with dashed lines. Residues subjected to mutagenesis study are indicated. Residues crucial for nucleolytic activity are highlighted with red fonts. Side view (90° rotation around vertical axis to the right) of the top structure. (C) Mutagenesis study. Mutations were introduced to sSLFN11-NTD; the resulting mutants (indicated on top of the gels) were incubated with tRNA-Ser substrate, and the reactions were resolved by denaturing urea−PAGE. Residues located in different charged sites are indicated with solid lines, using the same color scheme as in panel (B). Irrelevant mutations are indicated with asterisk. (D) Residue E42 inhibits nucleolytic activity sSLFN11-NTD, which is attributed to its charge. Various substitutions of E42, E42D, E42A, E42Q, E42K and E42I (indicated on top of the gels) were assayed for nucleolytic cleavage of tRNA-Ser (type II tRNA, top), tRNA-His (type I tRNA, middle) and tRNA-Sec (bottom). (E) Biolayer interferometry analysis of the binding between sSLFN11-NTD mutants and tRNA-Ser UGA. Biotinylated tRNA was immobilized on SA sensor chip for binding with sSLFN11-NTD or mutants (12.5 μM) in solution. On-rate (kon) of binding is plotted for each protein. KD (calculated by dividing kon with koff) values are indicated on top of the plot. Numbers below gels in panels (A), (C) and (D) indicate the percentage of uncleaved RNA substrates in each reaction.
To identify key residues governing the RNase activity of sSLFN11-NTD, we carried out a thorough mutagenesis analysis. A surface electrostatic potential plot of sSLFN11-NTD revealed three charged (negatively or positively) regions across the inner side of the pincer-shaped molecule (i.e. the area between the two pincer jaws), namely, site-1, site-2 and site-3 (Figure 4B). We then expressed a panel of sSLFN11-NTD mutants containing mutations in these sites. We chose primarily charged residues for mutagenesis because charged residues are often involved in RNase activities. For controls, we randomly selected three charged residues located on the outer sides of the pincer-like molecule. We measured the nucleolytic activity of these mutants with tRNA-Ser UGA substrates (Figure 4C). While three irrelevant mutations D262A, D264A and D349A elicited negligible effects on the RNase activity of sSLFN11-NTD, we identified seven key mutations that dramatically affected tRNA cleavage: four key mutations (K38, R41, E42 and K45) located in site-1, two (E204, E209) in site-2, and one (K216) in site-3. Surprisingly, in site-1, whereas alanine substitution of positively charged residues K38A, R41A and K45A severely impaired or abolished the RNase activity of sSLFN11-NTD, E42A stimulated its RNase activity. In site-2, either E204A or E209A obliterated RNase activity of sSLFN11-NTD, suggesting that these residues are essential for nucleolytic activity. Finally, K216A (site-3) severely impaired RNase activity.
Among the seven charged residues important for sSLFN11-NTD RNase activity, the role of E42 is distinct. E42 lies in the N-domain, on the inner side of the pincer. The counterparts of E42 in many SLFN members are often positively charged lysine or arginine residues (Supplementary Figure S1), suggesting residue 42 may be a key determinant for the activity various SLFNs. To test this idea, we substituted E42 with aspartate (E42D), alanine (E42A), glutamine (E42Q), lysine (E42K), and isoleucine (E42I) and compared the ability of the variants to cleave various tRNA substrates. The conservative E42D mutation had little effect on RNase activity, whereas all other mutations enhanced nucleolytic activity with both tRNA-Ser and tRNA-His substrates (Figure 4D top and middle gels). Among the mutations, the charge-reversal mutation E42K elicited the highest nucleolytic activity, and cleaved type I and II tRNAs indiscriminately. By stark contrast, none of these mutants could cleave the rare tRNA-Sec substrate (Figure 4D, bottom gel).
To gain mechanistic insight into the roles of these key residues in catalysis, we compared their binding affinity to tRNA substrates using biolayer interferometry (Supplementary Figure S3J; Figure 4E). We synthesized biotinylated tRNA-Ser UGA to label a streptavidin biosensor, which was then titrated against various sSLFN11-NTD mutants in solution. Divalent ions were excluded from the solutions to prevent nucleolytic reaction. While wild-type sSLFN11-NTD bound tRNA-Ser with a KD of 1.9 μM, mutants K38A and R41A led to decrease in tRNA binding affinity by 70 and 56 folds (KD = 132 μM and 106 μM, respectively), suggesting K38 and R41 are involved in RNA substrate binding. By contrast, mutant E42A displayed an elevated on-rate (kon) during the association step of the BLI titration, and the binding affinity (KD = 0.9 μM) of the E42A mutant with tRNA was also increased, which correlates with the enhanced RNase activity of E42. We speculate that the negative charge of E42 might repel negatively charged tRNA substrates, thus providing an explanation for the autoinhibitory role of E42. By contrast, mutations K45A, E204A, and E209A had little impact on tRNA binding affinity to sSLFN11-NTD (KD = 1.8, 1.5 and 1.4 μM, respectively), despite the same mutations severely damaging the nucleolytic activity of sSLFN11-NTD. In particular, the catalytic mutants E204A and E209A exhibited similar tRNA binding affinities to the wild-type enzyme, suggesting these residues contribute primarily to nucleolytic reaction rather than RNA substrate binding. K216A mutation led to a ∼6-fold decrease in tRNA binding affinity (KD = 10.7 μM), and this mutation was detrimental to RNase activity, suggesting that K216 may be involved in both RNA binding and cleavage.
Collectively, our results suggest that sSLFN11-NTD site-1 is the primary RNA binding site and governs substrate specificity. Residue E42 of site-1 is an unusual residue that negatively modulates RNA binding affinity and inhibits RNase activity. Site-2 is a putative RNase-active site for nucleolytic reaction. Site-3 is an auxiliary site assisting RNA binding. Since site-1 and site-3 are located opposite each other, they probably clamp on RNA substrates from both sides. Nevertheless, we cannot rule out the contribution of the C-terminal portion of sSLFN11 to substrate specificity or RNase activity.
Malone and colleagues identified that phosphorylation of hSLFN11 residue S219, T230 and S753 inhibits of its ribonuclease activity (42). Among these three phosphorylation sites, only S219 is conserved in sSLFN11, the residues S214 (Figure S1). S214 lies in the site-3 of sSLFN11-NTD. We demonstrate that site-3 harbors a cluster of positively charged lysines, and alanine substitution of the lysines abrogated or impaired the RNase activity (Figure 4B, C). Therefore, S214 phosphorylation might inhibit the RNase activity of sSLFN11 through neutralizing the positive charge of site-3, thus undermines its RNA binding.
sSLFN11-NTD cleaves cellular tRNA/rRNA
Synthetic RNAs are different from cellular RNAs in many ways, e.g. the lack of chemical modifications, some of which are essential for their functions. To investigate the activity of sSLFN11-NTD on cellular RNAs species, we extracted total RNAs from HEK-293T cells for use as RNase substrates. After incubating sSLFN11 or its variants with cellular RNAs, reaction mixtures were analyzed either by native agarose gel for separating large RNA species, or by denaturing urea−PAGE for separating small RNA species. sSLFN11-NTD cleaved both cellular ribosomal RNAs (28S,18S, and 5S rRNAs) and tRNAs (type I and II) in a concentration-dependent manner (Figure 5A, B). Consistent with the results on the synthetic tRNA substrate, sSLFN11-NTD exhibited a clear preference for type II tRNAs over type I tRNAs from cell extract because we observed cleavage products of type I tRNA species after type II tRNA species were consumed (Figure 5B). These results imply that sSLFN11 can inhibit translation by cleaving cellular tRNA/rRNA. The sSLFN11-NTD E42A mutant exhibited higher cleavage activity on cellular rRNAs (Figure 5C), confirming an autoinhibitory function of this acidic residue. Cellular tRNAs were also cleaved by sSLFN11-NTD E42A; however, we did not observe obvious difference in its cleavage pattern comparing with sSLFN11-NTD (Figure 5D). sSLFN11 FL cleaved cellular rRNA more efficiently than sSLFN11-NTD or sSLFN11-NTD E42A. The concentration of sSLFN11 FL achieving nearly complete cleavage (<5% uncleaved RNA substrate) of the 28S rRNA species was 0.5 μM, whereas that for sSLFN11-NTD and sSLFN11-NTD E42A were 16 and 2 μM, respectively (Figure 5A, C, E and Table 2). This suggests that the cleavage activity of sSLFN11-FL on rRNAs was ∼32-fold higher than that of sSLFN11-NTD and ∼ 4-fold higher than that of sSLFN11-NTD E42A. Therefore, it is possible that the C-terminal portion of sSLFN11 harboring the SWADL and helicase domains also contributes to its nucleolytic activity. Collectively, these results demonstrate that sSLFN11 can cleave cellular rRNA and tRNA, and exhibits similar substrate preference like the cleavage of synthetic RNA substates.
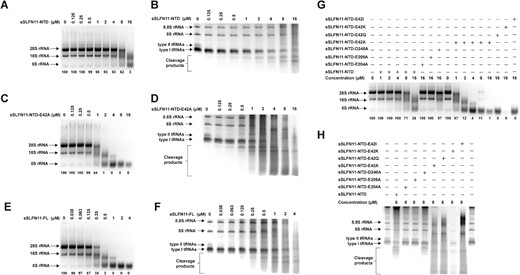
sSLFN11-NTD nucleolytic cleavage of cellular RNAs. (A–H) sSLFN11-NTD was assayed for cleavage of total cellular RNAs (extracted from HEK-293T cells) by increasing amount of sSLFN11-NTD; reaction mixtures were resolved using 1% agarose gel for analyzing cleavage products of rRNAs, or using 10% urea−PAGE for analyzing tRNAs or small RNAs. Enzyme concentrations are indicated on top of the gel. The gels were stained with SYBR Gold. Size of 28S, 18S and 5S rRNAs, type I and II tRNAs are indicated. (A) Analysis of sSLFN11-NTD cleaved cellular rRNAs using agarose gel. (B) Analysis of sSLFN11-NTD cleaved cellular tRNAs using urea−PAGE. (C) Analysis of sSLFN11-NTD mutant E42A cleaved cellular rRNAs using agarose gel. (D) Analysis of sSLFN11-NTD mutant E42A cleaved cellular tRNAs using urea−PAGE. (E) Analysis of sSLFN11-FL cleaved cellular rRNAs using agarose gel. (F) Analysis of sSLFN11-FL cleaved cellular tRNAs using urea−PAGE. (G) Cellular rRNAs cleaved by sSLFN11-NTD mutants harboring various E42 substitutions. (H) Cellular tRNAs cleaved by sSLFN11-NTD mutants harboring various E42 substitutions. Numbers below gels in panels (A), (C), (E) and (G) indicate the percentage of uncleaved 28S rRNA substrates in each reaction.
sSLFN11 and its variants exhibit different activity in cleaving cellular rRNAs
| Conc. (μM) Protein, % uncleaved RNA substrates | 0 | 0.038 | 0.063 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sSLFN11-FL | 100 | 97 | 98 | 87 | 35 | 2* | 0 | 0 | 0 | \ | \ |
| sSLFN11-NTD | 100 | \ | \ | 100 | 100 | 99 | 98 | 93 | 93 | 62 | 3* |
| sSLFN11-NTD E42A | 100 | \ | \ | 100 | 100 | 99 | 44 | 1* | 0 | 0 | 0 |
| Conc. (μM) Protein, % uncleaved RNA substrates | 0 | 0.038 | 0.063 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sSLFN11-FL | 100 | 97 | 98 | 87 | 35 | 2* | 0 | 0 | 0 | \ | \ |
| sSLFN11-NTD | 100 | \ | \ | 100 | 100 | 99 | 98 | 93 | 93 | 62 | 3* |
| sSLFN11-NTD E42A | 100 | \ | \ | 100 | 100 | 99 | 44 | 1* | 0 | 0 | 0 |
Numbers in the shaded cells indicate the percentage of uncleaved rRNA quantified from Figure 5 A,C,E; \: not determined;
*: achieving nearly complete cleavage, <5% uncleaved RNA substrate of the 28S rRNA.
sSLFN11 and its variants exhibit different activity in cleaving cellular rRNAs
| Conc. (μM) Protein, % uncleaved RNA substrates | 0 | 0.038 | 0.063 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sSLFN11-FL | 100 | 97 | 98 | 87 | 35 | 2* | 0 | 0 | 0 | \ | \ |
| sSLFN11-NTD | 100 | \ | \ | 100 | 100 | 99 | 98 | 93 | 93 | 62 | 3* |
| sSLFN11-NTD E42A | 100 | \ | \ | 100 | 100 | 99 | 44 | 1* | 0 | 0 | 0 |
| Conc. (μM) Protein, % uncleaved RNA substrates | 0 | 0.038 | 0.063 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sSLFN11-FL | 100 | 97 | 98 | 87 | 35 | 2* | 0 | 0 | 0 | \ | \ |
| sSLFN11-NTD | 100 | \ | \ | 100 | 100 | 99 | 98 | 93 | 93 | 62 | 3* |
| sSLFN11-NTD E42A | 100 | \ | \ | 100 | 100 | 99 | 44 | 1* | 0 | 0 | 0 |
Numbers in the shaded cells indicate the percentage of uncleaved rRNA quantified from Figure 5 A,C,E; \: not determined;
*: achieving nearly complete cleavage, <5% uncleaved RNA substrate of the 28S rRNA.
To further investigate the inhibitory function of E42, we compared the nucleolytic activities of a panel of sSLFN11-NTD mutants for cleaving cellular tRNAs and rRNAs (Figure 5G, H). Whereas the catalytically inactive sSLFN11-NTD mutants E204A and E209A failed to cleave cellular RNAs, the D248A mutant barely cleaved cellular RNAs, and the other mutants (E42A, E42K, E42Q and E42I) all displayed enhanced nucleolytic activity. Among these, E42K, a charge-reversal mutant, exhibited the highest activity. These results are consistent with the results obtained from the synthetic tRNA substrates.
sSLFN11 and hSLFN11 inhibit protein expression in cells based on codon usage
Finally, we investigated sSLFN11-mediated protein translation suppression in cells. Previous studies demonstrated that hSLFN11 targets viral protein expression based on codon usage bias (25). It was also shown that rSLFN13 inhibits HIV protein synthesis by cleaving cellular tRNA/rRNA (19). To characterize whether sSLFN11 has a similar function, we established sSLFN11-FL expression in HEK-293T cells by transfecting a plasmid carrying original sSLFN11 gene. Expression of wild-type sSLFN11 remained at a low level (Figure 6A) compared with the glyceraldehyde 3-phosphate dehydrogenase (GADPH) internal control. This might be because suboptimal codon usage of the original sSLFN11 gene, which has a Codon Adaptation Index (CAI) of 0.72.
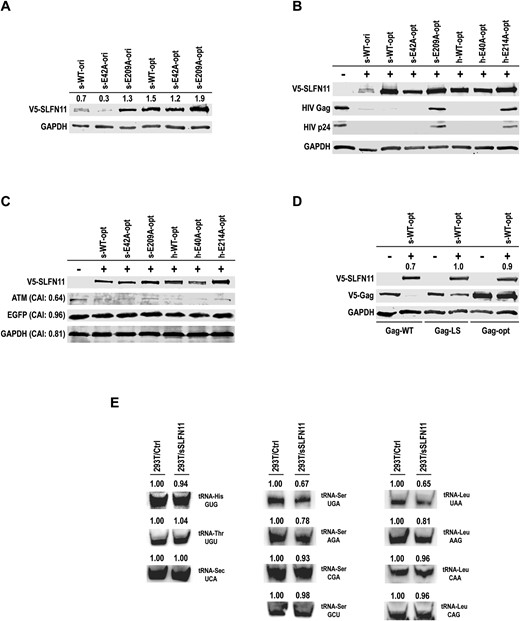
sSLFN11 and hSLFN11 inhibit protein expression in cells based on codon usage bias. (A) Expression of original sSLFN11 gene (s-WT-ori, CAI = 0.72) inhibited the expression of itself, and so was its hyperactive RNase mutant E42A (s-E42A-ori); whereas the inactive RNase mutant E209A (s-E209A-ori) did not inhibit its own expression. By contrast, the codon optimized sSLFN11 genes (s-WT-opt, s-E42A-opt and s-E209A-opt, CAI = 0.89) did not inhibit their own expression. Numbers above each lane indicate quantity of sSLFN11 band relative to that of GAPDH band. (B, C) Plasmid harboring codon optimized sSLFN11 gene, human Schlafen11 gene (hSLFN11, h-WT-opt CAI = 0.90 whereas original hSLFN11 gene has CAI = 0.71) or indicated SLFN11 mutants was transfected into HEK-293T cells with the plasmids (B) pNL4-3.Luc.R-E-, (C) pcDNA6-EGFP-V5-His B. Cell lysates were immunoblotted with the antibodies against V5-tag, HIV Gag-p24, EGFP, ATM or GAPDH, respectively. CAI values of ATM, EGFP and GAPDH are indicated on the left of the gels. (D) Co-expression of V5-tagged Gag-WT, Gag-LS or Gag-opt with WT sSLFN11 in HEK-293T cells. Protein level was visualized by western blot. The genes encoding sSLFN11 and hSLFN11 or their mutants used here were all codon optimized (with high CAI). Gag-WT: wild-type gag gene sequence corresponding to pGag-EGFP (NIH AIDS Research & Reference Reagent Program, No. 11468), in which only the inhibitory RNA sequence (INS) was eliminated through introduction of silent mutations while otherwise retaining the original viral codon usage. Gag-LS: codon optimized, in which all serine or leucine residues were encoded by a single codon, (Ser) AGC and (Leu) CUG, respectively. Gag-opt: gag gene sequence of this construct was optimized for the expression in human with high CAI. Numbers above the lanes containing sSLFN11 indicate quantity of sSLFN11 band relative to that of GAPDH band. (E) tRNA northern blot analysis of total RNA isolated from HEK 293T cells ectopically expressing pcDNA6-V5-His B vector or pcDNA6-sSLFN11(opt)-V5-His B. Numbers on top of the gels indicate the quantified band intensity of tRNAs relative to controls. tRNA-Ser or tRNA-Leu isoacceptors detected in the experiments are indicated on the right of the gels.
CAI is a measure of the synonymous codon usage bias for a given nucleic acid sequence, which quantifies codon usage similarities between a gene and a reference set. CAI ranges 0–1; higher the value more frequent a gene uses synonymous codons in the reference set (43). We measured the CAI values for our genes of interests by using a web-based server CAIcal (http://genomes.urv.es/CAIcal). The average CAI for all human genes is 0.76, and GADPH has a higher-than-average CAI of 0.81. Therefore, it is plausible that sSLFN11 cleaves the tRNAs required for its own expression based on codon usage. To test this hypothesis, we compared the expression level of a hyperactive RNase sSLFN11-FL mutant E42A and an inactive mutant E209A with the wild-type sSLFN11. Whereas the expression level of E42A was barely detectable, the expression level of E209A exceeded that of wild-type sSLFN11 (Figure 6A). Next, we co-transfected an HIV-1 reporter plasmid (pNL4-3.Luc.R-E-) with the plasmid harboring original sSLFN11 gene or its mutants E42A and E209A in HEK-293T cells (Supplementary Figure S4). Expression of HIV proteins Gag and p24 was dramatically inhibited in the presence of wild-type sSLFN11, which is consistent with lower-than-average CAI values of Gag and p24 (0.60 and 0.58, respectively). Inhibition of the expression of HIV Gag and p24 was strengthened when the sSLFN11 E42A mutant was co-expressed in cells, even though the E42A mutant was considerably less abundant than wild-type sSLFN11. By contrast, inhibition of Gag and p24 expression was fully released when the E209A mutant was co-expressed in cells (Supplementary Figure S4).
To investigate whether the protein translation inhibition we observed here was attributed to codon usage-based tRNA cleavage by SLFN11 or through other nonspecific factors, e.g. general toxicity caused by ectopic protein expression, we carried out further experiments with the codon optimized genes (Figure 6A–D). We synthesized codon optimized sSLFN11 and hSLFN11 genes with higher CAI, namely sSLFN11-opt and hSLFN11-opt. sSLFN11-opt and hSLFN11-opt had CAI of 0.89 and 0.90, whereas their original genes had CAI of 0.72 and 0.71. As anticipated, the expression of codon optimized sSLFN11 or hyperactive sSLFN11-E42A mutant no longer inhibited their own expression (Figure 6A, B). When co-expressed sSLFN11-opt or hSLFN11-opt with HIV-1 reporter plasmid pNL4-3.Luc.R-E-, we observed strong inhibition of Gag and p24 expression, and the inhibition correlated well with the RNase activity of SLFN11 proteins and their mutants (Figure 6B). Similar to sSLFN11 mutants, hSLFN11 harboring D40A mutation (equivalent to sSLFN11 E42A) was highly effective in suppressing Gag and p24 expression, whereas hSLFN11 E214A (equivalent to sSLFN11 E209A) lost the activity. It worth noting that, the expression of codon optimized sSLFN11 and hSLFN11 genes inhibit neither their own expression, nor the expression of the endogenous GAPDH with high CAI (0.81), which severed as an internal control. Nevertheless, this may not reflect the action of SLFN11 on an ectopically-expressed protein. To develop a better control, we co-expressed codon optimized sSLFN11 or hSLFN11 with EGFP, an unrelated protein with CAI of 0.95. As anticipated, the expression of the ectopically-expressed EGFP was not inhibited by SLFN11 proteins or their mutants (Figure 6C). Finally, we detected the expression of an endogenous gene, the ataxia-telangiectasia mutated gene (ATM), with low CAI (0.64). Consistent with previous studies (39), ATM expression was inhibited by sSLFN11 and hSLFN11 (Figure 6C).
Next, we investigated whether HIV Gag gene could become resistant to SLFN11-meditated translation inhibition if its sequence was optimization. Instead of using the entire HIV-1 reporter plasmid, we synthesized original Gag gene, Gag-WT (CAI = 0.64), and two codon optimized genes Gag-LS (CAI = 0.66) and Gag-opt (CAI = 0.90), which were inserted into pcDNA6-V5-His B vector. Whereas all serine and leucine codons (decoded by type II tRNAs) used the (Ser) high frequent AGC and (Leu) CUG codon in Gag-LS, the entire sequence in Gag-opt was optimized to achieve high CAI (Table S1). As shown in Figure 6D, in the absence of sSFLN11, all Gag genes were expressible in HEK293T cells, in which the expression level of Gag-opt was the highest, confirming codon optimization favored gene expression. Upon co-expression with sSLFN11, only the expression of Gag-WT was greatly inhibited, and the expression of Gag-LS and Gag-opt was unaffected.
To confirm whether type II tRNAs were selectively cleaved by sSLFN11 in cells, we carried out Northern blot analysis of the RNAs extracted from HEK 293T cells expressing sSLFN11 gene (codon optimized). As shown in Figure 6E, whereas type I tRNAs (tRNA-His GUG, tRNA-Thr UGU and tRNA-Sec UCA) remained unaffected, type II tRNAs were more efficiently cleaved by sSLFN11. Consistent with our RNase assays of sSLFN11-NTD in cleaving synthetic tRNAs, different cellular tRNA-Ser or tRNA-Leu isoacceptors were also cleaved by sSLFN11 with different efficiencies, and the tRNA-Ser and tRNA-Leu isoacceptors (tRNA-Ser UGA and tRNA-Leu UAA) were the most vulnerable tRNAs cleaved by sSLFN11.
Collectively, our results demonstrate that sSLFN11 inhibits protein expression in cells based on codon usage frequency, in which the codon usage of the type II tRNAs is critically important, and the nucleolytic activity of sSLFN11 is essential for its inhibitory function.
Identification of the RNase-active site
We identified seven residues that are important for the RNase activity of sSLFN11-NTD through our mutagenesis studies, and our crystallographic investigations revealed that all seven residues are located on the inner sides of the pincer-like sSLFN11-NTD, between the two pincer jaws, suggesting this must be the location of the RNase-active site. We subsequently analyzed the structural counterparts of these seven residues in other characterized SLFN structures to identify determinants governing nucleolytic activity.
In site-1 of sSLFN11-NTD, the unusual E42 residue is close to three neighboring basic residues K38, R41 and K45 (Figure 7A, B). Any mutation of E42 that neutralized or reversed its negative charge led to enhanced RNase activity of sSLFN11-NTD, whereas alanine substitution of any of these three basic residues greatly undermined the activity (Figure 4C). Hence, the net positive charge of site-1 is likely relevant to RNase activity. To test this theory, we compared the architecture of site-1 in hSLNF12, rSLFN13 and hSLFN5 (Figure 7B−E). Site-1 of rSLFN13-NTD contains R39 (the counterpart of sSLFN11-NTD E42) which is neighbored by K36, K38 and K42. Site-1 of hSLFN12-NTD contains K38 (the counterpart of sSLFN11-NTD E42) which is neighbored by K35, R37 and N41. Site-1 of hSLFN5-NTD contains E37 (the counterpart of sSLFN11-NTD E42) which is neighbored by R34, R36 and N40. The presence of E37 in site-1 of hSLFN5, as well as its low positive charge, correlates with the lack of RNase activity of hSLFN5-NTD and its poor RNA binding affinity (12). It is plausible that site-1 is responsible for RNA substrate recognition, and its net positive charge may govern RNA binding affinity through electrostatic attraction. This hypothesis is also supported by our biochemical results showing that while E42 of sSLFN11 autoinhibited its nucleolytic activity, the E42K charge-reversal mutation resulted in hyperactivity (Figure 4C, D, Figure 5G, H).
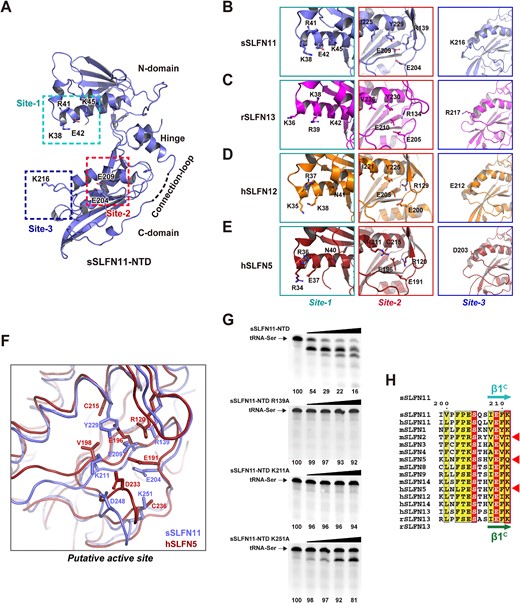
Structural determinant governing RNase activity of SLFN-NTDs. (A) Ribbon model of sSLFN11-NTD; catalytically important residues identified in mutagenesis studies are shown with stick model and labeled. They locate in three distinct sites on inner side of the pincer-shaped molecule. Connection-loop is located on the opposite side. (B–E) Structure of sSLFN11-NTD was superimposed with the structures of rSLFN13-NTD, hSLFN12 and hSLFN5. Sites topologically equivalent to three catalytically important sites identified in sSLFN11-NTD are shown in (B) sSLFN11, (C) rSLFN13, (D) hSLFN12 and (E) hSLFN5. Structural counterparts of catalytically important residues identified in sSLFN11 are shown with stick model and labeled. (F) Structural superimposition of sSLFN11-NTD (light blue ribbon) and hSLFN5 (red ribbon). Residues constitute the putative RNase active sites are shown with stick model and labeled. (G) RNase activity assays the mutants of sSLFN11-NTD (labeled). Increasing amount of enzyme (from left to right: 0, 0.25, 0.5, 1.0 and 2.0 μM) were incubated with tRNA-Ser substrate at 37°C for 30 min and the reaction mixtures were analyzed with urea−PAGE. Numbers below the gels indicate the percentage of uncleaved RNA substrates in each reaction. (H) Structure-based multiple sequence alignment showing residue K211 of sSLFN11-NTD are invariant in the RNase active SLFNs (red box), and unavailable in the RNase inactive SLFNs (red triangles).
Site-2 of sSLFN11-NTD contains the three-carboxylate cluster E204-E209-D248 that is topologically equivalent to the three-carboxylate cluster E205-E210-D248 originally identified in rSLFN13-NTD; the three-carboxylate cluster is essential for rSLFN13-NTD RNase activity (19). However, our mutagenesis results showed that although E204A and E209A mutations abolished the RNase activity of sSLFN11-NTD, D248A had less effect on the activity (Figure 4C). Furthermore, a recent study showed that hSLFN5-NTD is devoid of RNase activity despite containing the intact three carboxylate cluster E191-E196-D233 (12). Therefore, it is possible that the three carboxylates do not constitute the entire active site. To explore this further, we analyzed the neighboring regions of the three-carboxylate cluster and found three basic residues (R139, K211 and K251) that form salt bridges with E204, E209, and D248, implying their involvement in nucleolytic activity (Figure 7F). To prove this, we carried out additional mutagenesis studies and found that mutations R139A and K211A abolished the RNase activity of sSLFN11-NTD, and K251A greatly ablated the activity (Figure 7G). Multiple sequence alignment demonstrated that R139 is invariant in all SLFN proteins, and K211 and K251 are more specific for the RNase-active SLFN proteins. For example, sSLFN11 K211 is invariant in hSLFN11, hSLFN12, rSLFN13 and hSLFN13, but unavailable in the RNase-inactive mSLFN2, mSLFN5 and hSLFN5 (Figure 7H, Supplementary Figure S1).
From a structural perspective, we compared the conformations of the three-carboxylate between RNase-active SLFN-NTDs and RNase-inactive SLFN-NTDs. In the RNase-active enzymes, the catalytically essential residue sSLFN11 E209 and its structural counterparts E210 in rSLFN13 and E205 in hSLF12 all adopted a ‘down’ conformation (Figure 7B−F). By contrast, in the RNase-inactive hSLFN5, E196 (the counterpart of sSLFN11 E209) adopts an ‘up’ conformation. The up conformation of hSLFN5 E196 is stabilized by salt bridging with a unique R211, a feature not shared by RNase-active SLFNs (Figure 7B−E). Furthermore, the bulky Y229 residue of sSLFN11-NTD is located on top of E209, thereby preventing E209 from adopting the up conformation. Intriguingly, other RNase-active SLFN proteins all contain a tyrosine at this position (Y230 in rSLFN13, Y225 in hSLFN12), but hSLFN5 has a smaller cysteine (C215) in this position that allows E196 to adopt the up conformation.
CONCLUSION
Our structural characterizations revealed one striking feature of sSLFN11-NTD, it lacks the zinc-finger. The fourth zinc-coordinating residue becomes a glycine (G318) in sSLFN11, whereas its human ortholog hSLFN11 harbors all four zinc-coordinating residues; therefore, hSLFN11 probably contains a canonical CCCH-type zinc-finger similar to other SLFN proteins. This finding also provides an exception that the zinc-finger is not indispensable for the folding and stability of SLFN proteins. This is reminiscent of picornaviral 2C proteins. Despite sharing a similar fold, some enteroviral 2Cs contain the CCCC-type zinc-finger, others contain the CCC-type zinc-finger (44,45), and many non-enterovirus 2C lacks the entire zinc-finger motifs. Zinc-fingers are small and compact structural units that often function as binding modules for nucleic acids or proteins. Metzner et al., identified two basic residues R271 and R326 close to the zinc-finger of hSLFN5, which are important to nucleic acid binding (12). This feature is probably conserved in hSLFN12 and rSLFN13, but unavailable in sSLFN11 or hSLFN11 (Supplementary Figure S5). Therefore, SLFN11-NTD may recognize RNA substrates in a distinct manner, which underlies its unique nucleolytic activities.
In summary, we report the structural and biochemical characterization of an important SLFN protein, S. scrofa SLFN11, with a focus on its NTD. Although SLFN11 is among the most studied Schlafen proteins due to its role in cancer therapy and virus-host interactions, structural characterization of SLFN11 was lacking. We provide not only the structure for sSLFN11-NTD, but also biochemical characterization of its nucleolytic activity and RNA binding affinity, revealing features distinct from other SLFN members characterized to date. A thorough mutagenesis analysis and structural comparison with other SLFN members revealed key residues governing the nucleolytic activity of various SLFN proteins, and we identified key differences in features between RNase-active and RNase-inactive SLFN members. The findings expand our understanding of the functions of the Schlafen protein family.
Data Availability
The tRNA sequences used in this study were chosen based on the predictions from GtRNAdb (http://gtrnadb.ucsc.edu/GtRNAdb2/index.html). The codon-usage analysis was done using the web application (http://www.kazusa.or.jp/codon/). The atomic coordinate and structure factors of sSLFN11-NTD-Δ have been deposited in the Protein Data Bank under the accession codes: 7FEX.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank the staffs from BL19U1 beamline of National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility for assistance during data collection. We thank the staffs from the Core Facility of Institute of Pathogen Biology, Chinese Academy of Medical Sciences.
FUNDING
Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (2021-I2M-1-037, China); Fundamental Research Funds for the Central Universities (3332021092); Swiss National Science Foundation, the National Research Program Covid-19 (NRP78) from the Swiss National Science Foundation (grant number 4078P0_198290). Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments