





Abstract
DNA chemical modifications, including methylation, are widespread and play important roles in prokaryotes and viruses. However, current knowledge of these modification systems is severely biased towards a limited number of culturable prokaryotes, despite the fact that a vast majority of microorganisms have not yet been cultured. Here, using single-molecule real-time sequencing, we conducted culture-independent ‘metaepigenomic’ analyses (an integrated analysis of metagenomics and epigenomics) of marine microbial communities. A total of 233 and 163 metagenomic-assembled genomes (MAGs) were constructed from diverse prokaryotes and viruses, respectively, and 220 modified motifs and 276 DNA methyltransferases (MTases) were identified. Most of the MTase genes were not genetically linked with the endonuclease genes predicted to be involved in defense mechanisms against extracellular DNA. The MTase-motif correspondence found in the MAGs revealed 10 novel pairs, 5 of which showed novel specificities and experimentally confirmed the catalytic specificities of the MTases. We revealed novel alternative specificities in MTases that are highly conserved in Alphaproteobacteria, which may enhance our understanding of the co-evolutionary history of the methylation systems and the genomes. Our findings highlight diverse unexplored DNA modifications that potentially affect the ecology and evolution of prokaryotes and viruses in nature.
INTRODUCTION
DNA chemical modifications are found in diverse prokaryotes and viruses as well as eukaryotes. DNA methylation is a representative DNA modification that is catalyzed by DNA methyltransferases (MTases), wherein S-adenosylmethionine (SAM) provides the methyl group (1). In prokaryotes, three types of methylation (i.e., N6-methyladenine [m6A], C5-methylcytosine [m5C] and N4-methylcytosine [m4C]) have been investigated in detail (2). DNA methylation plays a role in regulating gene expression and DNA mismatch repair (3–5). These systems have various physiological functions, including asymmetric cell division (6,7), ultraviolet (UV) tolerance (8), motility (9) and virulence of pathogens (10–12). DNA methylation also facilitates cell protection from extracellular DNA (e.g., viral infection and plasmid transfection), known as restriction-modification (RM) systems (13,14). The RM systems are classified into four types based on subunit composition and cofactor requirements. Type I, II and III are composed of both MTase and restriction endonuclease (REase) and specify non-methylated DNA, while Type IV consists of only MTase and specifies modified DNA substrates (15). Some viruses may possess MTases and modify their genomic DNA to escape the host Type I, II and III RM systems. In contrast, Type IV systems have evolved to counter the viral anti-RM system, which results in a ‘co-evolutionary phage-host arms race’ (2,3). Moreover, evidence for gene duplication and loss, horizontal gene transfer within and between domains, and changes in MTase sequence specificity, has been frequently noted in the evolution of prokaryotes (16,17). In addition to methylation, other epigenetic modifications, such as phosphorothioation, have recently been reported to have significant effects on cells, including the maintenance of cellular redox homeostasis and epigenetic regulation (18). There has been a growing interest in exploring the various epigenomic systems amongst diverse prokaryotes and viruses owing to their importance in microbial physiology, genetics, evolution and disease pathogenicity (19–21). However, most studies rely on a small number of culturable prokaryotic strains, whereas the majority of microbes have yet to be cultured. This limited sample size skews our knowledge of microbial epigenomics, particularly in terms of diversity, distribution, and impact upon ecology and evolution.
Recent technological advances have led to the development of single-molecule real-time (SMRT) sequencing technology as a useful method for detecting DNA modifications. Its implementation in PacBio sequencing platforms has yielded an array of DNA modifications amongst prokaryotic (22–27) and viral strains (28,29). The ability of this technology to generate long reads with few context-specific biases (e.g., GC bias) (30) allows the circler consensus sequencing (CCS) method to generate accurate high-fidelity (HiFi) reads; a process facilitated by error correction within multiple ‘subread’ sequences in each single read (31). Based on the innovative SMRT sequencing technique, we conducted culture-independent shotgun metagenomic and epigenomic analyses of freshwater microbial communities. This allowed us to determine their natural DNA modification systems and metaepigenomics (32). Apart from PacBio, nanopore sequencing platforms, produced by Oxford Nanopore Technologies (ONT), can also achieve longer reads that potentially improve metagenomic assembly with high diversity (33). Accordingly, a hybrid approach with HiFi and ONT reads is an ideal way to improve metaepigenomic analysis by enhancing the accuracy of identifying organismal modification in highly diverse microbial communities.
Here, we conducted metaepigenomic analysis of pelagic microbial communities, using the SMRT sequencing technology, to reveal the epigenomic characteristics of diverse marine prokaryotes and viruses whose epigenomic status remains largely unknown. The diverse DNA modifications were successfully characterized in numerous metagenomic-assembled genomes (MAGs) from both prokaryotes and viruses, which were obtained using a combination of PacBio Sequel, ONT GridION and Illumina MiSeq sequencing platforms. Our computational prediction and experimental assays determined several MTases responsible for the detected methylated motifs, including the novel ones. In particular, a highly conserved methylation system with varied specificity was identified in Alphaproteobacteria, suggesting co-evolution between the methylation systems and the genomes.
MATERIALS AND METHODS
Seawater sampling
Seawater samples were collected at two close pelagic stations of the Japan Agency for Marine-Earth Science and Technology (JAMSTEC). Work at these stations, located in the northwest Pacific Ocean, yielded these samples during the JAMSTEC KM19-07 cruises of the Research Vessel (R/V) Kaimei in September 2019 (Supplementary Figure S1 and Supplementary Table S1). The sampling stations were approximately 180 and 140 km offshore from the main island of Japan and 60 km from each other. Each 50–320 L of seawater was collected from 5 and 200 m below sea level (mbsl) at station CM1 (34.2607 N 142.0203 E) and 90 and 300 mbsl at station Ct9H (34.3317 N 141.4143 E) (referred to as CM1_5m, CM1_200m, Ct9H_90m, and Ct9H_300m, respectively). Sampling permits for expeditions in Japan’s exclusive economic zone were not required, as our work was centered in domestic areas and did not involve endangered or protected species. Seawater from 5 mbsl was directly sampled using a built-in pumping system from the bottom of the ship via an intake pipe of approximately 5 m, which was designed for continuous monitoring of sea surface hydrography. The valve of the pumping system was opened for at least 30 min before the start of sampling to thoroughly flush the internal water and rinse the pipe. Seawater from 90, 200 and 300 mbsl was sampled using 12-L Niskin-X bottles (General Oceanic, Miami, Florida, USA) in a CTD rosette system. Vertical profiles of temperature, salinity, and pressure data were obtained using the SBE9plus CTD system (Sea-Bird Scientific, Bellevue, Washington, USA). The vertical profiles of dissolved oxygen (DO) concentrations were obtained using an in situ DO sensor RINKO III (JFE Advantech, Hyogo, Japan) connected to the CTD. The vertical profiles of chlorophyll a concentrations were obtained using an in situ Fluorometer RINKO profiler (JFE Advantech). The seawater samples in the Niskin-X bottles were transferred to sterilized 20 L plastic bags and immediately stored at 4°C until further filtration. Filtration was performed with 0.22 μm Durapore membrane filters (Merck KGaA, Darmstadt, Germany) after pre-filtration with 5 μm Durapore membrane filters (Merck KGaA) onboard. The filters were then immediately stored at temperatures below −30°C.
Flow cytometric assessments of prokaryotic cell and viral-like particle abundances
Seawater samples were obtained for flow cytometric assessment of prokaryotic cells and viral-like particle (VLP) abundances. The samples were collected every 10–50 m at station CM1 and 10–100 m at station Ct9H, fixed with 0.5% (w/v) glutaraldehyde (final concentration) in 2 ml cryo-vials on board, and stored at −80°C until further analysis. To assess prokaryotic cell abundance, 200 μl of each sample was stained with SYBR Green I Nucleic Acid Gel Stain (Thermo Fisher Scientific, Waltham, Massachusetts, USA) (5 × of manufacturer’s stock, final concentration) at room temperature for >10 min. To assess VLP abundance, 20 μl of each fixed sample was diluted 10 times with TE buffer and stained with SYBR Green I (0.5 × of manufacturer’s stock, final concentration) for 10 min at 80°C. Total prokaryotic cells and VLP abundance in 100 μl samples were determined using an Attune NxT Acoustic Focusing Flow Cytometer (Thermo Fisher Scientific) via the green fluorescence versus side scatter plot (34,35).
DNA extraction and shotgun sequencing
Microbial DNA was retrieved using the DNeasy PowerSoil Pro Kit (QIAGEN, Hilden, Germany) according to the supplier’s protocol. The filters were cut into 3 mm fragments and directly suspended in a cell lysis solution provided with the kit. SMRT sequencing was conducted using a PacBio Sequel system (Pacific Biosciences of California, Menlo Park, California, USA) at the National Institute of Genetics (NIG), Japan. SMRT libraries for HiFi read via CCS mode were prepared with a 5 kb insertion length. Briefly, 4–6 kb DNA fragments from each genomic DNA sample were extracted using the BluePippin DNA size selection system (Sage Science, Beverly, Massachusetts, USA). The SMRT sequencing library of CM1_5m and the other three samples were prepared using the SMRTbell Template Prep Kit 1.0-SPv3 and SMRTbell Express Template Prep Kit 2.0, respectively, according to the manufacturer’s protocol (Pacific Biosciences of California). The final SMRT libraries were sequenced using four, three, three, and three Sequel SMRT Cell 1M v3 for CM1_5m, CM1_200m, Ct9H_90m and Ct9H_300m, respectively. Nanopore sequencing of CM1_5m was conducted using a GridION Mk1 platform with five flow cells according to the manufacturer’s standard protocols at NIG. ONT libraries were prepared and purified simultaneously by filtering out a small number of fragments using AMPure XP beads (Beckman Coulter, Brea, California, USA). Illumina sequencing (2 × 300 bp paired-end reads) was conducted using an Illumina MiSeq platform (Illumina, San Diego, California, USA) at JAMSTEC. Illumina libraries were prepared using the KAPA Hyper Prep Kit (Roche, Basel, Switzerland) and pooled with Illumina PhiX control libraries, as described previously (36).
Bioinformatic analysis of the sequencing reads and assembled genomes
CCS reads containing at least five full-pass subreads on each polymerase read and with >99% average base-call accuracy were retained as HiFi reads using the standard PacBio SMRT software package with default settings. Metagenomic HiFi read coverage was estimated using Nonpareil3 with default settings (37). For taxonomic assignment of HiFi reads, Kaiju (38) in Greedy-5 mode (‘-a greedy -e 5’ setting) with NCBI nr (39) and GORG-Tropics databases (40) were used. HiFi reads potentially encoding 16S ribosomal RNA (rRNA) genes were extracted using SortMeRNA (41) with default settings, and full-length 16S rRNA gene sequences were then predicted using RNAmmer (42) with default settings. The 16S rRNA gene sequences were taxonomically assigned using BLASTN (43) against the SILVA database release 128 (44), wherein the top-hit sequences with e-values ≤1E-15 were retrieved. Coding sequences (CDSs) with >33 aa length in HiFi reads were predicted using Prodigal (45) in anonymous mode (‘-p meta’ setting). For Illumina read data, both ends of reads that contained low-quality bases (Phred quality score < 20) and adapter sequences were trimmed using TrimGalore (https://github.com/FelixKrueger/TrimGalore) via default settings. The remaining paired-end reads were merged with at least 10 bp overlap using FLASH (46) with default settings.
HiFi and ONT reads were de novo assembled using wtdbg2 (Redbean) with the settings for PacBio CCS and ONT reads, respectively (47). The assembled contigs from ONT reads were polished using both HiFi and Illumina short reads and HyPo (48). For the polishing, HiFi and Illumina reads were mapped on the pre-polished contigs using pbmm2, an official wrapper software for minimap2 (49) with CCS reads settings, and Bowtie2 (50) with ‘-N 1’ setting, respectively.
The assembled contigs were binned using MetaBAT (51) based on genome coverage and tetra-nucleotide frequencies, as genomic signatures. The genome coverage was calculated with Illumina reads using Bowtie2 with ‘-N 1’ setting. The quality of bins was assessed using CheckM (52), which estimates completeness and contamination based on the taxonomic collocation of prokaryotic marker genes with default settings. Bins with <10% contamination were retrieved according to the metagenome-assembled genome (MIMAG) standards (53) and defined as prokaryotic MAGs (P-MAGs). We noted that the partial genome would be sufficient for detecting DNA modifications and modified motifs; completeness was not considered for P-MAG definition. Sequences of 16S rRNA genes in each P-MAG were retrieved using RNAmmer (42) with default settings. The taxonomy of P-MAGs was estimated based on the 16S rRNA gene sequences, CAT (54), and Kaiju (38). P-MAGs that were not assigned to prokaryotes or assigned with low reliance (<0.6 supported score) using CAT were excluded from further analysis. CDSs with >33 aa length in each P-MAG were predicted using Prodigal (45) with default settings. Functional annotations were achieved through HMMER (55) search against the Pfam database (56), with a cutoff e-value of ≤ 1E-5. Clustered regularly interspaced short palindromic repeat (CRISPR) arrays were predicted using CRISPRDetect3 (57).
For viral sequence collection, the assembled contigs were screened using VirSorter2 (58) with default settings. Quality assessment of the retrieved contigs and removal of flanking host regions from integrated proviruses was performed using CheckV (59). Contigs assigned to either ‘Complete’ or ‘High-quality’ or ‘Medium-quality’ were defined as viral MAGs (V-MAGs) and used for further analysis. Taxonomy gradations lower than the kingdom level were estimated using CAT (54). CDSs were predicted using Prodigal (45) in an anonymous mode (‘-p meta’ setting). Functional annotations were achieved in the same way as for the P-MAGs.
Bioinformatic analysis of modification systems
DNA modification detection and motif analysis were performed in each MAG independently according to the officially provided tool SMRT Link v8.0. Briefly, subreads were mapped to the assembled contigs using pbmm2, and the interpulse duration ratios were calculated. Candidate motifs with scores higher than the default threshold values were retrieved as modified motifs. Those with infrequent occurrences (<50 and <10 in P-MAGs and V-MAGs, respectively) or very low methylation fractions (<10%) in each MAG were excluded from further analysis. Motifs with several ambiguous sequences that were considered to have occurred by misdetection were manually curated. For example, HBNNNNNNVGGWCCNH was detected in CM1_5m.V59, where H = A/C/T, B = A/G/T, V = A/C/G and W = A/W, but this motif represents palindromic GGWCC. Further, the spurious partial sequences of former HBNNNNNNV and latter NH were likely due to incomplete detection of the motif. Notably, we frequently found candidate motifs showing this type of ambiguity in V-MAGs. This possibly results from the method's weak motif estimation power for small genomes. It may also reflect the possibility that low presence of motifs in the genomes negatively affected the motif-finding algorithm implemented in MotifMaker, a tool based on progressive testing for seeking longer motif sequences using a branch-and-bound search.
Genes encoding DNA methyltransferases (MTases), restriction endonucleases (REases), and DNA sequence-recognition proteins (S subunits) were searched using BLASTP (43). They were compared against an experimentally confirmed gold-standard dataset from REBASE (60) (downloaded on 9 February 2021), with a cutoff e-value of ≤1E-5. The sequence specificity information for each MTase and REase gene was retrieved from REBASE. Pairs of MTase and REase genes in the same genome were examined to determine whether they possessed the same specificity and constituted potential RM systems. The BREX (61) and DISARM (62) systems were sought based on Pfam domains.
For accurate analysis of methylome diversity, P-MAGs with >20% completeness were used for the phylogenetic analysis. A maximum-likelihood (ML) tree of the MAGs was constructed using PhyloPhlan3 (63) on the basis of a set of 400 conserved prokaryotic marker genes (64) with ‘--force_nucleotides --diversity high --accurate’ settings. The proteomic tree of V-MAGs was estimated using ViPTreeGen (65) with default settings.
To construct a robust phylogenetic tree of Alphaproteobacteria P-MAGs, those with higher quality (>25% completeness) were retrieved and used for ML tree reconstruction using PhyloPhlan3 with ‘--force_nucleotides --diversity low --accurate’ settings. To calculate the expected/observed (E/O) ratio of each motif sequence, the expected and observed counts of its presentation on the genome were computed using R’MES (66) and SeqKit (67), respectively. An ML tree of MTases was constructed using MEGA X (68) with LG substitution model with a gamma distribution (LG+G), which was selected based on the Bayesian information criterion (BIC) and 100 bootstrap replicates. Three pairs of the Proteobacteria genome and MTase homolog genes were retrieved from the NCBI database and REBASE, respectively, and used for the following outgroups: pairs of Campylobacter sp. RM16704 and M.Csp16704III, Haemophilus influenzae Rd KW20 and M.HinfI, and Helicobacter pylori 26695 and M.HpyAIV. Multiple sequence alignment was performed using the MTase sequences, in addition to M.CcrMI from Caulobacter crescentus CB15, using Clustal Omega (69).
For phylogenetic tree analysis of Alphaproteobacteria and SAR11 genomes, a total of 112 and 195 deposited genomes, described by Muñoz-Gómez et al. (70) and Haro-Moreno et al. (71), were retrieved from the NCBI database, respectively. For the analysis of Alphaproteobacteria, four Betaproteobacteria and four Gammaproteobacteria genomes were retrieved from the NCBI database and used as outgroups. For the analysis of SAR11, genomes of Rickettsia felis URRWXCal2, Rhodospirillum rubrum ATCC11170, Rickettsia bellii RML369-C, and Acidiphilium cryptum JF-5 were retrieved from the NCBI database and used as outgroups. Phylogenetic trees were constructed using PhyloPhlan3 with ‘--force_nucleotides --diversity low --accurate’ settings. Subclades of the SAR11 P-MAGs were inferred based on the topology of the phylogenetic tree, in accordance with a previous definition (71–73).
Experimental verification of MTase activities
To verify MTase specificity, selected MTase genes were artificially synthesized with codon optimization by Thermo Fisher Scientific. The genes were cloned into the pCold III expression vector (Takara Bio, Shiga, Japan) using the In-Fusion HD Cloning Kit (Takara Bio). Additional specific sequences were inserted downstream of the termination codon for the methylation assay if an appropriate sequence was absent from the plasmid vector. The constructs were transformed into Escherichiacoli HST04 dam−/dcm− (Takara Bio), which lacks the dam and dcm MTase genes. In addition, constructs of Ct9H90mP5_10800 and Ct9H90mP30_5500 were alternatively induced into the pET-47b(+) expression vector (Merck KGaA) using the In-Fusion HD Cloning Kit, and transformed into E. coli BL21 Star (DE3) (Thermo Fisher Scientific), owing to severe insolubilization of the expressed protein in the former manner. The soluble protein concentrations were measured via SDS-PAGE as needed. E. coli strains were cultured in LB broth supplemented with ampicillin or kanamycin. MTase expression was induced according to the supplier’s protocol for the expression vector. Plasmid DNA was isolated using the FastGene Plasmid Mini Kit (Nippon Genetics, Tokyo, Japan) or NucleoSpin Plasmid EasyPure Kit (Takara Bio). REase NdeI was employed for linearization of plasmid DNA. Methylation status was assayed simultaneously with linearizing digestion using the appropriate REases. All REases were purchased from New England BioLabs (NEB) (Ipswich, Massachusetts, USA). All digestion reactions were performed at 37°C for 1 h, except for the simultaneous digestion of HinfI and TfiI at 37°C for 30 min, followed by 65°C for 30 min. DNA fragments were separated via capillary electrophoresis using 5300 Fragment Analyzer System (Agilent Technologies, Santa Clara, California, USA) and the HS Genomic DNA Kit (Agilent Technologies).
We further verified MTases with novel motif specificities (i.e., Ct9H300mP26_1870, Ct9H90mP5_10800, CM1200mP2_32760, CM15mP129_7780, CM1200mP10_13750 and CM15mP20_30) via SMRT sequencing. The chromosomal DNA of E. coli HST04 dam−/dcm− strains in which target MTases were transformed was extracted using the DNeasy UltraClean Microbial Kit (QIAGEN), according to the supplier’s protocol, after induction of gene expression. Multiplex SMRT sequencing was conducted using PacBio Sequel II (Pacific Biosciences of California) according to the manufacturer’s standard protocols. Briefly, 12–50 kb DNA fragments from each genomic DNA sample were extracted using the BluePippin size selection system (Sage Science) for continuous long read (CLR) sequencing. SMRT sequencing libraries were prepared using SMRTbell Express Template Prep Kit 2.0 and Barcoded Overhang Adapter Kit 8A according to the manufacturer’s protocol (Pacific Biosciences of California). All final SMRT libraries were sequenced using Sequel II SMRT Cell 8M. Methylated motifs were detected using SMRT Link v9.0 against the E. coli K-12 MG1655 reference genome (RefSeq NC_000913.2).
For the in vitro assay of CM15mP111_3240 MTase and its point mutant, recombinant proteins were purified. The N-terminal 6 × His-tag fusion MTase and D49G mutant were constructed using PCR and cloned into the pCold III expression vector. E. coli cells (HST04 dam−/dcm−), transformed with the constructs, were grown at 37°C for 16 h in 20 ml of medium A (LB medium containing 50 μg/ml ampicillin) via shaking. The culture was inoculated into 2 L of medium A in a 5 L flask, incubated at 37°C for 2–3 h with constant shaking, and allowed to grow until the optical density at 600 nm reached 0.6. Then, MTase expression was induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 15°C, and the cultures were subsequently incubated for 16 h according to the manufacturer’s standard protocol of pCold. E. coli cells were lysed via sonication in Buffer A [20 mM HEPES-Na (pH 7.5), 150 mM NaCl, 5% glycerol, 1 mM DTT, and 50 mM imidazole]. The cell lysate was centrifuged at 12,000 rpm and 4 °C for 30 min and then passed through a GD/X syringe filter with a 0.45 μm pore size (Cytiva, Marlborough, Massachusetts, USA). The supernatant was subjected to two-column chromatography using the ÄKTA prime chromatography system (Cytiva). The presence of the desired protein was confirmed via SDS-PAGE. Thereafter, the sample was loaded onto a 5-ml HisTrap HP column (Cytiva) at a flow rate of 2 ml/min. The column was then washed with Buffer A. The His-tagged protein was eluted with Buffer B [20 mM HEPES-Na (pH 7.5), 150 mM NaCl, 5% glycerol, 1 mM DTT and 300 mM imidazole]. The eluted fractions were pooled and diluted 5-fold with Buffer C [20 mM HEPES-Na [pH 7.5], 150 mM NaCl and 1 mM DTT]. The diluted solution was concentrated to approximately 5 ml using a 30 kDa molecular weight cutoff Amicon Ultra centrifugal filters (Merck KGaA). It was then passed through a Millex-GP syringe filter with 0.22 μm pore size (Merck KGaA), and loaded onto a HiLoad 16/600 Superdex 200 pg column (Cytiva) pre-equilibrated with Buffer C. The protein was collected as a single peak and concentrated to 2.5 mg/ml (∼50 μM in monomer concentration). It was then aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C, or preserved with 50% glycerol at −30°C until further use.
The purified MTases were used for enzymatic methylation. The substrate unmethylated DNA was produced via PCR, with the pCold III vector transferred CM15mP111_3240 gene as a template to match with the in vivo assay of the MTase. Methylation reactions were carried out in a reaction buffer [20 mM HEPES-Na pH 7.5, 100 mM NaCl and 100 μg/ml BSA] containing 5 nM substrate DNA and 1 μM purified MTase, at 20°C for 1 h, unless specified otherwise. To investigate salt sensitivity, NaCl concentration was varied from 0 to 400 mM. To investigate thermal sensitivity, the reaction temperature was varied from 5 to 40°C. To investigate star activity, MTase and glycerol concentrations were varied between 1 and 15 μM and 0–10% v/v, respectively, and the reaction time was extended to 3 h. The reactions were initiated with 160 μM SAM (NEB) in solution and terminated by adding guanidinium thiocyanate solution buffer NTI (Takara Bio). After the methylation reaction and following DNA purification, the methylation status was assayed using HinfI digestion at 37°C for 30 min.
RESULTS
Shotgun sequencing and HiFi read analysis
Four seawater samples were collected from the epipelagic (5 and 90 mbsl) and mesopelagic (200 and 300 mbsl) (referred to as CM1_5m, Ct9H_90m, CM1_200m and Ct9H_300m, respectively) layers of two closely positioned stations in the Pacific Ocean (Supplementary Note S1, Supplementary Figure S1, Supplementary Table S1). For each sample, PacBio Sequel produced 0.66–1.1 million (3.2–4.8 Gb) HiFi reads with >99% accuracy with the average length range 4311–4926 bp (Supplementary Figure S2A–D, Supplementary Table S2). The HiFi reads were estimated to cover 42–63% of the community diversity per sample (Supplementary Figure S3). In addition to SMRT sequencing, we conducted shotgun sequencing of CM1_5m using GridION and obtained 25 million (67 Gb) ONT reads (Supplementary Table S2). Notably, SMRT and ONT sequencing each requires >10 μg of DNA as initial input for library preparation, which limited our ONT sequencing to only a sample from the surface layer (CM1_5m). The average ONT read length (2734 ± 2013 bp) was shorter than the HiFi reads with high deviation, likely because of the methods used for DNA extraction based on bead-beating technique, although N50 reached 3.5 kb, and the longest read achieved was 200 kb in length (Supplementary Figure S2E). Illumina MiSeq reads were also obtained for each sample (Supplementary Table S2). The taxonomic assignment of the HiFi reads was consistent with those of previous studies, suggesting that the HiFi reads reflected general pelagic microbial communities with small sequencing biases (Supplementary Note S2, Supplementary Figure S4).
The number of genes related to DNA methylation and RM systems in the samples was determined by systematic annotation of MTase and REase genes on the HiFi reads, using the REBASE Gold Standard database (60). In general, genes assigned to MTase (M), REase (R), protein fused with the MTase and REase domains (RM), and DNA sequence-recognition protein used in the Type I RM systems (S) showed similar compositions among the microbial communities (Figure 1A). Within the MTase proteins (i.e., M and RM), Type II was predominant, accounting for 76.5–78.6% of each sample (Figure 1B). The relative abundances of Type I (11.2–12.1%) and III (3.4–5.9%) were approximately 2–3 times lower than those identified in the genomes of prokaryotic isolates, reported as 27% and 8%, respectively (23). Among the detected MTases, the most abundant predicted modification type was m6A (56.9–62.9%), followed by m4C (15.6–19.6%) and m5C (14.6–19.6%) (Figure 1C).
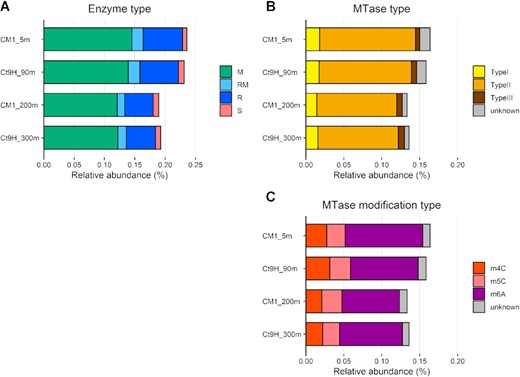
Relative abundance of genes encoding DNA restriction and modification enzymes in marine pelagic metagenomes. CDSs predicted from HiFi reads were used in this analysis. (A) Distribution of enzyme types: DNA methyltransferase (MTase; M), Restriction endonuclease (REase; R), protein fused with M and R domains (RM), and DNA sequence-recognition protein (S). (B) Distribution of MTase types. (C) Distribution of modification types.
Diverse DNA modifications in metagenomic assembled genomes
After HiFi and ONT read assembly and binning analyses, we obtained 233 and 163 prokaryotic MAGs (P-MAGs) and viral MAGs (V-MAGs), respectively (Supplementary Note S3, Supplementary Figure S5, Supplementary Table S3, Supplementary Data S1). From the reconstructed MAGs, a total of 178 and 42 candidate modified motifs were detected in 108 (46%) P-MAGs and 15 (9%) V-MAGs, respectively (Supplementary Data S2). Mapped subread coverages of the modified motifs were compatible with P-MAGs and V-MAGs that ranged from 30.6 to 508.9 × and 88.3 to 568.8 ×, respectively.
The detected motifs were composed of 59 unique motifs, including 32 motifs with palindromic sequences that allow double-strand modification. Among the said motifs, 27 and 23 were classified as m6A and m4C methylation types, respectively. Although current SMRT sequencing technology does not support detection of the m5C motif, we found four candidate m5C motifs with high subread mean coverage (259 × on average). Among the methylated motifs from P-MAGs, 57 (35%) showed <50% modification ratios on the genome, possibly because of the weak detection power of modification from subreads or the existence of strain-level epigenomic heterogeneity in the microbial communities. The modification types of the other five motifs were unclassified and possibly represented chemical modifications of the abovementioned three methylation types, such as phosphorothioation (27). The unclassified motifs showed low modification ratios (ranging from 14% to 45% with 30% on average), similar to previous observations of phosphorothioated motifs in E. coli (12%) and Thaumarchaeota (20%) strains (25,26).
Among the P-MAGs with methylated motifs, GATC was detected most frequently (41 P-MAGs), followed by GANTC (28 P-MAGs), CGCG (19 P-MAGs) and BAAAA (9 P-MAGs), where B = C/G/T and N = A/C/G/T, and the underlined characters indicate modified bases. Among the V-MAGs, RGCY (9 V-MAGs) was the most abundant motif, followed by CCNGG (4 V-MAGs), GGWCC (3 V-MAGs) and GGHCC (3 V-MAGs), where R = A/G, Y = C/T, W = A/T and H = A/C/T. Notably, even considering some vague motifs, at least 15 motifs (i.e., BAAAA, ACAAA, CAAAT, CTAG, GATGG, GATCC, GTNAC, GTWAC, SATC, TGNCA, TSAC, CTCC [m4C], GCGC [m4C], GGWCC [m4C] and TGGCCA [m5C], where S = C/G) did not match the known MTase motifs in the REBASE repository. In addition, methylated motifs likely catalyzed by Type I MTases, which are generally characterized as bipartite sequences with a gap of unspecified nucleotides (e.g., ATGNNNNNTAC), were undetected in all P-MAGs and V-MAGs. This result indicates that Type I RM systems are scarce in epipelagic and mesopelagic prokaryotes and viruses. Regarding vertical distribution of the modified motifs along with the water column, no clear relationship was observed between the frequency of the motif and habitat; 0.65, 1.2, 0.76 and 0.84 motifs were detected on average in CM1_5m, Ct9H_90m, CM1_200m and Ct9H_300m P-MAGs, respectively.
Prediction of MTases and corresponding methylated motifs
To identify MTases that catalyze methylation of the detected motifs, systematic annotation of MTase genes was performed. Sequence similarity searches against known genes stored in REBASE (60) identified 171, 43, and 7 of M, R, and RM genes, respectively, from 112 (48%) P-MAGs (sequence identity in the range 20–92%) (Supplementary Figure S6, Supplementary Data S3). M genes tended to be more frequently detected than R genes in each P-MAG (Supplementary Figure S7A). Only three S genes were found in the P-MAGs, and the small number was concordant with the results of HiFi read analysis (Figure 1A). Among the M and RM genes from P-MAGs, m6A (64%) was the most abundant modification type, followed by m4C (14%) and m5C (10%), as found in the HiFi read analysis (Figure 1C). Among the MTase types, Type II MTases were the most abundant (82%), and 9% and 6% of genes showed the highest sequence similarity to Type I and III MTases, respectively. This trend was consistent with the HiFi read analysis result in which Type I and III MTases were scarcely detected in the communities (Figure 1B). Most of the MTases were orphan, and only four pairs of Type II MTase and REase genes were predicted to possess the same motif sequence specificity and be adjusted on the genome, which may constitute intact Type II RM systems. Other known antiviral defense systems associated with DNA modification, BREX (61) and DISARM (62), were surveyed. However, no MTase genes likely associated with these systems were found in the P-MAGs. Moreover, neither the number of modified motifs nor MTase genes showed a clear association with the number of CRISPR arrays in the P-MAGs (Supplementary Data S1). Overall, these analyses highlight the previously unknown diverse MTases in epipelagic and mesopelagic prokaryotic communities and suggest that methylation systems play unexplored roles apart from their known role in the defense mechanisms against exogenous DNA.
A total of 58 (20%) MTase genes in P-MAGs showed the best sequence similarity to MTases, whose specificity was exactly matched to the motif identified in our metaepigenomic analysis (Supplementary Data S2 and S3). For example, CM1_200m.P15 contained one MTase that showed the best sequence similarity to those that recognized CCSGG, and this finding was perfectly congruent with the motif detected in the P-MAG. For CM1_200m.P39, two MTases similar to those that recognize either TTAA or CGCG were identified, and these motifs were congruently detected in the genome. In Ct9H_300m.P17, five MTases were predicted, two of which were similar to the known MTases that recognize either AGCT or GATC. All of the detected methylated motifs in the genome completely matched with them, suggesting that the two MTases were active.
At least one methylated motif was detected in 44 (19%) P-MAGs, whereas no MTase gene was found. We assumed that the corresponding MTase genes were missed because of insufficient completeness of genomes (including chromosome, plasmids, or multi-partite genomes such as chromid and megaplasmid) in the binning process. Alternatively, these MTase genes may have diverged considerably from known MTase genes. In contrast, at least one MTase gene was found in 30 (13%) P-MAGs, but no methylated motifs were detected. Among the 30 P-MAGs, 42 candidate MTase genes, comprising 1 m4C-type, 6 m5C-type, 27 m6A-type, and 8 type-unknown MTase genes were found. We anticipate that either the MTase genes were inactive, or the corresponding methylated motif went undetected owing to the low sensitivity of SMRT sequencing, especially for m5C modification (22,23).
Among the viral genomes, 82, 13 and 16 of the M, R and RM genes were identified in 49 (30%) V-MAGs (sequence identity in the range 23–73%) (Supplementary Figure S6, Supplementary Data S3). Similar to the case of P-MAGs, M genes tended to be more frequently detected than R genes in each V-MAG. Type II MTases were the most abundant (79%), followed by Type I (7%), with no Type III detected at all (Supplementary Figure S6, S7B). In contrast to that in P-MAGs, m4C (62%) was the most abundant modification type in V-MAGs, followed by m6A (30%) and m5C (1%). All MTases and methylated motifs were unmatched in V-MAGs, except for three pairs (GATC in CM1_5m.V34, GATC, and GTNNAC in Ct9H_90m.V1). This may reflect the very low number of viral MTases stored in the REBASE Gold Standard database, where 16 viral MTase genes were found out of a total of 1938 MTase genes.
Exploration and experimental verification of MTases with new specificity
Among the detected MTase genes, 132 (74%) and 94 (96%) MTases from P-MAGs and V-MAGs, respectively, showed inconsistency between the recognition motifs of their closest relatives and the methylated motifs identified in our metaepigenomic analysis (Supplementary Data S2 and S3). This result suggested that the homology-based estimation of MTase specificity was not sufficient, as in our previous metaepigenomic study of the freshwater microbiome (32). To reveal the catalytic specificity of these MTases, we selected potential pairs of MTase and methylated motifs as follows: (i) MTase and methylated motifs were present in the same genome, and novel correspondence was predicted, (ii) modification types (i.e., m4C, m5C and m6A) of MTase and methylated motifs were concordant and (iii) the complete sequence of the MTase gene was retrieved. Subsequently, the methylation specificities of selected MTases were experimentally verified by heterologous expression in E. coli. Briefly, plasmids with one artificially synthesized MTase gene were constructed and transformed into E. coli cells, and the methylation status of the isolated plasmid DNA was subsequently observed using REase digestion after heterologous expression. The artificially synthesized sequences are summarized in Supplementary Data S4.
In Actinobacteria, Ct9H_300m.P26, one orphan m6A MTase gene, and two m6A and m4C motifs were detected. However, none of the MTase and motif matched with each other. Thus, we predicted that Ct9H300mP26_1870, whose closest homolog encoded an MTase that exhibits CTCGAG methylation activity, would encode an MTase that recognizes BAAAA, whereas the motif sequence was not registered in REBASE and no MTase is currently reported to recognize the motif. The REase digestion assay result was consistent with the hypothesis that ScaI (AGTACT specificity) did not cleave the BAAAAGTACT sequence, which overlapped with BAAAA and AGTACT sequences on the plasmids, only when MTase was expressed in the cells (Figure 2A). We named this protein M.AspCt9H300mP26I, a novel MTase that possesses BAAAA specificity (Table 1).
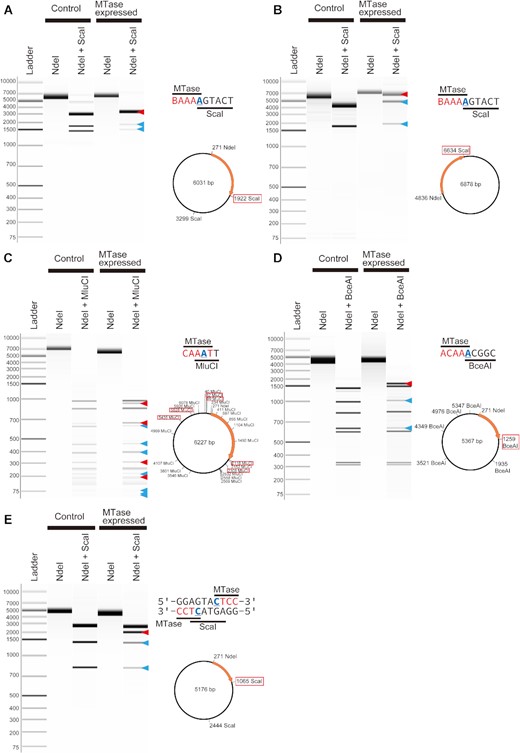
REase digestion assays of MTases with novel specificity. (A) Assay of the Ct9H300mP26_1870 gene. ScaI was used, where the plasmid contained two AGTACT target sites. Within the two sites, one of the target sites was BAAAAGTACT, where overlapped BAAAA and AGTACT were recognized by the MTase and REase. (B) Assay of the Ct9H90mP5_10800 gene. ScaI was used, where the plasmid contained one AGTACT target site in the BAAAAGTACT site. (C) Assay of the CM1200mP2_32760 gene. MluCI were used, where the plasmid contained 23 AATT target sites. Within them, the six target sites were CAAATT, where overlapped CAAAT and AATT were recognized by the MTase and REase, respectively. (D) Assay of the CM15mP129_7780 gene. BceI were used, where the plasmid contained six ACGGC target sites. Within them, one of the target sites was ACAAACGGC, where overlapped ACAAA and ACGGC were recognized by the MTase and REase, respectively. (E) Assay of the CM1200mP10_13750 gene. ScaI were used, where the plasmid contained two TCATGA target sites. Within them, one of the target sites was GGAGTACTCC, where a pair of CTCC and GGAG (comprehensive sequence of CTCC) and TCATGA were recognized by the MTase and REase, respectively. The pCold III (A, C–E) and pET-47b(+) (E) were used as expression vectors. The schematic representation and plasmid map are presented on the right side. The underlined characters indicate modified bases. The orange arrow represents the transferred gene, and the red framed digestion sites represent the location of the overlapped sequence. The band sizes were expected to emerge (red triangles) and disappear (blue triangles) when the induced MTase caused methylation. All plasmid DNAs were linearized using NdeI.
Novel MTases whose specificities were experimentally confirmed. The underlined characters indicate modified bases.
| Gene ID | Genome ID | Lineage | Top-hit protein in REBASE | Identity (%) | RM type | Recognition motif of the closest-match MTase | Modification position | Modification type | RM system | MTase name | Confirmed recognition motif | Novel specificity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM15mP129_7780 | CM1_5m.P129 | Bacteria; Chloroflexi; NA; NA; NA; NA; NA | M.Spn6BI | 35.5 | II | TCTAGA | (unknown) | (unknown) | No | M.CspCM15mP129I | ACAAA | Yes |
| Ct9H90mP5_10800 | Ct9H_90m.P5 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.Sgl8271II | 26.8 | II | ATTAAT | 5 | m6A | No | M.AspCt9H90mP5I | BAAAA | Yes |
| CM1200mP2_5150 | CM1_200m.P2 | Bacteria; Planctomycetes; Planctomycetia; Planctomycetales; Planctomycetaceae; NA; Planctomycetaceae bacterium | M.Sgl8271II | 24.5 | II | ATTAAT | 5 | m6A | No | M.PspCM1200mP2I | CAAAT | Yes |
| CM1200mP10_13750 | CM1_200m.P10 | Bacteria; Candidatus Marinimicrobia; NA; NA; NA; NA; Candidatus Marinimicrobia bacterium | M2.HpyAII | 44.2 | II | GAAGA | -2 | m4C | No | M.MspCM1200mP10I | CTCC | Yes |
| Ct9H300mP26_1870 | Ct9H_300m.P26 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.TliI | 25.0 | II | CTCGAG | 5 | m6A | No | M.AspCt9H300mP26I | BAAAA | Yes |
| CM15mP16_9820 | CM1_5m.P16 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; NA; NA; NA | M.CspNS6I | 62.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP16I | GANTC | No |
| CM15mP20_30 | CM1_5m.P20 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; Alphaproteobacteria bacterium | M.SmeI | 59.9 | ? | GANTC | 2 | m6A | No | M.AspCM15mP20I | GADTC | Yes |
| CM15mP30_3110 | CM1_5m.P30 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Pelagibacteraceae bacterium | M.Bba35685I | 49.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP30I | GAWTC | No |
| CM15mP57_4380 | CM1_5m.P57 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; NA | M.SmeI | 57.0 | ? | GANTC | 2 | m6A | No | M.AspCM15mP57I | GAWTC | No |
| CM15mP70_4410 | CM1_5m.P70 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Alphaproteobacterium | M.Bsp460I | 48.0 | II | GANTC | 2 | m6A | No | M.PspCM15mP70I | GAWTC | No |
| CM15mP111_3240 | CM1_5m.P111 | Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; NA; NA; NA | M.SstE37II | 55.6 | II | GANTC | 2 | m6A | No | M.RspCM15mP111I | GAWTC | No |
| Gene ID | Genome ID | Lineage | Top-hit protein in REBASE | Identity (%) | RM type | Recognition motif of the closest-match MTase | Modification position | Modification type | RM system | MTase name | Confirmed recognition motif | Novel specificity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM15mP129_7780 | CM1_5m.P129 | Bacteria; Chloroflexi; NA; NA; NA; NA; NA | M.Spn6BI | 35.5 | II | TCTAGA | (unknown) | (unknown) | No | M.CspCM15mP129I | ACAAA | Yes |
| Ct9H90mP5_10800 | Ct9H_90m.P5 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.Sgl8271II | 26.8 | II | ATTAAT | 5 | m6A | No | M.AspCt9H90mP5I | BAAAA | Yes |
| CM1200mP2_5150 | CM1_200m.P2 | Bacteria; Planctomycetes; Planctomycetia; Planctomycetales; Planctomycetaceae; NA; Planctomycetaceae bacterium | M.Sgl8271II | 24.5 | II | ATTAAT | 5 | m6A | No | M.PspCM1200mP2I | CAAAT | Yes |
| CM1200mP10_13750 | CM1_200m.P10 | Bacteria; Candidatus Marinimicrobia; NA; NA; NA; NA; Candidatus Marinimicrobia bacterium | M2.HpyAII | 44.2 | II | GAAGA | -2 | m4C | No | M.MspCM1200mP10I | CTCC | Yes |
| Ct9H300mP26_1870 | Ct9H_300m.P26 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.TliI | 25.0 | II | CTCGAG | 5 | m6A | No | M.AspCt9H300mP26I | BAAAA | Yes |
| CM15mP16_9820 | CM1_5m.P16 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; NA; NA; NA | M.CspNS6I | 62.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP16I | GANTC | No |
| CM15mP20_30 | CM1_5m.P20 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; Alphaproteobacteria bacterium | M.SmeI | 59.9 | ? | GANTC | 2 | m6A | No | M.AspCM15mP20I | GADTC | Yes |
| CM15mP30_3110 | CM1_5m.P30 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Pelagibacteraceae bacterium | M.Bba35685I | 49.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP30I | GAWTC | No |
| CM15mP57_4380 | CM1_5m.P57 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; NA | M.SmeI | 57.0 | ? | GANTC | 2 | m6A | No | M.AspCM15mP57I | GAWTC | No |
| CM15mP70_4410 | CM1_5m.P70 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Alphaproteobacterium | M.Bsp460I | 48.0 | II | GANTC | 2 | m6A | No | M.PspCM15mP70I | GAWTC | No |
| CM15mP111_3240 | CM1_5m.P111 | Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; NA; NA; NA | M.SstE37II | 55.6 | II | GANTC | 2 | m6A | No | M.RspCM15mP111I | GAWTC | No |
Novel MTases whose specificities were experimentally confirmed. The underlined characters indicate modified bases.
| Gene ID | Genome ID | Lineage | Top-hit protein in REBASE | Identity (%) | RM type | Recognition motif of the closest-match MTase | Modification position | Modification type | RM system | MTase name | Confirmed recognition motif | Novel specificity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM15mP129_7780 | CM1_5m.P129 | Bacteria; Chloroflexi; NA; NA; NA; NA; NA | M.Spn6BI | 35.5 | II | TCTAGA | (unknown) | (unknown) | No | M.CspCM15mP129I | ACAAA | Yes |
| Ct9H90mP5_10800 | Ct9H_90m.P5 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.Sgl8271II | 26.8 | II | ATTAAT | 5 | m6A | No | M.AspCt9H90mP5I | BAAAA | Yes |
| CM1200mP2_5150 | CM1_200m.P2 | Bacteria; Planctomycetes; Planctomycetia; Planctomycetales; Planctomycetaceae; NA; Planctomycetaceae bacterium | M.Sgl8271II | 24.5 | II | ATTAAT | 5 | m6A | No | M.PspCM1200mP2I | CAAAT | Yes |
| CM1200mP10_13750 | CM1_200m.P10 | Bacteria; Candidatus Marinimicrobia; NA; NA; NA; NA; Candidatus Marinimicrobia bacterium | M2.HpyAII | 44.2 | II | GAAGA | -2 | m4C | No | M.MspCM1200mP10I | CTCC | Yes |
| Ct9H300mP26_1870 | Ct9H_300m.P26 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.TliI | 25.0 | II | CTCGAG | 5 | m6A | No | M.AspCt9H300mP26I | BAAAA | Yes |
| CM15mP16_9820 | CM1_5m.P16 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; NA; NA; NA | M.CspNS6I | 62.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP16I | GANTC | No |
| CM15mP20_30 | CM1_5m.P20 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; Alphaproteobacteria bacterium | M.SmeI | 59.9 | ? | GANTC | 2 | m6A | No | M.AspCM15mP20I | GADTC | Yes |
| CM15mP30_3110 | CM1_5m.P30 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Pelagibacteraceae bacterium | M.Bba35685I | 49.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP30I | GAWTC | No |
| CM15mP57_4380 | CM1_5m.P57 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; NA | M.SmeI | 57.0 | ? | GANTC | 2 | m6A | No | M.AspCM15mP57I | GAWTC | No |
| CM15mP70_4410 | CM1_5m.P70 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Alphaproteobacterium | M.Bsp460I | 48.0 | II | GANTC | 2 | m6A | No | M.PspCM15mP70I | GAWTC | No |
| CM15mP111_3240 | CM1_5m.P111 | Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; NA; NA; NA | M.SstE37II | 55.6 | II | GANTC | 2 | m6A | No | M.RspCM15mP111I | GAWTC | No |
| Gene ID | Genome ID | Lineage | Top-hit protein in REBASE | Identity (%) | RM type | Recognition motif of the closest-match MTase | Modification position | Modification type | RM system | MTase name | Confirmed recognition motif | Novel specificity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM15mP129_7780 | CM1_5m.P129 | Bacteria; Chloroflexi; NA; NA; NA; NA; NA | M.Spn6BI | 35.5 | II | TCTAGA | (unknown) | (unknown) | No | M.CspCM15mP129I | ACAAA | Yes |
| Ct9H90mP5_10800 | Ct9H_90m.P5 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.Sgl8271II | 26.8 | II | ATTAAT | 5 | m6A | No | M.AspCt9H90mP5I | BAAAA | Yes |
| CM1200mP2_5150 | CM1_200m.P2 | Bacteria; Planctomycetes; Planctomycetia; Planctomycetales; Planctomycetaceae; NA; Planctomycetaceae bacterium | M.Sgl8271II | 24.5 | II | ATTAAT | 5 | m6A | No | M.PspCM1200mP2I | CAAAT | Yes |
| CM1200mP10_13750 | CM1_200m.P10 | Bacteria; Candidatus Marinimicrobia; NA; NA; NA; NA; Candidatus Marinimicrobia bacterium | M2.HpyAII | 44.2 | II | GAAGA | -2 | m4C | No | M.MspCM1200mP10I | CTCC | Yes |
| Ct9H300mP26_1870 | Ct9H_300m.P26 | Bacteria; Actinobacteria; Acidimicrobiia; Acidimicrobiales; NA; NA; NA | M.TliI | 25.0 | II | CTCGAG | 5 | m6A | No | M.AspCt9H300mP26I | BAAAA | Yes |
| CM15mP16_9820 | CM1_5m.P16 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; NA; NA; NA | M.CspNS6I | 62.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP16I | GANTC | No |
| CM15mP20_30 | CM1_5m.P20 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; Alphaproteobacteria bacterium | M.SmeI | 59.9 | ? | GANTC | 2 | m6A | No | M.AspCM15mP20I | GADTC | Yes |
| CM15mP30_3110 | CM1_5m.P30 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Pelagibacteraceae bacterium | M.Bba35685I | 49.9 | II | GANTC | 2 | m6A | No | M.PspCM15mP30I | GAWTC | No |
| CM15mP57_4380 | CM1_5m.P57 | Bacteria; Proteobacteria; Alphaproteobacteria; NA; NA; NA; NA | M.SmeI | 57.0 | ? | GANTC | 2 | m6A | No | M.AspCM15mP57I | GAWTC | No |
| CM15mP70_4410 | CM1_5m.P70 | Bacteria; Proteobacteria; Alphaproteobacteria; Pelagibacterales; Pelagibacteraceae; NA; Alphaproteobacterium | M.Bsp460I | 48.0 | II | GANTC | 2 | m6A | No | M.PspCM15mP70I | GAWTC | No |
| CM15mP111_3240 | CM1_5m.P111 | Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; NA; NA; NA | M.SstE37II | 55.6 | II | GANTC | 2 | m6A | No | M.RspCM15mP111I | GAWTC | No |
In Actinobacteria, Ct9H_90m.P5, two orphan MTase genes, and three methylated motifs were detected. While a pair of MTase and motif was concordantly matched, the other MTases did not match any motifs. The latter MTase gene Ct9H90mP5_10800 showed moderate sequence similarity (32%) to M.AspCt9H300mP26I using BLASTP search with a low e-value (1E-70), and either of the remaining motifs was m6A and m4C. Thus, we predicted that Ct9H90mP5_10800 MTase, whose closest homolog is an m6A MTase that exhibits ATTAAT methylation, would have BAAAA specificity. As expected, the REase digestion assay showed that ScaI resisted cleaving the BAAAAGTACT sequence only when the protein was expressed (Figure 2B). We named this protein M.AspCt9H90mP5I, a novel MTase that possesses BAAAA specificity (Table 1). Notably, another candidate orphan MTase gene, Ct9H90mP30_5500, was detected in Actinobacteria Ct9H_90m.P3. It was predicted to possess the same BAAAA specificity and showed moderate (33%) and high (87%) sequence similarity to M.AspCt9H300mP26I and M.AspCt9H90mP5I, respectively. However, this protein was insoluble in E. coli, resulting in no clear cleavage inhibition in our experiment.
A Planctomycetes CM1_200m.P2 had three orphan MTase genes and two methylated motifs. One of the MTases showed the highest sequence similarity to those recognizing TTAA with high similarity (64%). The other CM1200mP2_32760 and CM1200mP2_5150 MTases showed the highest sequence similarity to those catalyzing m6A modification and recognizing GTTAAC and ATTAAT, respectively, with low similarity (37% and 25%, respectively). The two detected motifs were GCGC (m4C) and CAAAT (m6A), the latter of which was not found in REBASE. Thus, we expected that either or both MTases would recognize and methylate the novel CAAAT motif. The construct CM1200mP2_32760 was not successfully prepared in our experiment, likely because the protein was toxic to E. coli. In contrast, CM1200mP2_5150 MTase showed that MluCI (AATT specificity) did not cleave all CAAATT sequences when only MTase was expressed. This clearly indicated that MTase recognizes CAAAT (Figure 2C). Accordingly, we named the protein M.PspCM1200mP2I, a novel MTase that possesses previously unknown CAAAT specificity (Table 1).
Chloroflexi CM1_5m.P129 had one orphan MTase gene, which showed the highest sequence similarity to those recognizing TCTAGA (whose modification type and position were not reported). However, the only methylated motif detected in the genome was ACAAA, which no MTase was currently reported to recognize. Thus, we hypothesized that CM15mP129_7780 MTase should recognize and modify this novel motif. The REase digestion assay result was consistent with the hypothesis that BceAI (ACGGC specificity) did not cleave the ACAAACGCG sequence when only MTase was expressed (Figure 2D). Accordingly, we named this protein M.CspCM15mP129I, a novel MTase that possesses previously unreported ACAAA specificity (Table 1).
In Candidatus (Ca.) Marinimicrobia CM1_200m.P10, one orphan MTase gene, and one methylated motif were detected. The reported recognition motif of the closest MTase is GAAGA (the modified base is the second position of the complementary sequence TCTTC), while the detected motif was CTCC. Thus, we hypothesized that the recognition motif of CM1200mP10_13750 MTase would be CTCC, a previously unreported methylated motif. The REase digestion assay showed that ScaI was inhibited from cleaving the GGAGTACTCC sequence site, where the ScaI-targeting site was complementally sandwiched by CTCC (Figure 2E). We named this protein M.MspCM1200mP10I (Table 1).
Furthermore, we conducted a re-sequencing analysis to examine the methylation status of the chromosomal DNA of E. coli in which each novel MTase gene was transformed and expressed. As a result, all five methylated motifs were successfully recalled in each of the E. coli genomes (Supplementary Table S4).
Phylogenetic distribution of modified motifs
To investigate the phylogenetic distribution of the DNA modification system in the MAGs, we used 117 P-MAGs (>20% completeness) and all 163 V-MAGs for robust phylogenetic tree reconstruction, and visualized the modification ratios of the detected motifs in each genome (Figure 3). Within the P-MAGs, modified motifs were sporadically distributed across the phyla, whereas some showed great concordance with the phylogenetic clades. For example, within the phylum Actinobacteria, CGCG and BAAAA were spread in the genomes of all organisms from the class Acidimicrobiia but were not detected in organisms from the class Actinobacteria. By contrast, AATT was found in three P-MAGs belonging to a subclade in Acidimicrobiia. TTAA was detected in four P-MAGs in Chloroflexi. GATC was detected with moderate to high modification ratios (19–99%) through archaeal P-MAGs, with two exceptions; no significant GATC signature was detected in Euryarchaeota Ct9H_90m.P24 (7%) and CM1_5m.P82 (0.4%) possibly due to the methylation activity being weak or absent in these organisms. AGCT was observed in both the Thaumarchaeota P-MAGs with high modification ratios (82–91%). CGCG was found in members from three phyla across the domain: Actinobacteria, Chloroflexi and Euryarchaeota. GANTC/GAWTC appeared in all 26 Alphaproteobacteria P-MAGs, with only one exception. In addition to methylation, AGCT modified motif showed weak modification ratios (2–19%) in the class Ca. Poseidoniia P-MAGs. However, this motif was detected only in Ct9H_300m.P10 in the motif prediction analysis. This result demonstrates that phylogeny-based modification ratio analysis is efficient for analyzing infrequently modified motifs.
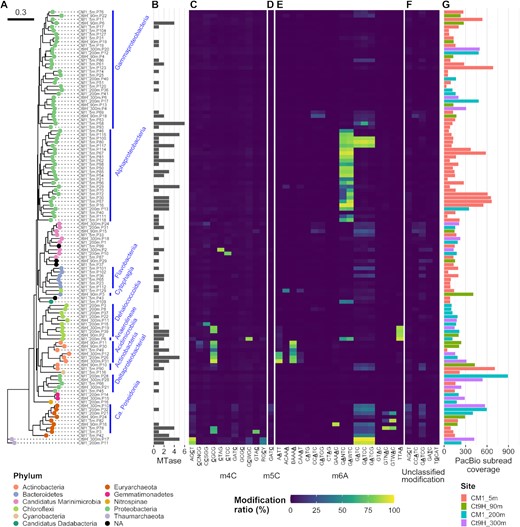
Methylomes of P-MAGs. (A) A phylogenetic tree was constructed based on a set of up to 400 conserved bacterial marker genes using the maximum-likelihood method. Node color indicates taxonomy at the phylum level. Nodes were grouped at class to family levels, if estimated (blue bars and texts). (B) Numbers of MTase genes identified in each genome. (C–F) Modification ratios of detected motifs per genome. (C) m4C, (D) m5C, (E) m6A and (F) unclassified modifications are shown individually. Motifs detected from P-MAGs without spurious sequence were used. The color range from blue over green to yellow represents modification ratios of motifs on each genome. The underlined characters indicate modified bases. Notably, modification ratios were affected by overlapped motif sequences; for example, GATCC was completely overlapped by GATC, and both motifs showed similar modification rates in their genomes except in Gammaproteobacteria CM1_5m.P58 where GATCC was detected on the genome as per the metaepigenomic analysis and concordantly the modification ratio of GATCC was higher than that of GATC. (G) Coverages of subread on each genome. The bar color represents the source of the genome.
By sharp contrast, many motifs showed no clear association with the phylogenetic topology. For example, GCWGC was solitary, with a high modification ratio in Chloroflexi CM1_200m.P6. Similarly, CTAG in Ca. Marinimicrobia Ct9H_300m.P2, CTCC in Ca. Marinimicrobia CM1_200m.P10, GTAC in Euryarchaeota CM1_5m.P3, ACAAA in Chloroflexi CM1_5mP129, and GAAAC in Euryarchaeota Ct9H_90m.P16 were found.
Within all the P-MAGs in this study, no methylated motif was detected in 125 (54%) P-MAGs with high subread coverage (ranging from 31.4–3305.7 × and 207.6 × on average); thus, this was not addressed by insufficient coverage depth for modification detection. The 125 P-MAGs were found to be dispersed across diverse phyla, such as Proteobacteria, Bacteroidetes, Ca. Marinimicrobia, Chloroflexi, Gemmatimonadetes, Cyanobacteria and Verrucomicrobia. Interestingly, neither methylated motifs nor MTase genes were detected in P-MAGs belonging to the following lineages: both members of Gemmatimonadetes and Nitrospinae, and all five members of Verrucomicrobia (Supplementary Data S1). Methylated motifs were also absent from all three Deltaproteobacteria P-MAGs, although two of them possessed the MTase gene. Within the Gammaproteobacteria P-MAGs, 31 of the 52 genomes lacked both methylated motifs and MTase genes. Taken together, these observations suggest the absence of a DNA methylation system in several clades. This finding contradicts that of a previous study, which reported the pervasiveness of DNA methylation among bacteria and archaea (23).
Methylated motifs were occasionally detected with low modification ratios in most V-MAGs, except for Phycodnaviridae and Myoviridae (Figure 4). Among the Phycodnaviridae V-MAGs, Ct9H_90m.V1 showed GATC and GTNNAC having a high modification ratio, whereas Ct9H_90m.V2 harbored TCGA. These results were consistent with previous findings, which reported that m6A is frequently found in Phycodnaviridae genomes (74). In 14 Myoviridae V-MAGs, 0–5 methylated motifs were detected. However, the proteomic tree showed numbers of V-MAGs that, though not taxonomically assigned, were closely related to the Myoviridae family (referred to as ‘Myoviridae-like’). The Myoviridae-like V-MAGs frequently appeared to share several m4C motifs (e.g., RGCY, CCWGG, GGWCC) with different combinations. Sometimes, they also harbored additional motifs, while a few numbers of modified motifs were detected in the motif prediction analysis (0.95 motifs per genome on average). This indicates that the taxonomic assignment of the viral genome was frequently missed due to the lack of viral genomes in the reference database and the severe underestimation of modified motifs in V-MAGs, likely due to their small genome size (see Materials and Methods). In contrast to the Phycodnaviridae and Myoviridae-like V-MAGs, methylation was scarcely detected in the other V-MAGs, including those of Siphoviridae and Podoviridae. Notably, the methylated motifs detected in the V-MAGs were rarely shared with those in the P-MAGs, and no modified motifs other than methylation were found. Five Myoviridae-like V-MAGs were predicted to be proviruses. However, no clear difference was observed in the modification ratio compared to the case for the other non-provirus V-MAGs. In 39 Myoviridae-like V-MAGs, several MTases were found to be encoded in their genomes (ranging from 0 to 6 and 2.2 MTase genes per genome on average) (Figure 4B and Supplementary Data S3). Yet, they were scarcely detected in the other V-MAGs (ranging from 0 to 3 and 0.1 on average).
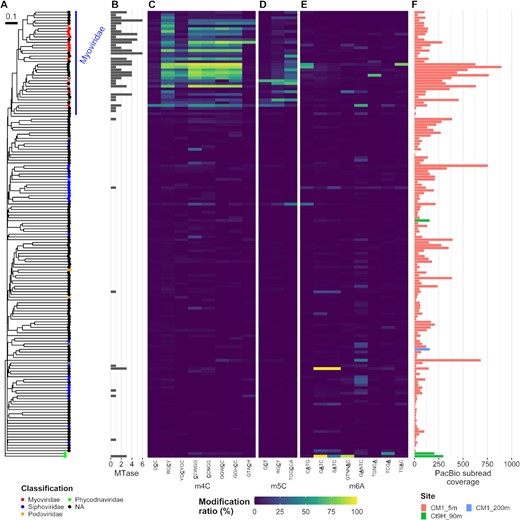
Methylomes of V-MAGs. (A) A proteomic tree was generated based on the global genomic similarities between viral genomes. Proviruses are indicated by circle cross. Node color indicates taxonomy at the family level. (B) Numbers of MTase genes identified in each genome. (C–E) Modification ratios of (C) m4C, (D) m5C and (E) m6A motifs. (F) Coverages of subread on each genome. Please also see Figure 3.
MTases that recognize GADTC/GAWTC motifs in marine Alphaproteobacteria
M.CcrMI is also known as ‘cell cycle-regulated MTase’ (CcrM) from Caulobacter crescentus. Along with GANTC specificity, it is one of the model protein of prokaryotic MTase that is well conserved in Alphaproteobacteria (23,75,76). Indeed, GANTC was previously identified in diverse lineages of Alphaproteobacteria isolates (23,77,78) and one MAG (32) using the modern SMRT sequencing technique, and no alternative motifs have been reported. In our metaepigenomic analysis, GANTC was concordantly detected in 26 of 40 Alphaproteobacteria P-MAGs (Supplementary Data S3). In addition, we detected similar but different motifs GAWTC, GADTC, and GAHTC from seven, four and one Alphaproteobacteria P-MAGs, respectively (where D = A/G/T) (Figure S8). This result strongly suggests the presence of unknown variations in the methylation system in the lineage. We should note that because all the detected modified bases on both DNA strands were used for motif prediction, the detection of non-palindromic GADTC and GAHTC motifs was not explained by the differences in the proportion of GACTC and GAGTC sequences in the genomic data (represented by one side of the DNA strand in the fasta file). It should also be noted that modifications other than m6A were rarely found on either of the strands in the motif. This suggests that the motifs did not result from the wrong prediction due to the modifications other than m6A. With regard to the Alphaproteobacteria P-MAGs, we predicted 13 complete gene sequences of MTase that were assumed to recognize either of the motifs. However, all of them showed the highest sequence similarity (47–80%) to those known to recognize GANTC (Supplementary Data S3).
Considering the correspondence of the methylated motifs and MTases, it was expected that four and one MTases would recognize GAWTC and GADTC, respectively, rather than GANTC (Supplementary Data S2 and S3). The REase digestion assay of the former four MTases (CM15mP30_3110, CM15mP57_4380, CM15mP70_4410 and CM15mP111_3240) showed that TfiI (GAWTC specificity) cleavage was completely blocked only when MTase was expressed in the cells, whereas HinfI (GANTC specificity) partly cleaved the plasmids (Supplementary Figure S9A–D). Despite exhibiting off-target effects under high concentrations of the enzyme, known as ‘star activity’ (79,80), assays of purified CM15mP111_3240 MTase protein suggested that it showed canonical specificity toward GAWTC (Supplementary Note S4, Figure S10A–C). The digestion pattern in the assay of CM15mP20_30 was also congruent with the hypothesized GADTC methylation and re-sequencing analysis successfully recalled the methylated motif, thus indicating its low efficiency with regard to GACTC methylation (Supplementary Note S5, Supplementary Figure S9E, Supplementary Table S4). By contrast, as expected, robust GANTC specificity was confirmed in the assay of CM15mP16_9820, which completely inhibited both TfiI and HinfI cleavage (Supplementary Figure S9F). Accordingly, we named the four (M.PspCM15mP30I, M.AspCM15mP57I, M.PspCM15mP70I, and M.RspCM15mP111I) and one (M.AspCM15mP20I) proteins as novel MTases that preferentially recognize GAWTC and GADTC, respectively, and the last protein (M.PspCM15mP16I), as one that recognizes GANTC (Table 1). Interestingly, we found that the temperature-activity profile of the purified M.RspCM15mP111I MTase (Supplementary Figure S10B) was concordant with the marine water temperature at the sampling sites (Supplementary Figure S1B), suggesting that MTase was thermally optimized in an epipelagic environment.
Based on the sequence alignment of the 13 MTases with M.CcrMI and its homologs, a glycine residue (corresponding to Gly40 in M.CcrMI) was roughly conserved in all MTases with GANTC specificity. By contrast, it was replaced with lysine or aspartic acid in all MTases with GAWTC specificity (Supplementary Figure S11). It has been reported that the M.CcrMI protein contains a substructure that forms a pocket to accommodate the third position of the recognized motif (i.e., nucleotide ‘N’ in GANTC); two hydrophobic residues Leu38–Leu42 stacks, and flexible Gly39 and Gly40 allow the acceptance of variable nucleotides in the position (81). This led to the hypothesis that a replacement of lysine/aspartic acid with glycine at the bottom of the fitting pocket would trigger physical interference in the third position of the motif sequence and change its sequence specificity (i.e., shift from GAWTC to GANTC). To test this hypothesis, we constructed a substitution mutant D49G of CM15mP111_3240 (the position corresponding to M.CcrMI Gly40) and performed the REase digestion assay. However, the mutant showed partial inhibition of HinfI cleavage as compatible with the original MTase, suggesting that another factor, other than merely the Gly40 residue, defined the third position of the motif as ‘W’ (Supplementary Figure S10D).
Evolutionary history and genomic impact of methylation systems in Alphaproteobacteria
To understand the evolutionary relationships among M.CcrMI homologs in Alphaproteobacteria, we analyzed the phylogenetic diversity of the methylated motifs and the frequencies of the motif sequences on each Alphaproteobacteria P-MAG (Figure 5A and C). In Rhodospirillales, SAR116, and Rhodobacteriaceae P-MAGs, all four subsets of GANTC (i.e., GAATC, GATTC, GACTC and GAGTC) showed high modification ratios. However, in one Rhizobiales and four SAR11 P-MAGs, GAWTC was methylated at higher modification ratios, whereas GASTC (i.e., GACTC and GAGTC) was almost unmethylated.

Methylomes and phylogenetic analysis of Alphaproteobacteria P-MAGs. Three homolog MTases, which were found in Proteobacteria isolates and previously confirmed to recognize GANTC were retrieved from REBASE and used as outgroups in this analysis; M.HpyAIV from Helicobacter pylori (Epsilonproteobacteria), M.Csp16704III from Campylobacter sp. (Epsilonproteobacteria), and M.HinfI from Haemophilus influenzae (Gammaproteobacteria). P-MAGs with >25% completeness were used in this analysis for robust phylogenetic tree prediction. (A) A phylogenetic tree of the Alphaproteobacteria P-MAGs. (B) Observed/Expected (O/E) ratio of the GANTC member. A pair of GAATC and GATTC sequences constitutes GAWTC, and all the four sequences (GAATC, GATTC, GACTC and GAGTC) constitute the GANTC motif, where W = A/T and N = A/C/G/T. (C) Modification ratios of each GANTC component. Blank rows indicate the outgroup whose methylation data were not available. (D) Phylogenetic tree of the MTase encoding genes. Supporting bootstrap values >40% are shown. The node shapes indicates MTases that were estimated (rectangle nodes) or experimentally confirmed (circle) whose specificity and outgroups are indicated by circle cross. The node colors indicate the specificity of each MTase.
The phylogenetic topologies of P-MAGs and MTase were matched in Rhodospirillales, SAR116, Rhodobacteraceae and Rhizobiales, suggesting the good conservation of the MTases in these clades (Figure 5A and D). By contrast, those in SAR11 showed incongruence with them, possibly because of the weak robustness of the phylogenetic inference of the MTases supported by low bootstrap values. Neither GANTC/GADTC/GAWTC methylation nor the corresponding MTase was detected in SAR11 CM1_5m.P40 (Supplementary Data S2 and S3), indicating that the organism lacked the methylation system. Regardless of the inconsistent topologies between organisms and proteins, the MTases with GAWTC or GADTC specificity were phylogenetically placed within those of GANTC with high sequence similarity and comprised a monophyletic group. Consequently, it is suggested that the methylation systems have been maintained in Alphaproteobacteria and the MTases, with GAWTC/GADTC specificity branching out from those with GANTC rather than being acquired from distant lineages (i.e., other from Alphaproteobacteria) by horizontal gene transfer.
Notably, the frequency of motif sequences in the genomes was less than expected when these motifs were methylated (Figure 5B and C). In Rhodospirillales, SAR116, and Rhodobacteriaceae P-MAGs, in which GANTC was highly methylated, the log2 Observed/Expected ratio (O/E ratio) of all subsets of GANTC sequences was -1.14 ± 0.53 (s.d.), on average. This means that GANTC sequences present with >2-fold lower frequency than that expected from the random distribution on their genomes, suggesting the existence of negative pressure against GANTC sequences. By contrast, in Rhizobiales and SAR11 P-MAGs, except for CM1_5m.P40, the GAWTC O/E ratio was significantly lower than that of GASTC (-1.73 ± 0.59 and 0.43 ± 0.42, respectively) (P < 0.05, U-test). This difference suggests the presence of a strong negative pressure on the GAWTC sequence, which was attenuated by GASTC. In SAR11 CM1_5m.P40, GANTC was free from methylation, and concordantly, the O/E ratios were approximately zero (-0.09 ± 0.08), suggesting a weak or no selective pressure on the GANTC sequences.
To gain a more global view of the GANTC sequence representation in the extensive Alphaproteobacteria class, we calculated the O/E ratios using 112 and 195 accessible genomes that covered diverse Alphaproteobacteria (70) and all major subclades (I–V) of SAR11 (71,82), respectively (Supplementary Data S5). All constituent GANTC sequences generally showed negative O/E ratios in Rhodospirillales, Sphingomonadales, Rhizobiales, Caulobacterales and Rhodobacterales (-1.78 ± 0.59) (Supplementary Figure S12). By contrast, those of Rickettsiales and Holosporales, including the numbers of endosymbiotic members, were temperate (-0.39 ± 0.20). Only in case of SAR11 were the O/E ratios of GAWTC significantly lower than those of GASTC (-1.30 ± 0.57 and -0.14 ± 0.37, respectively) (P < 0.05, U-test, Bonferroni correction). This was concordant with the P-MAG analysis (Figure 5B). These results indicated that all constituent sequences of GANTC were under negative pressure in Alphaproteobacteria, with prime exceptions included Rickettsiales and Holosporales, which evinced weak pressure, and SAR11, which showed signs of selectively attenuated pressure in GASTC constituents. Thus, the O/E ratio profile implied that the GANTC methylation system was not strictly conserved in all Alphaproteobacteria, compared to the GAWTC methylation system maintained in the exceptional group.
The estimated phylogenetic tree of SAR11 showed that one and seven P-MAGs belonged to subclades IV and V, respectively (Supplementary Figure S13). The O/E ratios of GAWTC were significantly negative (-1.30 ± 0.45), in contrast to those of GASTC (0.09 ± 0.30) (P < 0.05, U-test). However, they were not evenly distributed throughout the SAR11 subclades. For example, the GAWTC O/E ratios were higher in subclade Ic (-0.58 ± 0.10), whereas those of GASTC were comparatively lower in subclade Ia.1 (-0.51 ± 0.05). Notably, the GAWTC O/E ratios varied in subclade V (ranging from -2.7 to -0.1). Within subclade V, five minor subclades were identified based on the phylogenetic topology associated with the O/E ratio. One minor subclade, here referred to Va, showed the lowest GAWTC O/E ratio (-1.83 ± 0.59). By contrast, the subclades Vb, Vc and Vd showed comparatively higher GAWTC O/E ratios (-0.58 ± 0.28, -0.47 ± 0.06 and -0.62 ± 0.04, respectively). The other subclade, Ve, showed comparatively moderate O/E ratios (-1.00 ± 0.42), which is compatible with the other major subclades. Despite such variations, overall, the O/E ratio profile suggests that the negative selective pressure in GAWTC sequence is highly conserved but absent or weak in GASTC sequence throughout the SAR11 subclades. This may be driven by DNA methylation caused by MTases with GAWTC specificity. Further, fluctuating pressure among the subclades may be associated with ecological and evolutionary niches.
DISCUSSION
A possible function of DNA methylation in marine prokaryotic and viral communities
The crucial biological roles played by prokaryotic and viral DNA modification have long been emphasized. However, little is known about their diversity, ecological role and evolutionary history, especially in the environmental community. Several studies have conducted bisulfite sequencing to investigate prokaryotic m5C modifications using environmental samples (83,84). However, other m6A and m4C modifications that are more common in prokaryotes have not been investigated. Although community-level prokaryotic methylomes have recently been reported (32,85,86), the community-level viral methylome needs investigation. In the present study, we conducted a first metaepigenomic analysis of pelagic microbial communities. We then successfully identified a number of DNA modified motifs as well as MTase genes in diverse marine prokaryotes and viruses.
Most of the detected MTase genes in P-MAGs were neither adjusted with a cognate REase gene (Supplementary Data S3) nor associated with known physiological systems such as BREX (61) and DISARM (62). This implies that a large proportion of MTases in pelagic prokaryotes were orphan and inactive for protection against extracellular DNA and viral invasion. However, it should be noted that RM systems were possibly underestimated in our analysis. REase genes are typically diverse and could be overlooked in similarity-based searches. Furthermore, a pair of cognate MTase and REase genes is occasionally placed distantly on the same genome, making it difficult to distinguish them from the partial P-MAGs. Therefore, further validation is required. In addition, due to the similar relative abundance and compositional makeup of the MTase genes in the communities (Figure 1), it is possible that the effects of environmental factors changing with water depth (e.g., water pressure, temperature and viral abundance) are limited. The possible role of the methylation systems is a factor involved in gene regulation.
In addition, because solar UV radiation at the sea surface damages prokaryotic DNA (87,88), some DNA methylations may be associated with UV stress tolerance. In the DNA replication process of E. coli, DNA methylation functions as a marker of the original (parental) DNA strand and dictates mismatch repair on newly synthesized (daughter) unmethylated strands; a process known as methyl-directed mismatch repair (MMR) (4,5). Although the role of MMR in the repair of UV-induced DNA damage is unclear, MMR-deficient mutants have been reported to increase UV-induced mutation frequency and cell death in E. coli (89). In addition, MTase-suppressed Synechocystis mutants decrease tolerance against UV (8). It is anticipated that DNA methylation may play a key role in adaptation to the vast marine epipelagic and mesopelagic layers in prokaryotes, although further experimental and proteomic analyses (e.g., transcriptome and metatranscriptome) are required to confirm the epigenetic regulation of the genes involved.
Viruses, the most abundant biological entities in oceanic environments, play diverse roles in marine ecosystems (90). Among the V-MAGs, methylomes showed family level variance (Figure 4), suggesting the existence of strong selective pressure to maintain the methylation system in marine Myoviridae (and possibly Phycodnaviridae). Hence, DNA methylation among these members may be associated with the genetic roles and ecological strategies of these groups.
It has been hypothesized that the primary advantageous function of viral MTases is as a self-defense weapon against host-encoded defense systems (28,91,92). However, this hypothesis was not concordant with our finding that limited numbers of MTases constituted their known defense systems through the P-MAGs, as discussed above. Thus, the self-defense weapon may play a minor role in DNA methylation in marine viruses. One of the known roles of viral MTase is the initiation of DNA packaging during the late stages of viral infection, found in bacteriophage P1 (93). In this system, m6A modification labels the ends of the concatemeric viral DNA molecules, produced by rolling-circle replication. Further, the end points, where seven methylated motif sites are clustered in bacteriophage P1, are subsequently cut by an enzyme for DNA packaging into capsids. However, the Myoviridae V-MAGs possessed a variety of m4C motifs with different combinations in their genome (Figure 4), and the features are likely inefficient to use methylation as a delimiter of concatenated viral DNA replicons. Another possible role of viral methylation is to increase the stability of DNA for dense packing within a viral capsid, as well as alpha-putrescinylthymine modification in bacteriophage φW-14 (94). In addition, several viral genes are known to be transcriptionally controlled by a self-encoded MTase originally found in bacteriophage P1 (95). The possibility that viral MTase regulates host gene expression to facilitate viral genome replication cannot be ruled out. It would be interesting to explore the role of DNA modifications in the viral life cycle.
In contrast to Myoviridae and Phycodnaviridae, a few other V-MAGs, including members of Siphoviridae and Podoviridae, most of which lacked MTase genes (Figure 4), showed methylation. We anticipated that some viral DNA could be modified by MTases encoded in their host. However, we found no exact matches in the methylated motif pattern between P-MAGs and V-MAGs. We speculated that the potential hosts of the V-MAGs were rare in the communities and were missed in our P-MAG reconstruction. In addition, it is hypothesized that some unknown mechanisms in viruses may avoid modification by the host MTases or inhibit their enzymatic activities.
Evolutionary history and the genomic impact of methylation systems in Alphaproteobacteria
We found unprecedented M.CcrMI homologs that possess GAWTC and GADTC specificities from members belonging to Rhizobiales and SAR11. It is assumed that the methylation systems have significant importance in the cell cycle process as M.CcrMI, and thus have been under strong selective pressure for maintenance in that order. The MTase with GAWTC and GADTC specificity showed noncanonical GANTC specificity under unoptimized conditions as star activity (Supplementary Notes S4 and S5). This enzymatic feature resulted in a scenario in which these protein groups evolved from the ancestral MTase with GANTC specificity by depressing the affinity with GASTC or GACTC sequences. The assay of a mutant protein, in which we changed one residue at the bottom of the pocket, likely accommodated the third position of the motif and distinguished GANTC and GAWTC (Supplementary Figure S11), showed no obvious specificity shift from GAWTC to GANTC (Supplementary Figure S10C–D). This result suggests that either (a) the GASTC affinity is limited by other or additional residues, or (b) the MTase with GAWTC specificity forms a structure distant from M.CcrMI (81).
The O/E ratio analysis showed an underrepresented profile of GAWTC sequence present in the genome compared with GASTC in the Rhizobiales and SAR11 P-MAGs, in contrast to those of GANTC sequence. Furthermore, these were concordant with the detected methylated motifs and specificity of the MTases (Figure 5). This selection pressure suggests a significant (and possibly harmful) effect of methylation on biological processes such as gene expression, which is a critical regulatory change driven by methylation at the internal gene coding sequence and/or promoter region. The low frequency of the GANTC sequence in Alphaproteobacteria genomes has been previously reported (77). However, SAR11 (formerly classified in Rickettsiales) was not recognized as an individual group, and the frequency of GAWTC sequence was not evaluated in the study. Our analysis drastically expands our knowledge about methylation in the class, in that at least a part of SAR11 members possesses the GAWTC methylation system. Furthermore, O/E ratio analysis showed strong and specific negative pressure on the GAWTC sequence in their genomes, which presents a distinct contrast to the other Alphaproteobacteria orders (Supplementary Figures S12 and S13). Variance in GAWTC O/E ratios among SAR11 subclades showed that different methylation states were associated with the evolution of each subclade. In summary, we discovered diversification in MTase specificity that is likely associated with genomic evolution, though the molecular mechanism of the variation and its physiological and ecological benefits remain unclear.
Challenges of metaepigenomics in environmental microbiology
Variance in DNA modifications among lineages has already been explored in bioinformatics applications. For example, an approach of metagenomic binning based on the methylation patterns of assembled contigs has been proposed (96–98). However, our results indicate that sets of methylated motifs are frequently shared within phylogenetically close lineages at even higher taxonomic levels, such as phylum or order (Figure 3). This may render them worthless for distinguishing contigs into individual genome bins.
From another perspective, careful attention should be paid to the fact that metaepigenomic analysis is based on assembled ‘consensus’ genomes that may thus overlook epigenomic heterogeneity at lower taxonomic levels such as strain and species. Recent studies have reported possible variations in sets of methylated motifs and MTase genes at the genus to strain levels in wide prokaryotic lineages (24,99–101). Resolving the strain-level diversity of DNA modifications in complex metagenomic samples thus remains a challenge.
It is also challenging to reconstruct high-quality genomes of rare lineages in complex microbial communities. This is due, in part, to the sequencing read length and throughput of the PacBio platform; the HiFi reads covered only half of the microbial communities in this study (Supplementary Figure S3). Further sequencing efforts are required for higher-resolution analysis to reveal a complete picture of the epigenome in environmental microbial communities. Additionally, even using current SMRT sequencing technology, only a limited number of DNA modification types can be detected and classified with sufficient reliability (i.e., m4C and m6A), although a number of modifications occur in nature (2). Nanopore is a potential alternative approach for detecting DNA modifications, including m5C. Several bioinformatics tools have recently been proposed; however, most of them were either focused on specific contexts of DNA methylation (e.g., CpG methylated region) (102), required a priori knowledge of methylated motifs (103,104), or required additional sequencing of artificially demethylated genomes via whole genome amplification (97). Such tools were thus limited to metaepigenomic analysis targeted in environmental microbial communities, which typically harbor numbers of organisms and methylated motifs yet to be discovered. Further development of sequencing technology, accurate assembly tools, and reliable modification detection methods will be required for deeper evaluation of prokaryotic and viral DNA modifications in the environment.
CONCLUSION
To our knowledge, this is the first metaepigenomic analysis of pelagic microbial communities dominated by members not yet cultured with high complexity. We successfully acquired unprecedented DNA modifications and catalytic enzymes in diverse prokaryotes and viruses. Our findings demonstrate that metaepigenomics is effective for comparative analysis of DNA modifications within and between microbial populations. Moreover, the novel detection of variation of MTase specificity in Alphaproteobacteria and its impact on genomic signature illuminated the co-evolutionary relationship between the methylation system and genome, suggesting that the prokaryotic epigenome plays a greater role in genomic evolution than previously recognized. Despite several technical challenges in the accurate detection of DNA modifications as well as the analysis of strain-level variation in environmental microbes, further investigations are required to evaluate the relationships between molecular function, ecological benefit, and evolutionary impact of the prokaryotic and viral DNA modifications, including DNA methylation by M.CcrMI homologs in Alphaproteobacteria. We also anticipate that the metaepigenomics of prokaryotes and viruses under different ecological niches (e.g., sea area and water depth) and ecosystems (e.g., soil, gut and symbionts) will significantly deepen our understanding of the prokaryotic and viral epigenomes.
DATA AVAILAVILITY
The raw sequencing data and assembled genomes were deposited in the DDBJ Sequence Read Archive and DDBJ/ENA/GenBank, respectively (Supplementary Data S6). All data are registered under BioProject ID PRJDB11069 [https://ddbj.nig.ac.jp/resource/bioproject/PRJDB11069].
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank the captain, crew, and onboard scientists and technicians of the R/V Kaimei (JAMSTEC) during KM19-07 cruise. The SMRT and Nanopore sequencing were supported by NIG. We thank Keiko Tanaka, Eiji Tasumi, Akiko Makabe, Minoru Hamana, Masahito Shigemitsu, Hiroshi Uchida, Yusuke Tsukatani, Hidetaka Nomaki, Takeuchi Akinori, Shuhei Ota, Yuya Tada, Mancha Mabaso, Jarishma Gokul, and Thulani Makhalanyane for seawater sampling. We are grateful to Masami Koizumi for technical assistance with cell and viral-like particle counting and flow cytometry experiments, and Fumie Kondo and Miwako Tsuda for their helpful suggestions and support in the molecular experiments.
Author Contribution: S.H. conceived and designed the study, performed the sampling, molecular experiments, bioinformatics analyses and wrote the manuscript. T.S. performed the sampling, designed and performed the molecular experiments and protein purification, and wrote the manuscript. M.H. performed the sampling and DNA sequencing using Illumina. A.T. performed DNA sequencing using PacBio and Nanopore. S.K. designed the cruise. T.Y. designed and performed sampling and wrote the manuscript. T.N. wrote the manuscript and supervised the project. All authors read and approved the final manuscript.
FUNDING
The Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan [JP16H06429, JP16K21723, JP16H06437 to T.Y.; JP19H05684 within JP19H05679 to T.N.]; Japan Society for the Promotion of Science (JSPS) [JP18K11636, JP19H04246 to T.Y.; JP19K21203, JP20K15444 to S.H.; JP20H02020 to S.K.]; Institute for Fermentation, Osaka (IFO) (to S.H.). Funding for open access charge: Institute for Fermentation, Osaka (IFO).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments