





Abstract
Bacteria can acquire mobile genetic elements (MGEs) to combat infection by viruses (phages). Satellite viruses, including the PLEs (phage-inducible chromosomal island-like elements) in epidemic Vibrio cholerae, are MGEs that restrict phage replication to the benefit of their host bacterium. PLEs parasitize the lytic phage ICP1, unleashing multiple mechanisms to restrict phage replication and promote their own spread. In the arms race against PLE, ICP1 uses nucleases, including CRISPR-Cas, to destroy PLE’s genome during infection. However, through an unknown CRISPR-independent mechanism, specific ICP1 isolates subvert restriction by PLE. Here, we discover ICP1-encoded Adi that counteracts PLE by exploiting the PLE’s large serine recombinase (LSR), which normally mobilizes PLE in response to ICP1 infection. Unlike previously characterized ICP1-encoded anti-PLE mechanisms, Adi is not a nuclease itself but instead appears to modulate the activity of the LSR to promote destructive nuclease activity at the LSR’s specific attachment site, attP. The PLE LSR, its catalytic activity, and attP are additionally sufficient to sensitize a PLE encoding a resistant variant of the recombination module to Adi activity. This work highlights a unique type of adaptation arising from inter-genome conflicts, in which the intended activity of a protein can be weaponized to overcome the antagonizing genome.
INTRODUCTION
Bacteriophages (phages) are ubiquitous and predicted to outnumber their bacterial prey 10-fold (1). The constant competitive interactions between bacteria and their phage predators have driven the evolution of numerous anti-phage defense mechanisms that target different stages of the phage life cycle (2,3). To avoid extinction, phages must overcome bacterial strategies that hamper productive phage infection. The rapid acquisition of offensive and defensive mechanisms results in a co-evolutionary arms race that drives microbial evolution (4). Anti-phage mechanisms encoded on mobile genetic elements (MGEs) allow for rapid dissemination of defenses in microbial populations. Some MGEs can protect their bacterial host from phage predation by functioning as virus-like parasites that hijack phage products to promote their own spread (5–8). These subviral parasites, often referred to as satellites, are integrated into attachment sites in their bacterial hosts’ genomes. Upon induction during infection by a specific phage, a subviral parasite is triggered to excise (5,9,10), replicate (11,12), and hijack their inducing phage's structural components to package their own genome for transduction to naïve neighboring bacterial cells (7,8,13,14). For the bacterial host of this inter-viral conflict, the subviral parasite provides a benefit to the population because the inducing phage is inhibited. In response, the inducing phage may counteract the anti-phage mechanisms inherent to successful hijacking in order to avoid inhibition, resulting in a subcellular arms race.
Vibrio cholerae, the causative agent of the diarrheal disease cholera (15,16), is under constant attack by predatory phages (17–20). To defend against viral attack, strains of epidemic V. cholerae have acquired subviral parasites called the phage-inducible chromosomal island-like elements (PLEs) that restrict infection by their specific inducing phage, the dominant lytic phage ICP1 (13). Ten distinct PLEs have been identified to date, revealing these elements as a dynamic but persistent part of the MGE repertoire in V. cholerae isolated from cholera patients over the last century (20). PLEs share regions of nucleotide conservation but also contain variable modules often shared between two or more PLEs (13,20). Regardless of variability, all PLEs have a conserved mechanism to block ICP1 infection: PLEs excise from the chromosome upon infection (10,13), replicate to a high copy number (12), and package their genomes into modified ICP1 capsids to spread via transduction to neighboring V. cholerae cells (14) (Figure 1A). PLEs employ multiple mechanisms to interfere with different stages of the ICP1 life cycle that function synergistically to prevent successful phage propagation (14,19,21). These include decreasing phage DNA replication (12,21), manipulating virion assembly to inhibit packaging of the ICP1 genome (14), and other mechanisms that remain elusive. PLE’s parasitism of ICP1 completely abolishes phage progeny production and therefore enhances V. cholerae fitness by protecting the bacterial population from further phage attack (12,13,19,21).
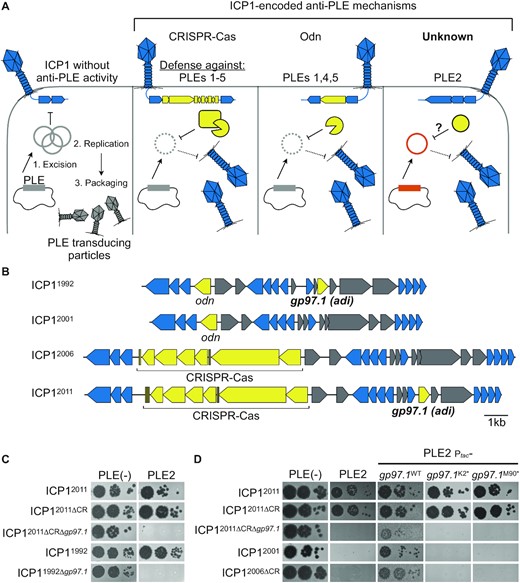
ICP1 isolates encoding adi (gp97.1) overcome PLE2-mediated anti-phage activity. (A) Schematic of the molecular warfare between Phage-inducible chromosomal island-Like Element (PLE) and the predatory bacteriophage, ICP1. ICP1 infection of PLE(+) V. cholerae triggers PLE excision and replication. PLE blocks phage production and packages its genome into transducing particles derived from ICP1 components that are released upon cell lysis. ICP1 overcomes PLE’s anti-phage activity by deploying nucleases: some isolates of ICP1 encode CRISPR-Cas which defends against PLEs 1–5 (13,18). Alternatively, ICP1 can encode the origin-directed nuclease (Odn) that defends against PLEs 1,4 and 5, but not PLE2 or PLE3 (22). A CRISPR(–) derivative of ICP12011 was shown to successfully replicate in the presence of PLE2 specifically (13), suggesting an unknown mechanism for ICP1 to defend against PLE2 (‘Unknown’ right panel). There are ten PLEs known, however, the models above are based on experimental host range information available for PLEs 1–5 (13,22). (B) Genomic organization of the regions of the ICP1 phage genome encoding anti-PLE mechanisms in representative isolates. Genes represented by blue arrows are conserved in all ICP1 isolates sequenced to date (23), while accessory genes in yellow have anti-PLE activity. Genes shown in gray are accessory genes that co-vary with the genes involved in countering PLE. (C) Ten-fold serial dilutions of various ICP1 isolates encoding gp97.1 and CRISPR (CR) and or their mutant derivatives spotted onto permissive PLE(–) or PLE2(+) V. cholerae (bacterial lawns in gray, zones of killing are shown in black). (D) Ten-fold serial dilutions of various ICP1 isolates spotted onto the V. cholerae host indicated (bacterial lawns in gray, zones of killing are shown in black). Ptac-gp97.1 is the plasmid expression construct to express Gp97.1 in trans, which was either wild-type (WT) Gp97.1 or engineered to possess a premature stop codon at amino acid position +1 or +89, resulting in K2* or M90*, respectively. Spot assays were performed in biological triplicate, and a single representative image is shown. For (B–D), the year of isolation for each ICP1 isolate is used as shorthand to differentiate unique isolates (full isolate details provided in Supplementary Table S1).
As an obligate intracellular parasite of V. cholerae, ICP1 risks extinction unless it acquires counter-defense(s) against PLE-mediated inhibition. ICP1 isolates, which have been recovered continuously since 1992, share a core genome and differ by accessory components, including those that allow ICP1 to compete against PLE (22,23). Of these accessory components, all ICP1 isolates encode one of two anti-PLE mechanisms: either a CRISPR-Cas system or a stand-alone nuclease, Odn (18,22–24) (Figure 1A). CRISPR-Cas uses PLE-derived spacers transcribed from the CRISPR array to guide the effector Cas complex to cleave PLE at various genomic locations, while the nuclease Odn specifically targets the PLE origin of replication. Both of these anti-PLE adaptations directly target the PLE genome, suggesting that the integrity of the PLE genome is integral to combating PLE’s restriction of ICP1. When ICP1 phages are armed with either CRISPR-Cas or Odn, PLE replication and transduction are limited, thereby restoring ICP1 phage production (18,22). However, the PLE replication module has sufficient allelic variation that allows some PLEs to escape nuclease attack by Odn (12,18,22). In this way, it appears that one of ICP1’s counter-defenses against PLEs has directly contributed to PLE diversification. Since CRISPR-Cas and Odn occupy the same genomic locus across all known ICP1 isolates, with a phage encoding either one or the other, ICP1 isolates encoding odn would be seemingly incapable of overcoming certain PLEs, like PLE2, which harbors an origin of replication not recognized by Odn (22). Therefore, there may be selection for an additional anti-PLE2 factor that could work synergistically with Odn to inhibit a larger suite of PLEs. Interestingly, an ICP1 isolate with mutations that inactivated its CRISPR-Cas system was still able to replicate and form plaques on V. cholerae harboring PLE2, but not other PLEs (13) (Figure 1A). This suggests that ICP1 phage isolates may have additional mechanisms to antagonize certain PLE variants that await discovery.
A conserved aspect of PLE activity is excision from the V. cholerae chromosome immediately following ICP1 infection (10,13). The mobilization of PLE is integral to its life cycle, allowing for its dissemination to naïve V. cholerae cells, which then gain the capacity to restrict ICP1 (10,13). This mobilization is mediated by integrases, which are site-specific recombinases that fall into two protein families based on their catalytic residue: tyrosine or serine (25). Like other MGEs, PLEs rely on integrases to mediate integration and excision in response to specific cues. PLEs encode large serine recombinases (LSRs), which are simple, requiring only the integrase and specific attachment (att) sites to carry out integration (25,26) To mediate excision, LSRs bind to a cognate recombination directionality factor (RDF) (25,26). In both integration and excision, LSRs possess specific DNA binding activity to recognize attachment sites and catalytic activity to introduce a double-stranded break for precise recombination. Due to the specificity and functionality of LSRs, they have been exploited for applications in genetic engineering (27–34).
PLEs encode one of two types of LSRs, referred to simply as the PLE1-type and PLE2-type (20), which differ in their integration site and cognate RDF required for excision (10). So far, only the PLE1-type LSRs have been characterized experimentally (10), though we infer attachment site recognition for PLE2-type LSRs from the conserved integration site of PLEs encoding this LSR type. For all PLEs, the integrase catalyzes PLE excision out of the chromosome in response to ICP1 infection, resulting in a circularized PLE intermediate. While PLE maintains its anti-phage activity against ICP1 in the absence of excision, PLE is unable to excise and transduce without the integrase (10,13). Therefore, integrases play a vital role in the PLE life cycle by allowing for the spread of PLE to neighboring cells (10). As for many PLE genes where divergent gene products carry out a conserved activity, it is unclear what may have contributed to the divergence of the PLE LSRs. The observation that PLEs encode divergent replication modules that can afford an evolutionary advantage upon encountering certain ICP1-encoded counter-defenses suggests that other variations observed between PLEs may be attributable to the ongoing molecular arms race with ICP1.
Motivated by the observation that PLE2 did not restrict a CRISPR-deficient ICP1 isolate, we set out to identify the CRISPR-independent mechanism by which ICP1 can antagonize PLE2 and understand why it does not afford protection against other PLEs tested (13). We found that ICP1 isolates can encode an effector protein whose activity results in degradation of the PLE2 genome. Further, we observed that the anti-PLE activity of this accessory protein is dependent on the PLE2 integrase and the specific target sequence generated by the LSR upon PLE2 mobilization. Unlike known anti-PLE mechanisms used by ICP1, this effector does not appear to be a nuclease itself but rather exploits the satellite-encoded recombinase to execute successful counter-defense. This work broadens our understanding of the innovations resulting from ongoing molecular arms races by revealing that weaponizing the enzymatic activity of other proteins can provide a means of adaptation.
MATERIALS AND METHODS
Strains and culture conditions
A complete list of strains used throughout this study can be found in Supplementary Table S1. V. cholerae strains used in this study are derivatives of E7946. Bacteria were grown on LB agar plates with appropriate antibiotic(s) at the following concentrations where appropriate: 75 μg/ml kanamycin, 100 μg/ml spectinomycin, 2.5 μg/ml chloramphenicol (for V. cholerae on plates and in 0.5% LB soft agar), 1.25μg/ml chloramphenicol (for V. cholerae in liquid), 25 μg/ml chloramphenicol (for Escherichia coli).
Phage spot plates
From LB agar plates, 2 ml cultures of each V. cholerae strain were grown to OD600 >1, then back-diluted into fresh media to OD600 = 0.05, and grown to OD600 = 0.2. For V. cholerae strains with adi under an inducible promoter on a plasmid, cultures were induced with 0.15 mM theophylline for 20 min at 37°C with aeration. Then 150 μl of mid-log V. cholerae was added to 4 ml of molten 0.5% LB soft agar (supplemented with 2.5 μg/ml chloramphenicol and 0.15 mM theophylline), poured onto 100 mm × 15 mm LB agar plates and allowed to solidify. For V. cholerae strains with int or intS12A under an inducible promoter on a plasmid, cultures were induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 20 min. Then 150 μl of mid-log V. cholerae was added to 4 ml of molten 0.5% LB soft agar (supplemented with 2.5 μg/ml chloramphenicol and 1 mM IPTG), poured onto 100 mm × 15 mm LB agar plates and allowed to solidify. Once the LB agar plates solidified, 2 μl of 10-fold serially diluted phages were spotted onto the plates and incubated for 4 h at 37°C. Following incubation, the plates were taken out of the incubator and imaged the next day. Assays were done in triplicate and a single representative image is shown. Replicates have been deposited on Mendeley Data.
Efficiency of plaquing infection assays
From LB agar plates, 2 ml cultures of each V. cholerae strain were grown to OD600 > 1, then back-diluted into fresh media to OD600 = 0.05, and grown to OD600 = 0.2. For V. cholerae strains with the empty vector control, or int or intS12A under an inducible promoter on a plasmid, cultures were induced with 1 mM IPTG for 20 min at 37°C with aeration. Then 10 μl of 10-fold serially diluted phages were added to 50 μl of mid-log V. cholerae and incubated for 5 min to allow for phage attachment to cells. Infected cells were added to 4 ml of molten 0.5% LB soft agar (supplemented with 2.5 μg/ml chloramphenicol and 1 mM IPTG), poured onto 60 mm × 15 mM Petri dishes and allowed to solidify. Once the LB agar plates solidified, the plates were incubated overnight at 37°C and plaque forming units were counted and compared to the permissive strain to calculate efficiency of plaquing.
Generation of mutant strains and constructs
For V. cholerae mutants containing chromosomal expression constructs, PCR products were created using splicing by overlap extension (SOE) PCR and introduced by natural transformation as described previously (35). Ectopic expression plasmids are derivatives of pMMB67EH containing a theophylline inducible riboswitch E as described in (10). Gibson assembly or around-the-horn cloning was used to assemble plasmid-based expression constructs which were then introduced into V. cholerae through conjugation with E. coli S17. Mutant derivatives of PLEs were constructed by inserting a selectable marker containing arms of homology to allow for recombination using the same process detailed above. ICP1 mutants were created through CRISPR-Cas gene editing as described previously (36). Briefly, an IPTG-inducible type 1E CRISPR-Cas system was engineered in V. cholerae and used to target various regions of the ICP1 genome. To generate clean deletions, a targeting plasmid was engineered with a recombination template to delete only the gene product targeted by the spacer. Escape phage that are able to form plaques on the targeting host have acquired random mutations or clean deletions due to recombination events. All constructs were confirmed via Sanger sequencing. The cloned attP sequences are provided in Supplementary Tables S3 and S4.
Circularization assay
From liquid cultures, V. cholerae PLE2(+) or PLE2 mutants were infected with ICP12006ΔCR phage at a multiplicity of infection (MOI) of 1 and incubated with aeration at 37°C for the time indicated before 100 μl of the sample was removed and boiled for 10 min. Alternatively, agar stabs of plaques (successful ICP1 infection on V. cholerae) were placed in 100 μl water and boiled for 10 min. 2μl of each boiled sample (from liquid or a plaque) was used as the template for PCR using primers indicated in Supplementary Table S2. The resulting reactions were run on 2% agarose gel with GelGreen (Biotium) before imaging using an EZ Dock Imager (Bio-Rad). For ectopic expression of Int, V. cholerae containing the plasmid expressing Int was induced with 1 mM IPTG and 1.5 mM theophylline at 37°C with aeration for 20 min at OD600 = 0.2 prior to phage infection. For ectopic expression of adi in miniPLE2, V. cholerae containing the miniPLE2 construct and plasmid encoding adi was induced for 20 min beginning at an OD600 = 0.2 prior to being boiled and used as templates for PCR, as described above.
Toxicity assays
From LB agar plates, 2ml cultures of each V. cholerae strain were grown to OD600 > 1, then back-diluted into fresh media to OD600 = 0.05, grown to OD600 = 0.2 (if slightly greater, normalized to OD600 = 0.2) and induced with 1 mM IPTG for 20 min at 37°C with aeration. Induced cultures were plated on LB agar plates supplemented with 1 mM IPTG and appropriate antibiotics.
Transduction assays
A V. cholerae PLE2(+) donor with a kanamycin resistance cassette downstream of orf27 (as in (13)) was grown to OD600 = 0.3 and infected with ICP1 at an MOI of 2.5. Cultures were incubated with aeration at 37°C for 5 min, centrifuged at 5000 × g for 2 min, and washed twice with fresh LB to remove unbound phage. Infected cell pellets were then resuspended in 2 ml LB supplemented with 10 mM MgSO4 and incubated for 30 min at 37°C with aeration. Following incubation, during which time lysis occurred, lysates were treated with chloroform and centrifuged at 4000 × g for 10 min to remove debris. 10 μl of the transducing lysate was added to 100 μl overnight culture of a PLE(−) spectinomycin-resistant (ΔlacZ::spec) recipient strain and incubated for 20 min at 37°C with aeration before plating on LB plates supplemented with kanamycin and spectinomycin to select for transductants. Assays were performed in biological triplicate.
Deep sequencing of phage-infected cultures and reads mapping
From LB agar plates, 2 ml cultures of each V. cholerae strain were grown to OD600 > 1, then back-diluted into fresh media to OD600 = 0.05, grown to OD600 = 0.3, and infected with ICP1 at an MOI of 1 and then incubated at 37°C with aeration. 1 ml aliquots at t = 8 and 16 min were taken and immediately pipetted into 1 ml ice-cold methanol. Samples were centrifuged at 5000 × g for 3 min and total DNA was extracted using the QIAGEN DNeasy Blood and Tissue Kit. Illumina sequencing was performed by the Microbial Genome Sequencing Center (Pittsburgh, PA) at a 200 Mb depth. Then, Illumina sequencing reads for t = 8 and t = 16 min were mapped to the relevant reference files to calculate read coverage and replicates were averaged. The 1 bp position for PLE2 in our mapping reference is equivalent to nucleotide position 8064 in the deposited PLE2 sequence (accession KC152961). Read coverage across all genomic elements (V. cholerae chromosomes, ICP1 and PLE2) was reported as percent of total reads normalized by element length. A ratio of averaged coverages for the PLE2 genome was then determined at each nucleotide position between experimental infections with ICP1ΔCRΔadi divided by ICP1ΔCR. This ratio calculation was also performed for the plasmid reference with intS12A divided by WT int. These calculations were performed for each timepoint variable and presence/absence of attP on the plasmid vector and then log2 transformed. The resulting coverage ratios across each reference for every experiment were then scanned using the find_peaks function in SciPy (37) with the following settings: height = none, threshold = none, distance = 250, prominence = 0.2, width = 500, wlen = none, rel_height = 0.5, plateau_size = none. The five most prominent inverse peaks (i.e. ‘valleys’), which equate to coverage dropouts, were plotted centered on the peak with 250 bp on either side.
Quantification of PLE genome replication by real-time quantitative PCR
From LB agar plates, liquid cultures were grown to OD600 > 1, back-diluted into fresh media to OD600 = 0.05, grown to OD600 = 0.3, infected with ICP1 at an MOI of 2.5 and incubated at 37°C with aeration. 100 μl samples were taken and immediately boiled for 10 min at t = 0 (uninfected), then at 4-min increments post infection until the final time point at t = 20 min. Boiled samples were diluted 1:1000 and subsequently used as templates for IQ SYBR (Bio-Rad) qPCR reactions. All conditions were tested in biological triplicate, and each reported data point is the mean of two technical replicates. The fold change was calculated by comparing the Cq value to the standard curve of known concentrations of PLE2 genomic DNA. A single primer set (in Supplementary Table S2) that amplifies a region 4.8 kb away from attP is used to detect PLE2 replication by qPCR.
Green fluorescence protein (GFP) reporter assays
The attP sequence (as in Supplementary Tables S3 and S4) was engineered between a constitutive promoter and GFP in a neutral locus (lacZ) in the V. cholerae chromosome. Plasmids containing Int, IntS12A or empty vector control were mated into the GFP-expressing V. cholerae strain. From LB agar plates, liquid cultures were grown to OD600 > 1, back-diluted into fresh media to OD600 = 0.05, grown to OD600 = 0.3. Cultures were induced with 1 mM IPTG and 1.5 mM theophylline for 20 min to induce expression of int or intS12A. Cells were spun down at 5000 × g for 3 min. Supernatant was removed and the pellet was washed with 2 ml 1× PBS twice. The pellet was resuspended in 2 ml 1× PBS and OD600 was read and standardized to OD600 = 0.3 across all cultures. 200 μl of cultures were transferred to a black 96-well plate and read with an excitation of 485 nm and emission of 535 nm. Relative fluorescence of V. cholerae with int or intS12A was calculated compared to the engineered GFP(+) V. cholerae strain containing an empty vector control plasmid. All conditions were tested in biological triplicate, and each reported data point is the mean of two technical replicates.
Protein purification
E. coli BL21(DE3) cells containing a plasmid-based 6xHis-SUMO fusion to int or adi, or 6xHis-SENP2 were grown to OD600 = 0.5 at 37°C and induced with 0.5 mM IPTG and grown for 16 h at 16°C. Cells were centrifuged at 4000 × g for 15 min and resuspended in 25 ml lysis buffer (50 mM Tris–HCl pH 7.8 150 mM NaCl, 1 mM BME, 0.5% Triton-X, 50 mM imidazole, Pierce™ protease inhibitor) and sonicated. Cells were centrifuged to remove cell debris. The resulting supernatant was filtered through a 0.2 μm cellulose filter (GE Life Sciences). Lysate was applied to an equilibrated HisTrap (Sigma) Ni-sepharose column and washed with 10 mL wash Buffer A (50 mM Tris–HCl, 150 mM NaCl, 50 mM imidazole, 1% glycerol, 2 mM BME, pH 7.8), followed by 10 ml wash Buffer B (50 mM Tris–HCl, 2.5 M NaCl, 50 mM imidazole, 1% glycerol, 2 mM BME, pH 7.8), then finally 10 ml wash Buffer A. The protein was eluted in 1 ml aliquots of imidazole elution buffer (50 mM Tris–HCl, 150 mM NaCl, 250 mM imidazole, 1% glycerol, 2 mM BME, pH 7.8). To remove the tag, the eluted protein was dialyzed against wash Buffer A + 5% glycerol overnight in 50K MWCO (Int and IntS12A) and 10K MWCO (Adi) Slide-A-Lyzer™ Dialysis Cassettes (Thermo Scientific™) with 20 U/mg of 6xHis-SUMO protease. To remove the SUMO protease and the cleaved tag for Int or Adi, the protein was added to Ni-NTA resin, resulting in the SUMO protease and the cleaved tag binding to the resin and leaving the protein of interest in the unbound buffer. Protein concentration was measured using a BioPhotometer D30 (Eppendorf). For storage, glycerol was added to 20% final concentration and samples were flash frozen for storage at –80°C.
Electrophoretic mobility shift assays (EMSAs)
Primers were annealed in water using a thermocycler from 95°C to 25°C with an incubation of 1 min per decrease in 1°C. Primers used as probes are in Supplementary Table S2. 80nM probes were incubated with 500 nM purified Int or IntS12A at 30°C for 20 min in 20 μl reactions (150 mM Tris–HCl, 10 mM MgSO4, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.8). The full reaction volume was then loaded onto an 8% acrylamide 0.5× Tris–borate (TB) gel and ran for 60 min at 100 V and stained with 0.5× TB buffer with GelGreen (Biotium) before imaging using an EZ Dock Imager (Bio-Rad). Assays were done in triplicate and a single representative image is shown. Replicates have been deposited on Mendeley Data.
In vitro recombination and nuclease assays
Probes were amplified from purified genomic DNA of PLE2(+) V. cholerae (attL and attR), PLE2(+) V. cholerae infected with ICP1 phage (attP), and PLE(–) V. cholerae cells (attC). Primers used to amplify probes are available in Supplementary Table S2. PCR products were PCR purified using Monarch® PCR DNA Cleanup Kit (New England BioLabs) and eluted with water. The concentration of probes was measured using a BioPhotometer D30 (Eppendorf). 100 ng probes were incubated with purified Int and/or Adi in 20 μl reactions (150 mM Tris–HCl, 10 mM MgSO4, 50 mM NaCl, 1 mM DTT, pH 7.8). Reactions were incubated at 30°C for 30 min, then 1μl of proteinase K was added to the reaction and incubated at 37°C for 30 min, followed by 10 min at 75°C to heat inactivate. 10 μl of reaction was loaded onto 2% agarose gel with GelGreen (Biotium) and ran for 30 min at 100 V and visualized using an EZ Dock Imager (Bio-Rad). Assays were done in triplicate and a single representative image is shown. Replicates have been deposited on Mendeley Data.
Microscopy
V. cholerae was grown in LB at 37°C to OD600 = 0.2, then split to half volumes. Half of each culture was induced with 1mM IPTG, 1.5 mM theophylline and 0.1% arabinose for 20 min at 37°C with aeration. Cultures were pelleted and resuspended in half volume 4% formaldehyde in 0.5× Instant Ocean® supplemented with 20 uM Hoechst dye (Thermo Scientific). Samples were incubated in the dark for 10 min at room temperature, then pelleted and resuspended in 1/20 volume 0.5× Instant Ocean. 10 μl sample volume was mixed with Prolog Gold Antifade mountant (Invitrogen), mounted on a glass slide with a coverslip, and imaged on a Zeiss Axioimager fluorescent microscope with Hamamatsu camera. Experiments were performed in biological triplicate and slides were mounted in technical duplicate and blinded prior to imaging. Exposure times were approximately 30 ms for DIC images and 15 ms for Hoechst-stained DNA fluorescence images. Images were prepared in ImageJ. Assays were done in triplicate and a single representative image is shown. Replicates have been deposited on Mendeley data.
RESULTS
ICP1 encodes adi to overcome PLE2 anti-phage activity
To identify ICP1-encoded gene(s) that allow phage to antagonize PLE2, we took advantage of our collection of ICP1 mutants and began screening for ICP1 phage mutants in the ICP12011 ΔCRISPR background that no longer formed plaques on PLE2(+) V. cholerae. Through this screen, we identified gp97.1 (gene product AXY82195.1) as necessary for ICP12011 to form plaques on PLE2(+) V. cholerae (Figures 1B and C). Gp97.1 is a small protein (147 amino acids) of unknown function unique to ICP1 isolates and has no sequence similarity to proteins of known function. Intriguingly, gp97.1 is encoded by phages encoding CRISPR-Cas or odn (Figure 1B), suggesting it may provide an additional fitness advantage to phages independent of either of the known anti-PLE mechanisms. Accordingly, gp97.1 was also required for an Odn(+) phage isolate, ICP11992, to form plaques on PLE2(+) V. cholerae (Figure 1C). Notably, our initial attempts to complement Δgp97.1 phage mutants were confounded by the observation that expression of gp97.1 was toxic to PLE2(+) V. cholerae (elaborated on further below) (Figure S1). With decreased inducer concentrations (see methods) that allowed the bacterial lawn to form, we found that expression of gp97.1 in trans restored plaque formation for engineered Δgp97.1 phage mutants on PLE2(+) V. cholerae (Figure 1D). Although gp97.1 is encoded alongside other unique accessory genes (Figure 1B), expression of gp97.1 in trans was also sufficient to allow ICP12001 and the ΔCRISPR derivative of ICP12006 form plaques on PLE2(+) V. cholerae (Figure 1D). ICP12001 and ICP12006 are isolates that do not encode gp97.1 or associated flanking genes (Figure 1B), demonstrating that gp97.1 functions independently of co-varying accessory genes. To evaluate whether the Gp97.1 protein or its transcript had the relevant activity to allow ICP1 to form plaques on PLE2 (+) V. cholerae, we introduced premature stop codons at amino acid positions +1 (K2*) or +89 (M90*) while maintaining the rest of the gp97.1 coding region. Consistent with the relevant activity being attributable to the Gp97.1 protein, we found that ICP1 was unable to plaque on PLE2(+) V. cholerae expressing gp97.1(K2*) and gp97.1(M90*) (Figure 1D). Since this gene product is both necessary and sufficient for ICP1 to overcome PLE2, and as informed by subsequent experiments, we named Gp97.1 attachment site directed inhibitor or Adi.
Genome replication dynamics following ICP1 infection of PLE2(+) V. cholerae are indicative of Adi-mediated nuclease activity targeting attP
Both known mechanisms that ICP1 uses to counter PLE activity (CRISPR-Cas and Odn) harness nucleases to restrict the robust levels of PLE replication seen during ICP1 infection (12,22,24). Therefore, we evaluated the replication of PLE2 during infection by ICP1 ± adi. Moving forward, we performed experiments with ICP12011, which we will refer to as ICP1 for simplicity (unless otherwise noted). We opted to assess replication using deep sequencing of total DNA during an infection time course to provide insight into replication dynamics (12) and reveal potential regions of nuclease cleavage by looking for areas with sudden drops in reads coverage (21). To evaluate total DNA content in infected cells at different stages of infection, we collected samples at relatively early (8 min) and late (16 min) time points post-infection, consistent with the established kinetics of ICP1 replication and PLE’s response to infection (12,38). Total DNA from samples at each time point was sequenced, and the resulting sequencing reads were mapped to the V. cholerae, ICP1 and PLE genomes. Consistent with previous observations measuring replication of a different ICP1 isolate in a permissive PLE(–) V. cholerae host (12), ICP1 encoding adi infecting a PLE2(+) host led to the robust accumulation of phage DNA late in infection (Figure 2A). As expected for this infection setup wherein PLE2 is antagonized by Adi, robust phage DNA replication coincided with minimal PLE replication. In contrast, during infection by ICP1 Δadi, PLE replication increased ∼7-fold, although replication of PLE2 in these experiments was not as robust as observed previously for PLE1 following infection by a different ICP1 isolate (12). Nevertheless, the increase in PLE replication following infection with ICP1 Δadi coincided with decreased phage replication (Figure 2A), consistent with the established link between PLE replication and inhibition of ICP1 DNA replication (12,21). The decrease in reads mapping to the PLE genome suggested that PLE transduction would also decrease in the presence of Adi. To test this, we evaluated PLE2 transduction following infection by ICP1 ± adi. As expected, PLE transduction efficiency decreased ∼10-fold following infection by adi(+) ICP1 compared to the Δadi derivative (Figure 2B). Taken together, the observed replication dynamics and transduction assays are consistent with Adi-mediated interference of PLE replication through nuclease activity directed against the PLE genome itself.
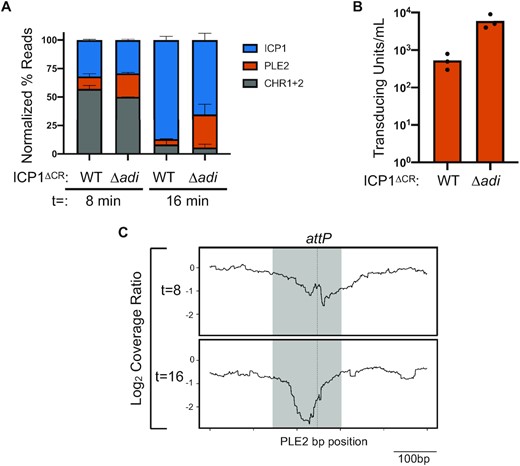
Genome replication dynamics following ICP1 infection of PLE2(+) V. cholerae are indicative of Adi-mediated nuclease activity targeting the PLE2 mobilization product, attP. (A) Percent DNA read abundances normalized to the element's length for PLE2, both V. cholerae chromosomes (CHR1 + 2), and ICP1 at the time point indicated (in minutes) following infection by ICP1 ΔCRISPR (ΔCR) harboring wild-type adi (WT) or the Δadi derivative. Bar height is the mean of two biological replicates. (B) Transduction of PLE2 following infection with ICP1 or the Δadi derivative. Bar height represents the mean of three biological replicates and each dot is a measurement from an independent assay. (C) The log2-transformed coverage ratio across the PLE2 genome (relative position shown on the X axis) following infection by Δadi or WT ICP1 over the course of infection. The largest coverage ratio dropout is focused on the 500bp region surrounding attP and represents the average of two replicates. The region comprising the circularization junction, attP, is highlighted in grey with the dashed vertical line indicating the crossover junction between attL x attR to form attP.
Having observed diminished PLE replication during infection with adi(+) ICP1, we next determined if there were localized drops in reads coverage in the PLE genome during infection that appeared in an Adi-dependent manner. To generate cleavage maps across the PLE genome, we calculated the coverage ratio of PLE reads obtained following infection with ICP1 Δadi relative to wild-type ICP1 at both 8- and 16-min post-infection and analyzed coverage drops, representing a depletion of reads (Figure 2C, Figure S2A, and Figure S2B). Intriguingly, we identified that the reads surrounding the PLE2 circularization junction (attP – attachment site PLE) at both 8- and 16- min post-infection showed the largest drop in coverage when ICP1 encoded adi (Figure 2C), suggesting nuclease activity targeting this region. Collectively, these data suggest that upon ICP1 infection, PLE2 excises and circularizes to form the attP junction, which is then targeted by phage-encoded Adi for degradation, thereby compromising the integrity of the PLE2 genome and favoring productive ICP1 infection.
Phage-encoded Adi requires the attP target sequence and catalytically active PLE-encoded integrase for anti-PLE2 activity
Following phage infection, the formation of the PLE circularization junction, attP, is dependent on the PLE-encoded large serine recombinase (LSR) and recombination directionality factor (RDF) (10). Based on our results, we predicted that Adi mediates PLE interference through nucleolytic cleavage of attP; as such, we predicted that PLE2’s LSR would be required for Adi-mediated interference. The PLE1-type LSR (Int) (WP_002040296) was previously shown to be required for PLE1 excision but was not necessary for PLE replication or for PLE to block ICP1 plaque formation (10,12). To experimentally confirm the requirement of PLE2’s predicted LSR (WP_053027292.1) (10,13) for PLE excision in response to ICP1 infection, we constructed a PLE2 Δint derivative and challenged it with ICP1. Consistent with the predicted role of the PLE2 LSR, circularization was not detected in the Δint derivative and was restored upon expression of Int in trans (Figure 3A). These results were further confirmed by in vitro assays using purified PLE2 Int, in which we observed the recombination of attP and attC sites to form recombinant attL and attR sites (Figure 3B and C), as is characteristic of LSRs (26).
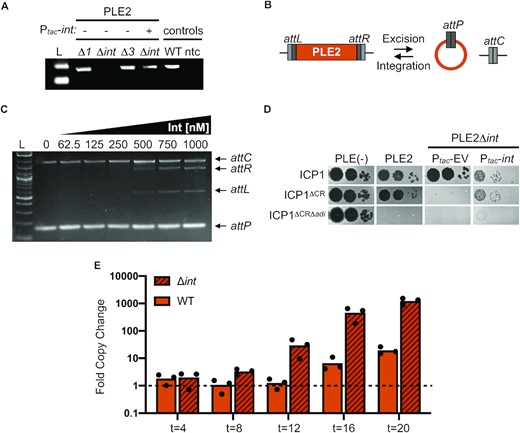
The PLE2 integrase is necessary for adi(+) ICP1 to antagonize PLE2. (A) Agarose gel showing the circularization PCR of PLE2 derivatives lacking ORFs and complemented with a plasmid expressing int as indicated, following phage infection (ORFs 1 and 3 flank the integrase and were also tested). Controls on the right show circularization of wild-type PLE2 following ICP1 infection (WT) and a no template control (ntc) for the PCR. (B) Schematic of PLE2 integrated into the chromosome with attL and attR denoting the attachment sites flanking the PLE. Excision of PLE2 upon ICP1 infection results in formation of attP and attC for the attachment sites on the PLE and the chromosome, respectively. For excision, Int, att sites, and recombination directionality factor (RDF) are required. For integration, only the Int and att sites are required. (C) Agarose gel showing products from in vitro DNA recombination assays of attC and attP (integration) at increasing Int concentrations (as indicated above the gel). The input templates and recombination products (attL and attR) are labeled on the right. (D) Ten-fold serial dilutions of adi(+) ICP1 and the ΔCRISPR (CR), ΔCRΔadi derivatives spotted on PLE(–), PLE2(+) and PLE2Δint V. cholerae containing an empty vector (EV) or plasmid expressing int under an inducible promoter. Complemented PLE2Δint V. cholerae were grown and plated with inducer (bacterial lawns in gray, zones of killing are shown in black). (E) Replication efficiency of wild-type (WT) PLE2 and the Δint derivative in V. cholerae host strains calculated as the fold change in PLE2 DNA copy at the time indicated (in minutes) following infection with adi(+) ICP1ΔCR as assessed by qPCR. For (A) and (C), L indicates the ladder.
In the absence of Int, PLE2 does not excise to form the circularization junction attP (Figure 3A). Following these observations, we predicted that Adi would require Int to mediate excision of PLE2 to form the target sequence, attP, allowing for ICP1 plaque formation. In support of this, we observed that ICP1 encoding adi could no longer form plaques on the PLE2 Δint derivative (Figure 3D). ICP1’s plaquing phenotype was restored by expressing Int in trans, revealing that PLE2 excision sensitizes it to Adi-mediated anti-PLE activity (Figure 3D). Replication of PLE1 following phage infection was previously shown to reach wild-type levels in the absence of its Int (12) or RDF (10). Therefore, we next evaluated if Int negatively impacted PLE2 replication following infection by ICP1 expressing Adi. Consistent with the role of Int in sensitizing PLE to Adi-mediated interference, we observed that upon adi(+) ICP1 infection, replication of wild-type PLE2 was impaired ∼100-fold relative to PLE2 Δint (Figure 3E). This observation demonstrates that dependence on the PLE2 LSR is a critical facet of Adi-mediated anti-PLE activity.
Having observed a drop in reads coverage consistent with cleavage of attP in the presence of Adi (Figure 2C), we next sought to test the hypothesis that attP alone was sufficient for Adi-mediated targeting of PLE. To do this, we engineered the target sequence attP directly into PLE2 Δint (Figure 4A), reasoning that the integrase would now be dispensable for ICP1 plaque formation. Surprisingly, we observed that adi(+) ICP1 was unable to plaque on the attP(+) strain, indicating that attP was not sufficient to direct Adi activity (Figure 4B). Furthermore, Adi does not possess domains predicted to confer DNA binding or nuclease activity, consistent with in vitro assays in which we detected neither activity for purified Adi (Figure S3A and Figure S3B). Therefore, we next considered the possibility that the requirement of PLE2 Int for Adi-mediated anti-PLE activity (Figure 3D) extended beyond its role in generating the attP junction. Notably, ICP1 plaque formation was restored on the attP(+) PLE2 Δint host with the complementation of Int in trans (Figure 4B), suggesting that the integrase protein itself is required, and ICP1’s inability to plaque on PLE2 Δint is not solely due to the lack of the attP target sequence.
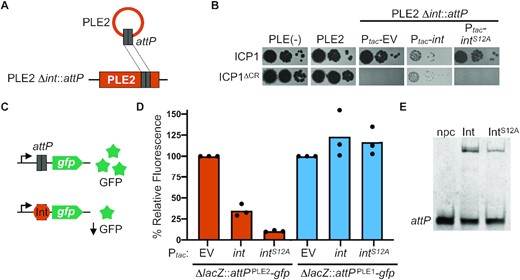
PLE2 attP and catalytic activity of the PLE integrase are required for adi(+) ICP1 to antagonize PLE2. (A) Schematic of PLE2 Δint::attP construct (bottom) with attP cloned into PLE2 Δint. The cartoon of circularized PLE2 is shown as a reference to indicate the formation of attP upon ICP1 infection, which was then cloned into PLE2 Δint. (B) Ten-fold serial dilutions of adi(+) ICP1 and the ΔCRISPR (ΔCR) derivative spotted on PLE(–), PLE2(+) or PLE2Δint::attP containing an empty vector (EV) or plasmid expressing int or intS12A under an inducible promoter grown and plated with inducer (bacterial lawns in gray, zones of killing are shown in black). (C) Schematic of the in vivo green fluorescence protein (GFP) reporter assay. Steric hindrance by proteins that bind DNA in the region between the constitutive promoter and gfp result in lower levels of GFP expression. The DNA used to test protein binding is the excision product attP of either PLE2 or PLE1, denoted as attPPLE2 and attPPLE1 in panel (D), respectively. (D) V. cholerae cells with either attPPLE2 or attPPLE1 containing an empty vector (EV) or plasmid expressing int or intS12A under an inducible promoter were grown with inducer. Relative fluorescence of int and intS12A to the empty vector control is shown. Bar height is the mean of three biological replicates and each dot is a measurement from an independent assay. The empty vector control (set at 100%) is shown for reference. (E) Electrophoretic mobility shift assay (EMSA) using a probe harboring attP incubated without protein (npc), with purified Int or IntS12A.
Previous work has shown that the catalytic activity of LSRs is decoupled from DNA binding activity (26,39–41). Therefore, we considered the possibility that Adi may exploit the DNA binding activity of Int to localize to attP and target it for degradation. To test this, we engineered a mutation in the predicted catalytic serine residue of PLE2 Int (S12A). We verified that IntS12A was catalytically inactive using an in vitro recombination assay where, as expected, purified IntS12A did not recombine attP and attC (Figure S3B). To verify that IntS12A retained its DNA binding activity, we performed in vivo and in vitro assays. To assess DNA-binding in vivo, we cloned attP in between a constitutive promoter and green fluorescence protein (GFP) in the V. cholerae chromosome, reasoning that expression of a protein that binds attP would result in steric hindrance and decreased fluorescence (Figure 4C). We found that expression of either Int or IntS12A resulted in decreased fluorescence compared to empty vector control in the strain containing PLE2’s attP, consistent with DNA binding activity for both proteins. In contrast, no decrease in fluorescence was observed following expression of either protein in the strain containing PLE1’s attP (Figure 4D), consistent with the anticipated specificity of an LSR for its cognate att site. To confirm our in vivo assay, we assessed if purified Int and IntS12A could bind attP directly using an electrophoretic mobility assay (EMSA) and found that both purified proteins demonstrated binding to attP in vitro (Figure 4E, Figure S3C, and Figure S3D). With the catalytically inactive IntS12A we were then able to test whether Adi relies solely on Int's DNA binding activity, reasoning that complementation of PLE2 Δint::attP with the IntS12A mutant would restore plaque formation if Adi only required Int to localize to attP. However, we observed that adi(+) ICP1 was still unable to form plaques even with the target sequence attP and the DNA binding activity of IntS12A (Figure 4B). Together, these results indicate that ICP1-encoded Adi requires the catalytic activity of the PLE2 LSR and attP to exert anti-PLE activity.
Recombination directionality factors (RDFs) are the only group of proteins described to bind directly to LSRs, resulting in a conformational change in the LSR to favor the excision reaction and the formation of the circularized junction, attP (26,42,43). The RDF for the PLE1-type LSR has been identified as ICP1-encoded PexA (10); however, the RDF for PLE2-type LSRs has not been identified. Our results up to this point indicate that Adi may interact directly with PLE2 Int to induce a conformational change and modulate its activity, similar to the role of an RDF. To address if Adi functions as an RDF for PLE2, we first constructed a minimal platform to assess PLE2 excision, referred to as ‘miniPLE2’, as was previously published for PLE1 (10). MiniPLE2 encodes Int under the control of its endogenous promoter but no other PLE2 genes and is integrated into the same attachment site as full-length PLE2. Upon ICP1 infection, miniPLE2 excised and circularized in the absence of other PLE products, demonstrating that no other PLE2 genes are required for PLE2 excision (Figure S4A). To evaluate if Adi can function as the RDF for PLE2 Int, we expressed adi in V. cholerae carrying miniPLE2 and found Adi was not sufficient for excision and circularization (Figure S4A). In addition, we observed PLE2 circularization following infection with ICP1 Δadi (Figure S4B), supporting our conclusion that Adi is indeed not the RDF. Having identified at least some of the necessary components for Int/Adi mediated anti-PLE activity, we were motivated to recapitulate the predicted att-directed nuclease activity of purified Int and Adi in vitro. However, we observed that while Int maintained its recombinase activity in vitro in the presence of Adi, there was no obvious aberrant nuclease activity when the purified proteins were combined (Figure S5). Adi also did not inhibit the integration reaction as an RDF would (41,44,45), further confirming it is not the RDF. The inability to reconstitute destructive nuclease activity in vitro could be due to a missing factor (e.g. other proteins or co-factors in the nuclease buffer). Although we predict that Adi may share features with the PLE2 RDF, namely its ability to interact with and perhaps modulate Int-directed DNA manipulation, these data demonstrate that Adi is not functionally equivalent to the RDF but instead represents a novel factor also capable of altering LSR activity.
PLE2 Int and attP are sufficient to sensitize PLE1 to Adi-mediated anti-PLE activity
Previously, CRISPR-deficient adi(+) ICP1 was shown to overcome PLE2 but could not overcome PLE1 encoding the PLE1-type LSR (13). This observed host range aligns with the differences between the mobility module (comprised of Int and cognate attachment sites) of PLE1 and PLE2. Specifically, the integrases and excision products of PLE1 and PLE2 are divergent (25.7% amino acid identity and 50% nucleotide identical, respectively) (Figure S6A). Therefore, we hypothesized that expression of PLE2 Int in trans would sensitize PLE1 to adi(+) ICP1, but only if the putative target site, attPPLE2 (Figure S6B), was cloned into PLE1. To test this, we cloned attP directly into PLE1 (Figure 5A) and assessed the sensitivity of this strain to phage infection in the presence and absence of ectopic expression of Int. The attP(+) PLE1 strain retained the ability to restrict adi(+) ICP1, confirming that attP is not sufficient for Adi-mediated anti-PLE activity (Figure 5A). Likewise, PLE1 lacking attP retained the capacity to block plaque formation by this phage with PLE2 Int expressed in trans (Figure 5A). In contrast, the PLE1 strain bearing the attP sequence was no longer inhibitory to ICP1 with PLE2 Int expressed in trans (Figure 5A), confirming that both attP and the PLE2 integrase are required to mediate sensitivity to adi(+) ICP1. Further, attP was not sufficient to sensitize PLE1 to ICP1 antagonism even in the presence of Int if attP was cloned outside of PLE in the V. cholerae chromosome (Figure 5B). The requirement of attP to be encoded in cis (within PLE) is consistent with attP serving as the target sequence for Adi-mediated Int-dependent anti-PLE activity.
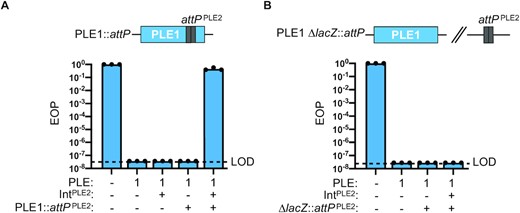
PLE2 Int and attP are required to sensitize PLE1 to ICP1 phage infection. (A) Schematic showing relevant construct design of PLE1::attP wherein PLE2 attP was engineered into PLE1. Efficiency of plaquing (EOP) of adi(+) ICP1 ΔCRISPR on V. cholerae with the PLE indicated. The presence of an induced plasmid expressing Int from PLE2 is indicated by +/–Int, and the presence of PLE2 attP in PLE1 (as in schematic) is indicated by +/–attP. EOPs were calculated relative to a permissive PLE(–) host (shown at left for reference). Bar height is the mean of three biological replicates and each dot is a measurement from an independent assay. The dashed line indicates the limit of detection (LOD) for this assay. (B) Schematic showing relevant construct design for PLE2 attP cloned in a neutral location (lacZ) in the V. cholerae chromosome. EOP (as in panel A) except PLE2 attP is cloned into lacZ. Bar height is the mean of three biological replicates and each dot is a measurement from an independent assay. The dashed line indicates the limit of detection (LOD) for this assay.
Co-expression of Int and Adi results in attachment site DNA degradation
Having established that Int, Adi, and the target sequence attP are sufficient for anti-PLE activity that results in impaired replication of the PLE genome, we wanted to directly assess the fate of DNA in the presence of both Int and Adi. Since we could not recapitulate DNA degradation in vitro, we turned to experiments in V. cholerae in the absence of phage infection. This was partly motivated by the observation that co-expression of int, but not the catalytically inactive mutant intS12A, and adi was toxic to V. cholerae (Figure 6A). We anticipated this toxicity was due to nuclease activity driven by Int and Adi. To assess this possibility, we performed light and fluorescence microscopy on cells after a short induction of int, adi, or both. Congruent with the toxicity assay results, we observed that expression of either int or adi alone resulted in normal DNA staining, while co-expression of int and adi resulted in a dramatic loss of DNA signal by Hoechst staining (Figure 6B). By light microscopy, all induced cells appeared physiologically normal, suggesting that the specific observed loss of DNA signal during Int and Adi co-expression was not due to apparent cell lysis, membrane damage, or cell division defects. Interestingly, the strain used in these assays is PLE(–) and therefore does not contain attP, but it has attC, the chromosomal integration site also recognized by Int, suggesting that Int and Adi will destroy any DNA for which Int has inherent specificity.
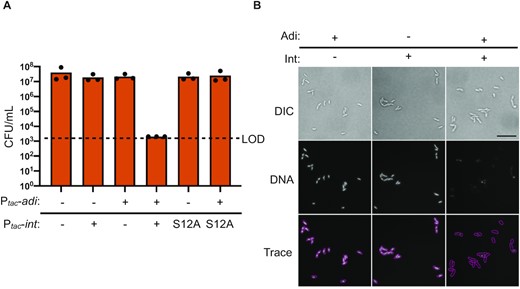
Co-expression of int and adi results in destructive nuclease activity in V. cholerae. (A) Cell viability as measured by colony forming units (CFU) per ml of V. cholerae expressing int, intS12A, or an empty expression construct (–) in the chromosome with an empty vector plasmid (–) or plasmid expressing adi as indicated. Dashed line indicates the limit of detection (LOD). Bar height is the mean of three biological replicates and each dot is a measurement from an independent assay. (B) Representative images of differential interference contrast (DIC) light microscopy and fluorescence microscopy of V. cholerae cells induced to express either int, adi, or both for 20 min followed by aldehyde fixation and staining with Hoechst DNA stain. Cells were imaged at 100x magnification with white light (DIC) or DAPI filter (DNA). Scale bar represents 10 microns. Traces were generated manually from matched overlayed DIC and DNA images. Slides were prepared and imaged from three independent biological replicates in technical duplicate and blinded prior to imaging.
The microscopy data and cell survival assays are suggestive of destructive nuclease activity upon co-expression of int and adi in V. cholerae in the absence of other PLE and phage products. However, we reasoned that such nuclease activity must be directed to not be self-destructive for ICP1. Therefore, we were curious whether co-expression of int and adi was sufficient to mediate DNA degradation specifically of the attP sequence, recapitulating the depletion of PLE2’s attP observed during phage infection (Figure 2C). To assess this, we co-expressed int or the intS12A mutant with adi in V. cholerae containing an empty vector control or a plasmid containing the attP target sequence and performed deep sequencing of the total DNA before and after induction. The resulting sequencing reads were mapped to the relevant plasmid sequence to look for evidence of plasmid degradation. Strikingly, the proportion of total reads mapping to the attP(+) plasmid decreased ∼10-fold only when catalytically active int was co-expressed with adi (Figure 7A). Importantly, we did not observe depletion of the empty vector plasmid upon co-expression of int and adi, indicating that the attP sequence is required for targeting. We compared the coverages between V. cholerae expressing the catalytically active int or its mutant derivative intS12A and scanned for dropouts in the coverage ratio as was performed for PLE2 previously. As expected, we observed that the largest drop in the coverage ratio was localized at the target sequence, attP (Figure 7B). Further, we observed no drop in coverage ratio when comparing the induced empty vector plasmid controls, suggesting that nuclease activity specifically targets the attP site (Figure S7). Altogether, these data suggest that Int and Adi are sufficient for attP targeting in the absence of phage infection and that no other PLE or ICP1 products are required for the depletion of attP.
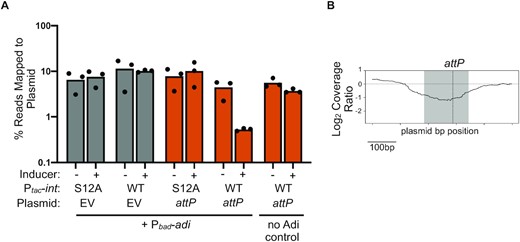
Co-expression of int and adi targets plasmids in an attP-dependent manner. (A) Percent reads abundance mapped to the plasmid indicated prior to induction (–) and 20 min post-induction (+) in V. cholerae strains induced to express either int or intS12A and adi. Strains had either an empty vector control (EV) or plasmid containing the PLE2 attP target sequence. Bar height is the mean of three biological replicates and each dot is a measurement from an independent assay. (B) Log2-transformed sequence coverage ratio across plasmid containing attP (relative position shown on the X axis) following induction of int and adi by showing the most prominent coverage drop detected compared to induction of intS12A and adi 20 min post-induction. The largest coverage ratio dropout is focused on the 500bp region surrounding attP and represents the average of three replicates. The region comprising the circularization junction, attP, is highlighted in grey with the dashed vertical line indicating the crossover junction between attL x attR to form attP.
Overall, our data support a model in which adi(+) ICP1 infects a PLE2(+) V. cholerae host, resulting in Int-mediated PLE2 excision and the formation of the PLE mobilization product, attP. Concomitantly, the anti-PLE factor Adi alters Int activity such that attP, a DNA sequence which Int has inherent specificity for, serves as the target site for its destructive nuclease activity, resulting in loss of integrity of the PLE genome and restored production of ICP1 progeny phage (Figure 8A).
![Overall model of how ICP1 weaponizes PLE’s recombinase to subvert phage restriction. (A) Upon ICP1 infection, Int excises PLE2 from the chromosome, Adi exploits Int's catalytic activity resulting in PLE DNA degradation through targeting of attP. ICP1 can then successfully replicate and release phage progeny following cell lysis. (B) A model representing a possible mechanism for Adi activity based on the conserved mechanism of LSRs (25,26,39). For simplicity, the RDF is not shown. [1] The Int subunits (shown here as orange circles) are bound to double-stranded DNA (shown as bars), with each bar denoting the different att units (two half attL units, attL and attL’ are light and dark grey bars, respectively; two half attR units, attR and attR’ are dark and light grey bars, respectively). The change in color highlights the crossover junction between the att sites upon recombination (attL and attR’ form attC; attR and attL’ form attP). [2] The dinucleotide 3’ overhangs (shown here as thin lines) are formed upon double-stranded cleavage of the DNA (26,29). [3] Rotation and [4] ligation of the Int subunits form new att sites: attC (shown as a light grey bar) and attP (shown as a dark grey bar). Adi (shown as a yellow star) binds to Int and prevents ligation, resulting in exposed dinucleotide 3’ hydroxyl overhangs that can be targeted by nucleases for processive DNA degradation.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/50/19/10.1093_nar_gkac845/1/m_gkac845fig8.jpeg?Expires=1750231193&Signature=SVCwXXuXrLGX4F9lPvVn2ROZHSIXVRWTgSUl04FRwnMeo0NATsaLSrNvKdNwFMrYeXvN6ZFOdJ3nZBf6Sv9rLpg9eXo6yAcHxaO2dOcPd9AXdKxOi7U-p3k47gBCG-H1-6GK2WBUHnkiaJRu87EndAyqn1fxw3~m-xfzJ9XxschrifJMkXZWgTBtw0aJ8Blq~~esz3ffufbhFvpJJqKUgYuzD3oGQqpADDwsJC0QpxKV8IiGD28pYxZ2sXvtJZIdCS0dlPy1qi96-wlpCgF04U4u6Sigsoczouu5bUqHegklV~lGlFaCWUWv~hnXiT4mw4GQ8buzB3nT9x5O~v7rWw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overall model of how ICP1 weaponizes PLE’s recombinase to subvert phage restriction. (A) Upon ICP1 infection, Int excises PLE2 from the chromosome, Adi exploits Int's catalytic activity resulting in PLE DNA degradation through targeting of attP. ICP1 can then successfully replicate and release phage progeny following cell lysis. (B) A model representing a possible mechanism for Adi activity based on the conserved mechanism of LSRs (25,26,39). For simplicity, the RDF is not shown. [1] The Int subunits (shown here as orange circles) are bound to double-stranded DNA (shown as bars), with each bar denoting the different att units (two half attL units, attL and attL’ are light and dark grey bars, respectively; two half attR units, attR and attR’ are dark and light grey bars, respectively). The change in color highlights the crossover junction between the att sites upon recombination (attL and attR’ form attC; attR and attL’ form attP). [2] The dinucleotide 3’ overhangs (shown here as thin lines) are formed upon double-stranded cleavage of the DNA (26,29). [3] Rotation and [4] ligation of the Int subunits form new att sites: attC (shown as a light grey bar) and attP (shown as a dark grey bar). Adi (shown as a yellow star) binds to Int and prevents ligation, resulting in exposed dinucleotide 3’ hydroxyl overhangs that can be targeted by nucleases for processive DNA degradation.
DISCUSSION
Here, we uncover a novel anti-PLE mechanism present in some ICP1 isolates that appears to function as a counter-defense mechanism by exploiting PLE’s mobility. Thus far, known ICP1-encoded anti-PLE counter-defenses rely on nuclease activity to target the PLE genome: CRISPR-Cas provides anti-PLE defense against a broad range of PLEs (13,18,24), while Odn targets the origin of replication of PLEs 1, 4 and 5 (22). Because Odn does not target all PLEs, Adi serves to expand a given phage's capacity to defend against all PLEs. This is exemplified by ICP11992, which is armed with both Odn and Adi, allowing it to target multiple PLEs: Odn targets the origin of replication of PLEs 1, 4, and 5, while Adi restricts PLE2 by targeting the PLE2-specific attP site. Intriguingly, many ICP1 isolates encoding CRISPR-Cas also encode Adi (23), signifying that there is a fitness benefit for ICP1 to maintain Adi, the anti-PLE2 effector, even in the presence of a broader anti-PLE defense system. Although CRISPR-Cas restores ICP1 plaque formation on a PLE2(+) host (13), this defense alone may not be enough to completely abolish PLE replication and transduction, as was observed for PLE1 (18,24). By encoding CRISPR-Cas and Adi, the combined action of two anti-PLE counter-defense mechanisms could help to compensate for differences in interference seen with different CRISPR spacers (24). Further, encoding two anti-PLE systems would require multiple counter-adaptations by PLE to escape, ensuring ICP1 maintains the upper hand. Alternatively, the nuclease activity stimulated by Adi and Int may help to generate substrates for adaptation of the phage CRISPR system, analogous to what occurs in bacteria with CRISPR and restriction-modification systems (46). Previous analyses show that proteins with similar functions (e.g. anti-defense systems) cluster together in specific genomic locations or hotspots (22,47–49). Now that the adi locus has been functionally identified, genes in the same genomic context in adi(–) ICP1 isolates are compelling candidates to explore as possible additional counter-defense mechanisms.
It is interesting as to why PLEs would encode PLE2-type integrases when doing so would sensitize PLE to Adi. Previous work has shown that PLE1-type integrases exploit ICP1-encoded PexA as the RDF to favor the excision of PLE1 from the chromosome (10). However, pexA is not essential and can be mutated in ICP1 (10). While PLE2’s RDF has not yet been identified, it is possible that PLE2-type integrases evolved to exploit an essential ICP1 gene product as their RDF. Therefore, if PLE2-type integrases evolved to exploit an essential gene, then in a tit-for-tat model, perhaps ICP1 evolved to exploit this integrase to PLE’s demise. While the evolutionary path that led to the divergence of mobility modules in PLEs can only be speculated, it is clear that the modular diversity observed across PLEs can provide a means to escape the molecular warfare with their inducing phage while maintaining their fundamental life cycle. Innovations arising from the conflict between these antagonizing genomes continue to inform the investigation of the so-called microbial dark matter, genes and systems whose functions cannot be bioinformatically inferred but become apparent when examined in the context of inter-microbial conflict.
A question remains as to the specific mechanism of Adi activity that renders PLE2 unable to restrict ICP1. Although it is still unclear how Adi modulates Int activity, consideration of how LSRs function may provide insight into the possible mechanism employed by Int/Adi targeting. A hallmark of the LSR mechanism is its concerted action in double-stranded cleavage (26,39,45). During excision, LSRs bind to their DNA substrates and form a synaptic complex leading to activation of the catalytic serine residue and cleavage of the DNA substrates at the crossover dinucleotide (26,39,45). An intermediate of this recombination is the formation of dinucleotide 3’ overhangs (26,39,45). One of the possible mechanisms by which Adi could act to damage the PLE2 genome is by preventing the integrase from re-ligating the dsDNA break while the integrase is mediating recombination, thereby resulting in free 3’ hydroxyl groups on DNA ends that nucleases could target for processive degradation of the DNA (Figure 8B). This is consistent with Int/Adi-dependent phenotypes observed across different assays including impaired PLE replication (Figure 2A and Figure 3E), depletion of reads mapping to the plasmid bearing attP (Figure 7A), and visible loss of chromosomal DNA (Figure 6B). While this proposed mechanism suggests a direct interaction between Int and Adi, we did not see evidence of an interaction using pulldown assays or bacterial adenylate cyclase two-hybrid assays. Our attempt to recapitulate the protein-protein interaction may be confounded by the transience of the interaction or the lack of RDF (or an additional factor) that could stabilize the interaction between Int and Adi. Notably, however, expression of Int and Adi in V. cholerae is sufficient for attP targeting, suggesting that nuclease activity targeting the PLE does not require other PLE or ICP1-encoded factors (including the putative RDF) but does not rule out requirements for other V. cholerae host-encoded factors. Lastly, another way in which Int activity can be modulated is through modification of the protein itself. However, we are not aware of any LSRs that undergo modification, except during recombination in which the LSR is covalently bound to the 5’ DNA ends through a phosphoseryl linkage (26,39–41). Thus, further study of the mechanism by which Adi exploits Int activity to target PLE2 may reveal more widespread modifications to LSR activity than are currently appreciated.
To our knowledge, RDFs are the only known proteins that interact with LSRs to modulate their activity. In the context of phage infection, Int binds to the RDF resulting in PLE excision. At the molecular level, RDF-Int binding alters the activity of the LSR, modulating Int's activity to favor excision resulting in the recombination between attL and attR (44,50,51), and inhibiting attP and attC recombination (integration) (44). Because of the simple requirements and specificity of directional recombination, LSRs are used as tools for genetic engineering (27–34). The discovery of Adi and the data presented here suggest that other factors could modulate the activity of LSRs in unexpected ways (i.e. to have destructive nuclease activity) and implicates an additional barrier in the development of LSRs as tools for genetic engineering.
Our model of Adi activity raises the possibility that other factors, including known defenses, could be exploited in inter-genome conflicts. Nucleases are particularly pervasive effectors mediating conflicts between hosts, mobile elements, and viruses (52). Such nucleases could be manipulated to the detriment of the organism deploying them, in a similar manner to how Adi exploits Int for anti-PLE activity. For instance, perhaps some anti-CRISPRs bind Cas proteins to modulate their activity in such a way that instead of cleaving the antagonizing genome, Cas targets its host genome for degradation instead. Likewise, restriction-modification systems could be exploited in such a way that the self-genome usually protected from nuclease destruction becomes targeted. Moreover, such proteins that alter the activity of other enzymes could be challenging to predict, perhaps explaining why such mechanisms have yet to be discovered. Still, encoding small proteins like Adi that weaponize existing proteins or even large protein complexes may allow additional pathways to survival for mobile elements like phages that have constrained genome sizes.
DATA AVAILABILITY
The sequencing data from phage (± adi (gp97.1)) infected PLE2(+) V. cholerae and plasmid depletion assays generated in this study have been deposited in the Sequence Read Archive database under BioProject accession code PRJNA839547. Replicates have been deposited to Mendeley Data, DOI: 10.17632/2nzzpp69g9.1.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank members of the Seed lab, including former lab member Dr. Zach Barth, for critical feedback and thoughtful discussion regarding this project. Thanks to the Glaunsinger Lab for the plasmid encoding 6xHis-SENP2. Special thanks to Denise Schichnes and the UC Berkeley Biological Imaging Facility for advice and equipment. We are incredibly grateful for the Biology Scholars Program (Research Fellows Program) and the McNair Scholars Program for supporting MHTN as an undergraduate researcher, and allowing them to pursue scientific research. MHTN extends special gratitude to Ernesto Bonilla and Bear for all their support.
FUNDING
This project was supported by Grant numbers R01AI127652 and R01AI153303 to K.D.S. from the National Institute of Allergy and Infectious Diseases. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or NIH. K.D.S. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. Funding for open access charge: NIH [R01AI153303].
Conflict of interest statement. K.D.S. is a scientific advisor for Nextbiotics, Inc.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments