





Abstract
Covalent DNA interstrand crosslinks are toxic DNA damage lesions that block the replication machinery that can cause a genomic instability. Ubiquitous abasic DNA sites are particularly susceptible to spontaneous cross-linking with a base from the opposite DNA strand. Detection of a crosslink induces the DNA helicase ubiquitination that recruits NEIL3, a DNA glycosylase responsible for the lesion removal. NEIL3 utilizes several zinc finger domains indispensable for its catalytic NEI domain repairing activity. They recruit NEIL3 to the repair site and bind the single-stranded DNA. However, the molecular mechanism underlying their roles in the repair process is unknown. Here, we report the structure of the tandem zinc-finger GRF domain of NEIL3 and reveal the molecular details of its interaction with DNA. Our biochemical data indicate the preferential binding of the GRF domain to the replication fork. In addition, we obtained a structure for the catalytic NEI domain in complex with the DNA reaction intermediate that allowed us to construct and validate a model for the interplay between the NEI and GRF domains in the recognition of an interstrand cross-link. Our results suggest a mechanism for recognition of the DNA replication X-structure by NEIL3, a key step in the interstrand cross-link repair.
INTRODUCTION
DNA interstrand cross-links (ICLs) are toxic DNA lesions that can arise from various endogenous and exogenous sources (e.g. cis-platinum, mitomycin C, acetaldehyde and formaldehyde) and ICLs can also form spontaneously from an abasic site (1–3). Inevitably, such a diverse chemical nature of cross-linking agents is reflected by a multitude of different structures of individual ICLs (4–8).
Abasic site interstrand cross-links (Ap-ICLs) arise spontaneously from naturally occurring abasic (Ap) sites. An aldehyde group of the ring-opened ribose at C1′ forms an N-glycosidic bond with an amine group of a base in the opposite DNA strand (9,10). It is noteworthy that a stable and specific Ap-ICL forms between an Ap site and an adenine in the opposite strand within the 5′-Ap-dT-3′ sequence (11,12).
ICLs pose impenetrable barriers to DNA replication, and the ICLs must be removed by specialized DNA repair pathways to prevent their deleterious effects. Initially, the Fanconi anaemia (FA) DNA repair pathway was identified in the repair of the ICL (13–15). Mutations in one of the many FA genes lead to Fanconi anaemia, a rare genetic disease characterized by chromosomal abnormalities, bone marrow failure and a strong predisposition to cancer. The cellular and molecular hallmark of FA is a sensitivity to cross-linking agents which leads to its implication with the ICL repair (16–18). Later, with the emergence of the Ap-ICL, an alternative, non-canonical or NEIL3 (Nei endonuclease VIII-like 3) pathway was discovered (19). FA pathway described in J.C. Walter's laboratory was a prerequisite for the discovery of the NEIL3 pathway of ICL repair (19).
NEIL3 has a catalytic NEI domain and belongs to the Fpg/Nei DNA glycosylase family. The enzymes from this family are structural and functional homologues, and they are conserved from bacteria to humans (20,21). Their generic role in base excision repair (BER) is to remove single base lesions arriving mainly from oxidative stress (22). In mammals, three NEIL (Nei endonucleases VIII-like) DNA glycosylases were identified: NEIL1, NEIL2 and NEIL3 (23–26). In general, Nei enzymes are bi-functional glycosylases, with glycosylase and Ap-lyase activities. The first step of catalysis is a nucleophilic attack on an N-glycosidic bond of the damaged base and its release. The enzyme then forms a covalent link (Schiff-base) with the orphaned ribose of the damaged DNA strand. In the second and slower step, phosphodiester bonds of the DNA are broken due to a β or β-δ elimination mechanism (Ap-lyase activity), generating a toxic 3′-DNA lesion and a single-strand DNA break (27–30).
Initially, NEIL3 was reported to have poor Ap-lyase activity on substrates with double-stranded DNA (dsDNA). Surprisingly, higher activity and therefore specificity was observed for single-stranded DNA (ssDNA) substrates (20). It has been shown that NEIL3 binds to the telomere sequence via its C-terminus and protects telomere integrity in proliferating cells within the S/G2 phase (31). Interestingly, NEIL3 was found to participate in the repair of the Ap-ICL. In Xenopus egg extracts, the Ap-ICL was unhooked when one of the two N-glycosidic bonds forming the ICL was cleaved by NEIL3. N-glycosyl cleavage by NEIL3 is the primary unhooking pathway for the Ap-ICL (19).
NEIL3 is the only member of the Fpg/Nei glycosylase family with an elongated C-terminus and it contains three sets of zinc finger domains (NEI, NZF and GRF domains 1 and 2) (Figure 1A). The N-terminal NEI domain is responsible for the DNA glycosylase repair activity on the Ap-ICL (19). The NLP4-type zinc finger (NZF) is a ubiquitin-binding domain employed in the recruitment of NEIL3 glycosylase to an Ap-ICL via TRAIP-dependent ubiquitylation of CMG helicase. NZF is required for efficient Ap-ICL repair in Xenopus extracts ex vivo. However, the NZF domain is not essential for enzymatic unhooking of DNA cross-links on model substrates in vitro (32).
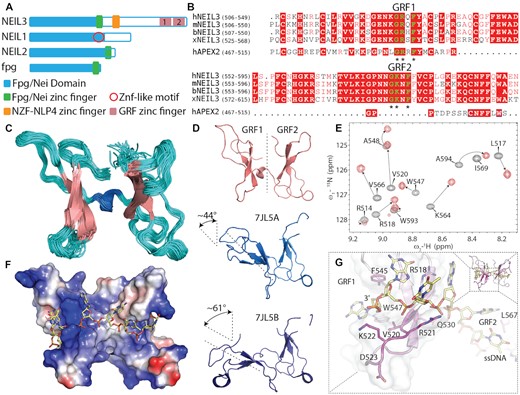
NMR-based structure of GRF domains in complex with ssDNA. (A) Schematics of the domain organisation of NEIL3, NEIL2, NEIL1, NEI/Fpg domain (blue), two GRF domains GRF1 and GRF2 (pink blocks) NEIL3 are located at the C-terminus, schematics bacterial fpg added for comparison. (B) Sequence alignment of GRF 1&2 from different organisms in comparison with single GRF domain from hAPEX2. Asterisks denote the GRF motif. (C) NMR ensemble of mouse GRF 1&2 pseudo dimer (loops in pink, b-sheets in magenta, and linking a-helix in cyan) (PDB ID 7OMK, Supplementary Table S1). (D) A comparison of inter-domain rearrangements of mouse NEIL3 GRF 1&2 (pink) and human NEIL3 GRF1&2 domains, two dimer molecules were present in the asymmetric unit of crystal structure PDB ID-7JL5 (light and dark blue) (35), human GRF1 was aligned to mouse GRF1, the angle difference for GRF2 (chain A) was 43.9° with lateral displacement of 9.5Å and GRF2 (chain B) was 61.4° with a displacement of 11.2 Å. (E) Selected 1H–15N HSQC spectral overlay of free mGRF (black cross-peaks) and GRF:ssDNA complex (red cross-peaks). (F) Electrostatic surface potential of GRF:ssDNA complex (ssDNA yellow sticks). (G) Detail of the residues responsible for GRF:DNA interaction (R518, V520, (R521) and W547 from panel (E) and highest chemical shift perturbations shown as the gradient of magenta (more detail is shown in Supplementary Figure S3).
The other two zinc finger domains (GRF1 and GRF2) are homologues with the ‘GRF’ zincfinger from APEX2 Ap endonuclease. GRFs are known to interact with ssDNA (33). The GRF name originates from the triad of conserved residues: Gly (G), Arg (R) and Phe (F). However, in GRF2, the Arg is replaced with Lys, which is distinct from the APEX2 GRF motif (Figure 1B). Interestingly, lysine (K553) in GRF2 disrupted the binding with short ssDNA (34). The crystal structure of the human GRF1-GRF2 dimer revealed a shape resembling a butterfly. These two GRF domains together were able to bind ssDNA with greater affinity than individual domains via their avidity (35). Experiments in Xenopus extracts have shown that GRF domains play a role in efficient Ap-ICL unhooking, and they are required for the efficient recruitment of NEIL3 to the stalled replication fork on the Ap-ICL. In the absence of GRFs, the unhooking reaction was compromised. This indicated that the GRFs’ role in Ap-ICL repair might be the proper recognition of the crosslinked substrate (32). In contrast, another study has revealed that GRF domains inhibit the enzymatic activity of NEIL3 in vitro (35). These seemingly contradictory observations suggest that the mechanistic role of the GRF domain in Ap-ICL repair is more complex, and it is yet to be explained.
In this study, we investigated the molecular architecture of recognition of the stalled replication fork by NEIL3 DNA glycosylase during Ap-ICL repair. We obtained two structures of GRF domains with and without ssDNA from restraints from NMR measurements. We observed that the GRF domains recognize not only ssDNA as reported previously but also very short replication-fork intermediates. The GRF1&2 domains bind to the 5′-ssDNA overhangs of the DNA duplex with a slight preference. These observations indicate that GRFs may play a role in the recognition of native Ap-ICL DNA substrates.
To understand the architecture of Ap-ICL repair, we crystallized and solved the structure of the NEI domain from mouse NEIL3, covalently trapped with the DNA substrate. We observed that, unlike GRF domains, the NEI domain recognises 3′-ssDNA overhangs on duplex or fork DNA structures. Based on our structural, biochemical and enzymological data, and with the use of AlphaFold, we constructed a model of NEIL3 bound to the replication fork in the intermediate state. This data shed light on the repair scenario of Ap-ICL repair on the stalled replication fork. In this model, we considered two collided DNA replication forks that arise from bidirectional replication, forming a letter X (14,19). Our data suggest a possible mechanism by which NEIL3 recognizes the DNA replication X-structure. The data also indicate the role of GRF and NEI domains in the recognition of the native Ap-ICL substrate.
MATERIALS AND METHODS
Cloning, expression and purification of the recombinant GRF and NEI protein domains
The GRF and NEI domains were amplified from the full-length mouse NEIL3 gene NM_146208 (Origene). PCR products were cloned using Gibson assembly (36). The tandem GRF1&GRF2 domains (505–597) were cloned into a pSUMO vector with an N-terminal His8x-SMT3 tag. The NEI domain (1–282) was cloned into a modified pET-24b vector with a C-terminal 3C protease (HRV) site and subsequent His6x tag. The proteins were expressed and purified using standard protocols (37,38). Briefly, the plasmids were transformed into Escherichia coli BL21 Star™ (DE3) cells (ThermoFisher). Small 5 ml cultures were grown overnight at 37°C in LB medium. Expressions were carried out after inoculation of ZY5052 autoinduction media supplemented with 50 μM ZnSO4. After reaching an optical density of OD600 = 0.6–1 at 37°C, the temperature was lowered to 18°C, and the culture was grown overnight. The harvested cells were lysed by sonication in a lysis buffer containing 20 mM Tris–HCl pH 8.0; 300 mM NaCl, 30 mM imidazole pH 8.0, 10% glycerol and 1 mM tris(2-carboxyethyl)phosphine (TCEP). The supernatant was separated by centrifugation, incubated with 2 ml of Ni-NTA resin (Machery-Nagel), and extensively washed with the lysis buffer using the batch technique. The proteins were eluted with the lysis buffer supplemented with 300 mM imidazole pH 8.0.
The proteins were desalted on a HiPrep 26/10 desalting column (GE Healthcare) and loaded on a cation exchange HiTrap SP HP column equilibrated with buffer A (20 mM Tris–HCl pH 8.0, 125 mM NaCl, 10% glycerol, 2 mM β-mercaptoethanol (β-ME)). In the case of the NEI protein, 70 mM NaCl was used for buffer A. The proteins were eluted by gradients of NaCl. The solubility tag GRF domain was cleaved by SUMO protease from yeast (Ulp1), and after overnight incubation at 4°C, the sample was desalted and reloaded on cation exchange as described above. The proteins were purified on size-exclusion Superdex 75 Increase GL HiLoad 10/300 (GE Healthcare) in buffer A. The purity of the proteins was verified on SDS-PAGE in a 15% polyacrylamide gel, stained with Coomassie brilliant blue. The proteins were concentrated and flash-frozen in N2(l) and kept at –80°C.
Isotopically labelled GRF protein for NMR experiments was expressed using the minimal medium containing [15N] NH4Cl, and [13C] glucose supplemented with 0.1 μg/ml of ampicillin, 20 μM ZnSO4, 1 mM MgCl2 and 10 μM FeCl2 in citric acid. Inoculated cells were incubated in a shaker at 37°C. After reaching an OD600 of 0.6, protein expression was induced with 600 μM isopropyl β-d-1-thiogalactopyranoside (IPTG), the temperature was lowered to 18°C and the culture was grown overnight. Labelled protein was purified as mentioned previously.
NMR spectroscopy
NMR spectra were acquired at 25°C using an 850 MHz Bruker Avance spectrometer, equipped with a triple-resonance (15N/13C/1H) cryoprobe. The sample volumes were either 0.16 or 0.35 ml, in 12.5 mM NaPhos pH 6.5, 50 mM NaCl, 1 mM TCEP, 5% D2O/90–95% H2O. A series of double- and triple-resonance spectra (39,40) were recorded to obtain sequence-specific resonance assignment. in NMRFAM-SPARKY (Lee et al., 2015). 1H–1H distance restraints were derived from the 3D 15N/1H NOESY-HSQC and 13C/1H NOESY-HMQC spectra using a NOE mixing time of 100 ms.
The structural calculation was carried out in CYANA (41) using NOESY data in combination with backbone torsion angle restraints, generated from assigned chemical shifts using the program TALOS+ (42). The combined automated NOE assignment and structure determination protocol (CANDID) was used for automatic NOE cross-peak assignment. Subsequently, five cycles of simulated annealing combined with redundant dihedral angle restraints were used to calculate a set of converged structures with no significant restraint violations (distance and van der Waals violations < 0.5Å and dihedral angle constraint violations < 5°). The 35 structures with the least restraint violations were further refined in explicit solvent using YASARA (43) and subjected to further analysis using the Protein Structure Validation Software suite (www.nesg.org). The statistics for the resulting structure are summarized in Supplementary Table S1-1. The structures, NMR restraints and resonance assignments were deposited in the Protein Data Bank (PDB ID 7OMK) and BMRB (accession code: 34630). The relaxation data were obtained using the 300 µM 15N labeled sample. The relaxation delays were 0.0448, 0.0672, 0.112, 0.179, 0.246, 0.381, 0.784, 1.23 s for R1 and 0, 0.0310, 0.0628, 0.0931, 0.124, 0.186, 0.279 s for R2 measurements. The recovery delay was set to 1.2 s with 4 or 8 scans per FID. The {1H}–15N steady state nuclear Overhauser enhancement (NOE) was measured in an interleaved manner using a 5 s long train of selective 180° pulses for proton irradiation, separated by 22 ms delays (44), and centered at 8.2 ppm in order to avoid water saturation. The offset of these pulses was shifted by 50 kHz in the reference experiment to ensure constant sample heating (44,45).
Gel-based and fluorescent anisotropy DNA binding assays
A specific three-letter code (NTX code) was used to name the oligonucleotides used in the binding assays. The NTX code indicates that N stands for the number of ssDNA nucleotides of the single-stranded arm of the DNA structure, T for thymidine, and X for the shape of ssDNA structure (Y fork, 3′ or 5′ overhangs).
Labelled (FAM) and unlabelled DNA oligonucleotides were custom synthesized (Microsynth, Switzerland), and the DNA sequences are listed in Supplemental Figure 1. Fluorescent dye FAM-dT was placed instead of adenine (x) in the apex of the hairpin 5′-GACGCGxAGCGTC-3′. DNA oligos were resuspended in TES buffer: 10 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM EDTA. Double-stranded DNA substrates were prepared by mixing equimolar amounts of complementary strands, which were heated to 95°C and allowed to anneal by slow cooling to room temperature. The hairpin substrates were heated to 95°C and then rapidly cooled on ice.
Native EMSA was performed as follows; 5 μM labelled or unlabelled DNA and 5 μM GRF were mixed and incubated for 30 min at 25°C in 20 mM NaH2PO4 pH 6.5, 100 mM NaCl, 10% glycerol and 1 mM TCEP. Samples were then separated by electrophoresis on 15% native polyacrylamide gels (1× TBE, 15% polyacrylamide/bisacrylamide 14:1). The gels with unlabelled DNA were stained with Coomassie Brilliant Blue to visualize the GRF:DNA complex and were analyzed using the commercial software ImageQuant TL. Gel lines of constant width were manually selected and background subtraction was performed using the rolling sphere method. Bands with constant dimensions containing the binding product were selected manually. The values for each gel band were plotted on a graph. All gels were scanned at Typhoon LASER imager (GE).
DNA and NEIL3 trapping assay
DNA oligonucleotides (sequences are shown in Supplementary Figure S1) were annealed as previously mentioned. For abasic site creation, 0.5 U uracil–DNA glycosylase (UDG) (New England Biolabs) was added per one sequence reaction and kept for 20 min at room temperature. 250 nM DNA substrates and 200 nM NEIL3 were incubated in the presence of 100 mM NaBH3CN in 20 mM HEPES 7.0, 50 mM NaCl and 1 mM TCEP at 25°C. At given time points, 30 μl of the reaction was taken and mixed with 5× SDS of sample buffer (60 mM Tris pH 6.8, 25% (v/v) glycerol, 2.9% (w/w) SDS, 0.1% (v/v) Bromphenol Blue, 714 mM β-ME) to terminate the reaction. These samples were resolved on a 15% SDS-PAGE.
Crystallization and structure determination
DNA hairpin substrates for crystallographic experiments were synthesized with the following sequences: 5′ overhang 5′-TTTTTTUACGCGAAGCGTG-3′ and 3′ overhang. The DNA (250 μM) hairpin substrates were annealed by being heated to 95°C for 5 min and then rapidly placed on ice. 300 μl of annealed DNA was treated with 3 μl of UDG (NEB) and incubated at RT to generate the Ap site. Subsequently, 300 μl of purified NEI domain (300 μM) supplemented with 200 mM NaBH3CN was added. The reaction was allowed to proceed for 1 h to form the trapped NEI:DNA complex. The progress of the reaction was checked on 15% SDS-PAGE gels. The reaction mixture was desalted on a 5 mL HiTrap Desalting column (Cytiva) and loaded onto a 5 ml HiTrap Q HP column (Cytiva) equilibrated with 20 mM Tris–HCl pH 8.0 and 50 mM NaCl, 1 mM TCEP. The column was eluted with a linear gradient of NaCl. The trapped complex was loaded on Superdex 200 ® Increase 10/300 GL (Cytiva). The fractions containing NEI:DNA intermediate were concentrated to roughly 200 μM (OD260 nm = 30).
The crystals were grown at 22°C using the sitting drop method where 200 nL trapped complex was mixed with 200 nL with a reservoir solution containing 50 mM PIPES pH 7.5, 4% (w/v) PEG 8000 20 mM MgCl2 and 1 mM spermine. Crystals were soaked for approx. one minute in 20% glycerol supplemented in the reservoir. The cryo-protective condition was prepared by mixing 8 μl of the reservoir with 2 μl of glycerol. Mounted crystals were flash-frozen in liquid nitrogen. All datasets were collected at synchrotron beamline MX 14.2 at BESSY II Helmholtz–Zentrum Berlin, Germany. They were indexed with XDS directly at the beamline (50). Data were scaled with Aimless (51). The molecular replacement was carried out in Phaser software using the structure of the mNEIL3 NEI domain as a model PDB ID = 3W0F (52). The structures were refined using Refine, and the models were manually edited in Coot. All three programs were implemented in the Phenix package (53,54). Data collection and model refinement statistics are listed in Supplementary Table S2. The structure figures were generated using PyMOL (http://www.pymol.org/).
RESULTS
The structure of GRF determined by NMR spectroscopy
We have obtained the NMR structure of GRF1 and GRF2 domains of mouse NEIL3. The overall fold of previously reported crystal structures of individual human GRF domains is in good agreement with our NMR structure (35). The RMSD between mouse and human GRF domains RMSD (GRF1) = 1.94Å and whilst the RMSD (GRF2) = 0.87Å. However, some structural differences were observed mainly in the loop regions. Each domain consists of three antiparallel beta-sheets with a zinc finger region. Two GRF domains are positioned into a butterfly-like structure connected with a short helix linker (Figure 1C). It is noteworthy that interdomain organisation is different for the crystal structure of GRFs from human NEIL3 and our structure (Figure 1C). In the case of the previously published structure of human GRF, two molecules of GRF1&2 occupy the asymmetric unit, and each of the GRF pairs makes a different angle between the domains (Figure 1D) (35).
The structure of single-stranded DNA bound to NEIL3 GRF pseudo-dimer
The ability of GRF domains to bind single-stranded DNA is a well-documented feature (33,55). We set out to understand the role of GRF domains in DNA repair and we have obtained the structure of mouse GRF1&2 pseudo-dimer in the presence of DNA using NMR spectroscopy.
We attempted to obtain this structure with ssDNA oligonucleotide. We carried out NMR titrations of short oligonucleotides ssDNA. The isotopically unlabelled DNA (7mer 3′-GTCATGA-5′) was titrated with 13C–15N isotopically labelled GRF protein. Overlay of the 1H–15N HSQC spectra of a free and DNA bound revealed residues that were significantly perturbed by DNA interaction (Supplementary Figure S3). We triangulated the position of the DNA backbone bound to GRF and generated a data-driven HADDOCK model of their complex (Figure 1). The lowest energy structure ensemble of GRF pseudo-dimer displays a relatively rigid structure without any significant interdomain rearrangements. In GRF:DNA complex structure, the DNA is oriented from its 5′ end, located at GRF2, to the 3′ end leaning over the GRF1. In the middle of the DNA, there is a junction of GRF dimers, and there is little to no interaction between the two (Figure 1).
We believe that there is a reason for the butterfly arrangement of NEIL3 GRF1&2. The crystal structure of E. coli Topoisomerase 1 (PDB ID 4RUL) contains five GRF-like ssDNA binding domains which are organized in a series that stimulates its enzymatic activity (Supplementary Figure S2) (55). The fold of these zinc-ribbon (ZR) domains (TOP1–ZR1–ZR5) resembles the fold of the GRF domain. Three ZRs contain zinc, while the other two are zinc-less. A structural overlay of these domains with our GRF:DNA structure reveals that TOP1 ZR domains are arranged along with DNA in parallel to each other (Supplementary Figure S2). On the other hand, the butterfly pseudo-dimer of GRF has an antiparallel arrangement. Moreover, the interlink joining GRF1&2 domains seems to be unique, and no such arrangement can be seen in the solved crystal structure of Topoisomerase 1 (E. coli). Neither pair of these TOP1 ZR domains has an interdomain arrangement that is similar to NEIL3 GRF1&2. The DNA binds to the GRF domains in such a way that the domains can bind to either strand or to even both strands, at the replication fork.
GRFs recognition of single-stranded DNA
First, the focus was to determine the minimal length of ssDNA oligonucleotide recognised by the pair of the GRF domains. NMR titrations have shown that the optimal length of an ssDNA oligo was ∼7–9 nt. To assess interactions with ssDNA, we employed a semi-quantitative electrophoresis mobility shift assay (EMSA). Due to the opposite charge of GRF, the GRF:DNA complexes with short ssDNA oligonucleotides up to 11 nt long did not migrate into the PAGE gel. Therefore, we used a dsDNA hairpin (DNAhp, 13 nt long) as a substrate with ssDNA 5′ and 3′ overhangs. Using DNAhp increased the overall negative charge of the complex and improved mobility within the gel. The DNAhp oligonucleotides contained ssDNA 5′ and 3′ oligo-dT overhangs that were 0, 2, 4, 6, 8, 10 or 12 nt long (Supplementary Figures S1 and S4). The breaking point where the complex formation was observed in the gel was at around 4 nt. Next, we performed qualitative EMSA assays to assess GRF binding with DNA forks and tested the effect of fluorescent dye on the DNA binding. We did not observe significant differences in the binding of FAM-labelled versus unlabelled DNA (Supplementary Figure S4). We then performed a fluorescence anisotropy binding assay to quantitatively compare DNA and compared GRF binding to 5′ and 3′ ssDNA overhangs. Repeatedly, the binding constants for oligonucleotides with 5′ and 3′ ssDNA overhang, were comparable. We observed a slightly greater affinity of GRF domains towards oligonucleotides with 5′ ssDNA overhang. In the control experiment, there was significantly weaker binding between the dsDNA helix and GRF domain, confirming specificity for ssDNA (Figure 2A).
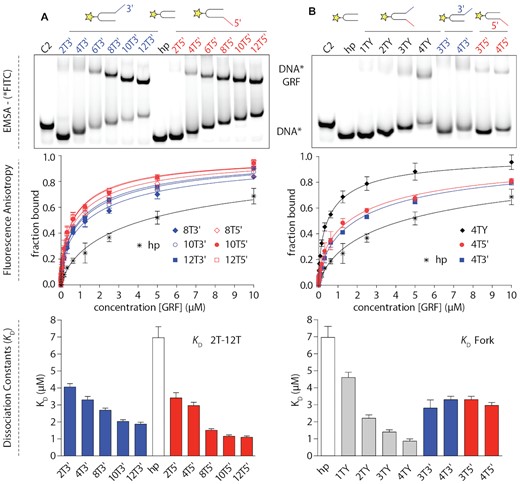
GRF domain, DNA binding from single-stranded DNA to recognition of DNA fork. Binding of GRF1&2 to FAM-labelled (star) DNA duplexes with 3′ (blue) and 5′ (red) ssDNA overhangs. (A) Comparative binding of GRF to DNA with different lengths of 3′ and 5′ overhangs. The KD value decreases with increasing 3′ and 5′ overhang length. (B) Binding of replication fork vs. ssDNA overhangs. Binding assays were performed in triplicates and data were plotted as mean ± SD. Native EMSA of GRF:DNA binding without a fluorescent dye is shown in Supplementary Figure S4 and further data; e.g. KD values and the remaining binding curves and raw fluorescence and anisotropies are shown in Supplementary Figure S5. The alternative fit with two-site model is shown Supplementary Figure S6.
GRF and interaction with replication-like structures
The architecture of GRF1&2 suggests that two different DNA chains could bind each domain, such as each arm of the DNA replication fork. To test this hypothesis, we tested the interaction of DNA replication-like structures with GRF1&2. We have designed small DNA forks with short ssDNA arms of identical length with DNA duplexes with hairpin ends to ensure all oligos were annealed (Supplementary Figure S1). The length of ssDNA arms at both 5′ and 3′ ends, arms of the fork, were 0, 1, 2, 3 or 4 nucleotides long. We performed qualitative EMSA assays to assess GRF binding with DNA forks and then performed a fluorescence anisotropy binding assay to quantitatively verify binding to the DNA forks and compared it to DNA oligonucleotides with ssDNA overhangs. GRF1&2 was able to bind well to a replication fork DNA substrate with short ssDNA arms (Figure 2B). In the case of all 2TY, 3TY, and 4TY GRF1&2 bound significantly better to these forks rather than to 3T3′ or 3T5′ oligonucleotides (and to 4T3′ and 4T5′ respectively) with ssDNA overhangs of the same length (Figure 2B). Moreover, the affinity GRF1&2 was greater for forks than for the oligos with ssDNA overhangs with an identical total number of nucleotides (e.g. 4TY versus 8T3′ or 8T5′). As GRF1&2 binds to the fork structures of a shorter length better than to the simple overhang, this suggests that both arms of the fork may be involved in the binding. Because the migration of GRF:fork-DNA on a native PAGE gel was similar to the GRF complex with an ssDNA overhang, we hypothesise that each GRF domain binds to one side of the fork.
GRF and the binding of replication fork with DNA damage
To further investigate the role of GRF in the Ap-ICL repair we tested how the presence of DNA damage (Ap site) affects the DNA-protein interaction. Similarly like in experiments in Figure 2A, DNAhp with ssDNA overhang at either end were used. Ap site as DNA damage was placed in the duplex close to the branching point as indicated in Supplementary Figures S4 nd S5. In native EMSA DNA binding experiments, we observed a preference of the GRF domains for the 5′ ssDNA overhang. However, from quantitative KD measurement, the binding of 5′ ssDNA overhang was only slightly increased by the presence of the damage site in DNA. This was observed in both cases with the damage located either on the top or the bottom DNA strand. This experiment demonstrated a subtle selectivity for the 5′ end, and it did not suggest that the presence of DNA damage alterations in the vicinity of the fork branching accentuate these differences (Supplementary Figures S4 and S5).
Single-stranded DNA at the 3′-end of the lesion is required for the efficient catalytic activity of NEIL3
The NEI domain of NEIL3 glycosylase has been reported to have a substrate (Ap-ICL) preference when catalysing the repair. The original Ap site of the crosslink is located on the 3′-arm of the replication fork (56). To understand the role of the individual arms of the fork, we carried out enzymatic assays with a simplified DNA replication fork with only one ssDNA overhang (fork) present. We have used the Ap site within the DNA duplex substrates with 3′ or 5′ overhangs 6 nt long (6T). The Ap sites were generated by UDG treatment of uracils placed in the duplex at the branching point. The reaction of the Ap site containing substrates with the NEI domain was terminated by the excess of NaBH3CN covalently cross-linking the NEI domain to the DNA, forming a DNA–protein crosslink. The reactions were resolved on an SDS-PAGE gel. NEI enzymatic assays revealed a 33-fold rate enhancement using the substrate with the 3′ overhang versus the 5′ overhang (Figure 3) (56). These experiments demonstrate that ssDNA is required in the 3′ direction of the Ap site for efficient catalysis.
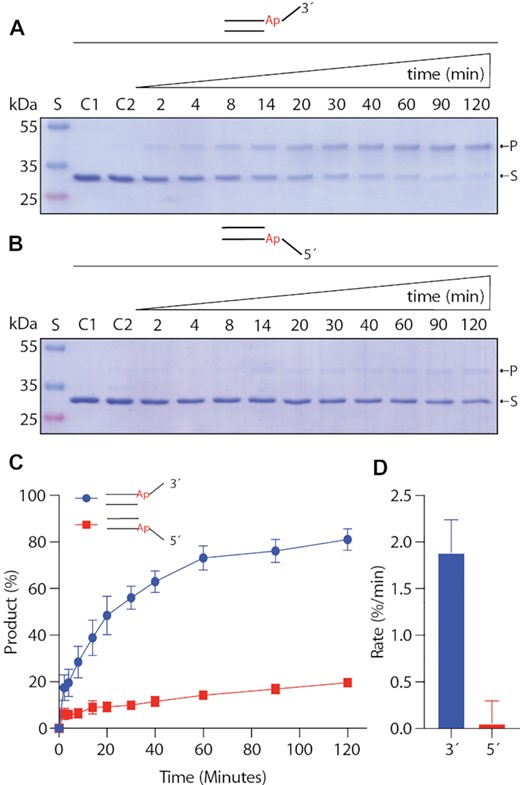
NEIL3 enzymatic assays with native Ap site within dsDNA with 3′- or 5′-overhangs. Enzymatic reactions of the mNEIL3 NEI domain with DNA with (A) 3′-overhangs (B) 5′-overhangs. Reactions were captured by the formation of the covalent intermediate by reducing the Schiff-base with NaBH3CN and resolved on 15% SDS-PAGE. (C) Gels bands corresponding to the substrate (S) and products (P) were quantified and plotted against reaction time. (D) Initial rates were calculated and plotted. Reactions were repeated in triplicates. The error is an SD (standard deviation).
Crystal structure of abasic site DNA intermediate with mNEIL3 NEI domain
To solve the structure of the complex of mNEIL3 with a DNA substrate, we expressed and purified the N-terminal domain of mNEIL3 1–282 (NEI domain). We prepared and set up crystallization plates with covalently trapped complexes of NEI-DNA. The stable trapped complex was prepared by reducing the Shiff-base intermediate of the repair reaction of the mouse NEI domain and the Ap site with NaBH3CN (57). We used several different DNA substrates, including DNA hairpins with ssDNA overhangs mimicking a simplified DNA replication fork with only one arm. Both of the DNA hairpin substrates were mirror images, with 6T long 3′- or 5′-overhangs. Despite the aforementioned preference of NEIL3 towards the Ap site before the 3′ overhang (Figure 3), we obtained well-diffracting crystals only for NEIL3:DNAhp with a 5′ overhang. The crystals diffracted to 2.28 Å resolution (PDB ID 7Z5A), and the structure was solved by molecular replacement using a mouse NEIL3 apo structure (PDB ID 3W0F) as a search model and refined to good geometry and R factors (please see Supplementary Table S2 for more details) (52).
The structure of the NEI domain consists of two subdomains, NTD and CTD. NTD domain contains catalytic residues with the reactive amino group of the N-terminus followed by eight anti-parallel beta-sheets that form a sandwich flanked by a small pair of helices and a large loop that is only partially visible in the electron density, likely due to its flexibility. The protein is connected via a further pair of α-helices, with CTD containing four helices and a zinc-finger with two antiparallel β-sheets. The overall fold is not only very similar to the apo form but also to the fold of the other members of the Nei/Fpg family (57). The orientation of NTD and CTD is identical to the published apo (unliganded) crystal structures of the mouse NEIL3 (52). This is in contrast with some Nei/Fpg enzymes that undergo conformational changes upon DNA binding (38). As previously reported, NTD and CTD are locked by a network of hydrogen and salt bridges between individual domains (Supplementary Figure S8E) (52). The lack of interdomain rearrangements and large interdomain interface suggests more interdomain rigidity in the NEIL3 glycosylase.
NEIL3 and the recognition of a trapped intermediate-like DNA hairpin with a 5′ overhang
The structural superimposition of apo and DNA:mNEIL3 structures of NEI domains revealed that DNA does not contribute to any significant conformational changes in the NEI domain (RMSD = 0.394 Å) (Supplementary Figure S7) (58). Several loops on both sides of the DNA strand relocate in order to accommodate the substrate.
The trapped DNA molecule is recognised exclusively in the vicinity of the active site residue and further on towards the hairpin and its 3′ end. At this point, residues K82, H96, R94 and N193 are responsible for the interaction with the damaged part of the strand. Furthermore, the active site methionine M1 is covalently linked with the Ap site that is stabilized by the hydrogen bridge E3 and further anchored by M99 and R272 (Figure 4). In contrast, besides the hydrogen bridge with Y243, there is an apparent lack of specific interaction between the protein and the ssDNA backbone on the 5′ end from the damaged site (Supplementary Figure S8).
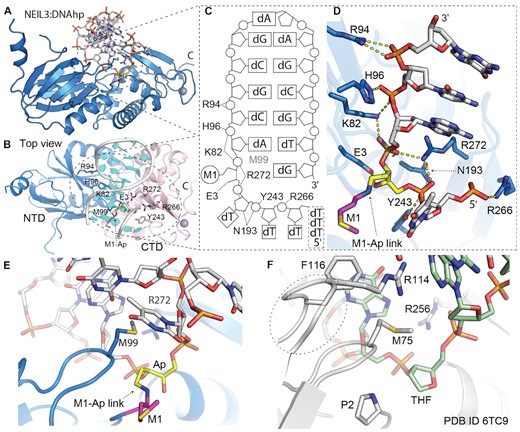
Crystal structure of trapped mouse NEIL3:ssDNAhp complex; DNA hairpin with 5′ overhang. (A) Overall structure of NEI glycosylase domain (light blue PDB ID 7Z5A) trapped in complex with DNA hairpin. (B) Top view on the NEIL3:DNA complex with highlighted residues involved in DNA interaction (sticks) N-terminal and C-terminal domains (NTD light blue and CTD in light pink). (C) Scheme of the interactions between DNA and NEI domain, M99 is intercalated between the Ap site and an orphan base. Structural representation of NEI residues directly involved in interaction with DNA. (D) Detail of the DNA binding pocket with reduced Shiff-base between methionine (M1) and Ap-site (M-Ap link). (E, F) A comparison of the active site of mouse NEIL3 glycosylase and bacterial fpg; fpg:dsDNA complex (PDB ID: 6TC9) with the Ap site analogue (THF), the typical residues for Nei/Fpg, here F116, R114, M75 inserted in the place of the damaged base. Two of these residues (R114&F116) are missing in the case of NEIL3 (38). For further comparison of mNEIL3 NEI domain and bacterial Fpg protein that undergoes a conformational change upon DNA binding the Supplementary Figure S9.
Structural aspects of NEIL3 preference for ssDNA
Overall, our data on the structure of the NEI domain with DNA has confirmed several predictions that were based on the apo structure (52). This work on the structure of mNEIL3 has suggested several differences between NEIL3 and other enzymes from Fpg/Nei family that our NEI:DNA structure has confirmed (52). There is a complete lack of contact between the complementary DNA strand and the enzyme. In addition, two acidic residues, E269 and D175, do not favour the recognition of the opposite strand of DNA (Supplementary Figures S7 and S8). Since, NEIL3 glycosylase is the Ap-ICL repairing glycosylase, the damaged oxidized base ‘capping loop’ (αF-β9/10 loop of Fpg/Nei enzymes) is completely missing, suggesting that the recognition and accommodation of the Ap-ICL and the entire associated DNA structures are carried out by different mechanisms. Interestingly, N-terminal methionine was involved in catalysis and not cleaved off, as it is for other members of the family (Figure 4 and Supplementary Figure S7).
NEIL3 and possible recognition of DNA replication fork intermediates
We conducted experiment-driven modelling to unravel the recognition of the replication fork structures by NEIL3 glycosylase. First, we took an AlphaFold (AF) generated model of the entire NEIL3 where GRFs are in contact with the NEI domain (Figure 5A). Then, using the AF model, we superimposed our structures of GRFs and NEI domains with DNA (Figure 5B). The structures matched relatively well to the AF model (RMSD (NEI) = 0.453 Å and RMSD (GRF1&2) = 1.980 Å). Interestingly, the orientation of GRFs and NEI is such that DNA from the NEI domain overlaps with the DNA of the GRF dimer in a shape letter Y, resembling the DNA replication fork (Figure 5B). We constructed a model of the DNA replication fork bound to NEIL3 glycosylase mid-repair. In this model, we used the interdomain arrangement from the AF and connected DNA strands from both the crystal structure of NEI:DNA and NMR-modelled structure GRF1&2:DNA. The hairpin turn in the dsDNA part of the model was maintained for clarity (Figure 5C, D).
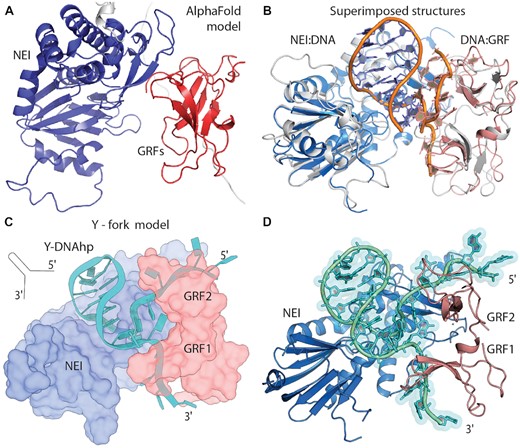
From the AlphaFold model and our NEI and GRFs DNA complexes to recognition of DNA replication fork. (A) AlphaFold (AF) model of NEI (blue) and GRF1&2 domains (red). (B) Superimposed crystal structure of NEI:hpDNA (light blue) and NMR model GRF1&2 (light red) with ssDNA on AF model (here white), DNA backbones (orange). (C) Schematics of an experimentally driven model where one replication DNA fork is recognised by NEI&GRFs based on AF interdomain positions. (D) cartoon representation of the model of Y-DNA:NEI&GRFs complex.
DISCUSSION
From GRF inhibition to a two-fork recognition mechanism
Using NMR spectroscopy, we obtained the structures of tandem GRF domains from mouse Neil3 (Figure 1C). The work reporting the crystal structure of human GRF domains suggests some interdomain motion (Figure 1D) (35). On the other hand, our data indicate restricted interdomain movement between GRF1&2, suggesting a relative rigidity between these domains. The GRF dimer interface is fortified by salt and hydrogen bridge interactions. These interactions include residues R560, D549, and R529, interactions with mainchain atoms A548 with Q530, R560 with F552. The residues at the N- and C- termini of the GRF domains are involved (E595 and S505, R514 and G597) (Supplementary Figure S9). Our NMR measurements do not support this behaviour in the case of the mouse protein homologue as for all ensembles for apo and DNA bound GRF dimer the structure is virtually identical. Moreover, our biochemical data indicate the ability of GRF to bind short replication forks, and the GRF dimer has an ideal arrangement to accommodate the branching point of the replication fork, with each domain binding one DNA strand. These data together suggest the possible role of GRFs in the binding of replication structures during the repair.
Recent work has demonstrated that GRF domains inhibit the enzymatic activity of the NEIL3 NEI domain (35). This observation might seem surprising, as other studies have shown that GRFs are known to stimulate the activity of the enzyme containing this domain (33,55). Competition of GRF and NEI for ssDNA may explain the observed decrease of the reaction rate catalysed by the NEI domain (35). This data could be explained as follows, both GRF1&2 and NEI domains efficiently bind Y forked substrates in their ssDNA parts, therefore they could simply be competing for the substrate of the NEIL3 enzyme. Therefore, the single Y-fork model scenario can explain the previously observed GRF inhibition of the NEI domain during the catalysis.
We predicted two different models for the role of GRF domains in Ap-ICL repair. Either GRF inhibits or regulates the enzymatic activity of the NEI domain, or GRF increases the selectivity of the NEIL3 enzyme towards structurally more complex substrates generated during the replication coupled repair of the Ap-ICL (19). Here, we propose a model that assumes that GRF1&2 bind a DNA replication fork at the interface with NEI, resulting in a Y-structured DNA replication fork model (Figure 5D). Two converging forks from each direction were observed during the efficient crosslink repair in Xenopus egg extracts (14,19,32). Therefore, we suggest a two-fork model, with the mechanism of its recognition accomplished by NEIL3 (Figure 6).
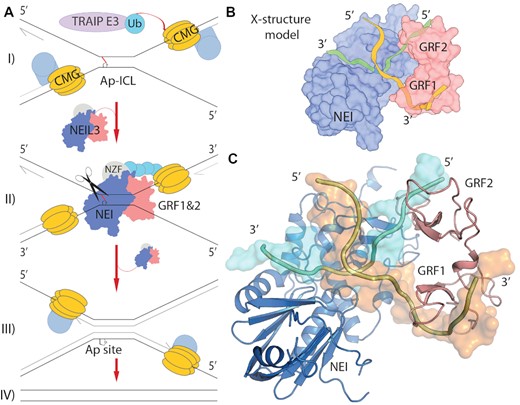
DNA X-structure of replication-coupled DNA repair of the Ap-ICL. (A) A model of replication coupled repair of the Ap-ICL (32). I) Two replication forks stall upon the crosslink. The CMG helicase (yellow) is ubiquitylated by TRAIP1 (light purple). Ubiquitin chains recruit NEIL3 (NEI, GRFs, NZF; blue, red, slate) II) NEIL3 recognition of the double fork, the DNA X-structure at Ap-ICL. III) The Ap-ICL has been unhooked, followed by further steps of the repair. IV) Finally, repaired daughter strands (19,32). (B) Schematics of design of the model. (C) Final model of NEIL3 bound to the X-structure, experimentally driven model of stalled replication fork on the Ap-ICL recognised by NEI (blue) and GRF1&2 (red) domains of NEIL3 glycosylase. Damaged DNA strand containing Ap-site leading to the crosslink (cyan), undamaged strand (orange)
The single-fork model and our NEI domain crystal structure both contain fork-like structures that are in an orientation that does not favour enzymatic catalysis (Figure 5). For efficient catalysis, the fork branching point or the 3′ ssDNA overhang mimicking the replication fork should be oriented in the opposite direction, away from the GRF domains, with the 3′ ssDNA branch of the fork located towards the DNA binding pocket and the residues K82, H96, and R94 (Figures 3 and 4) (56). If we suppose that the AlphaFold prediction of this GRF:NEI interdomain interaction is correct, the GRF would bind one fork to one end of the DNA, and the NEI domain would require a free 3′ ssDNA end for efficient catalysis. To fulfil the requirement of the NEI domain for ssDNA and to avoid competition for the substrate, the second fork must be present, forming a structure in the shape of the letter X (Figure 6). Both NEI and GRF domains bind the ssDNA part of the fork-like structures in a complementary manner. This is due to the preference of GRFs for 5′ ends and the preference of NEI for 3′ ends, while both bind replication forks. Together, biochemical data and predictions indicate that GRF recognizes one fork and the NEI domain recognizes the other fork of the X-structure (Figure 6) (56). While GRFs might bind one or both arms of the fork. An X-structure is commonly drawn in repair models as a prediction of the repair centre, as it was initially based on an electron micrograph from seminal work on ICL repair (14). Additionally, several seminal publications from J. C. Walter's laboratory proposed this model of two stalled replication forks (15,19). Overall, the two-fork model proposed here with NEIL3 glycosylase resembles an X-structure, one fork is recognized by NEI, and the other fork is recognized by GRF. The orientation is such that the Ap site is located in the NEI’s active site pocket (Figure 6). Our structural and biochemical data strongly support that two forks are required for the efficient Ap-ICL repair by NEIL3 glycosylase. Although many models are possible for binding of NEIL3, our data is consistent with a simple model whereby NEIL3 can engage a double-fork structure which might occur with converging replication forks at the ICL. Additional studies are necessary to evaluate it.
X-structure flexibility allows Ap-ICL flipping-out mechanism
If an X-structure is present, the rate of the repair will not be impeded by competition for the fork by the interaction with GRF. Instead, both NEI and GRFs would bind one of the forks in their preferred orientation, with the Ap site in the active site pocket. This would result in efficient crosslink repair.
However, the general mechanism of the Fpg/Nei family of enzymes involves the damaged base flipping outwards into the active site pocket (59). This movement allows for a nucleophilic attack of the N-glycosidic bond of the lesion. Such an action requires dramatic conformational changes within the duplex (60). No flip-out mechanism of the lesion seems possible in the case where dsDNA is crosslinked to another strand. It Is more likely that the entire DNA chain is rotated in the active site pocket.
The dsDNA is relatively rigid on its own, and the addition of an interstrand crosslink would restrict yet another degree of freedom. In the case of an X-structure, all four ssDNA arms could move relatively freely. We predict that once the substrate is recognised by the enzyme, the X-structure arrangement would allow one of the DNA strands to rotate with the crosslink in a ‘flip-out-like’ mechanism, positioning the N-glycosidic bond of the Ap-ICL towards the active site residue. This then allows for a nucleophilic attack, the formation of the Shiff-base, and unhooking of the crosslink, therefore liberating one strand from the other. Then the further steps of Ap-ICL repair can continue.
DATA AVAILABILITY
The structural data generated for this study have been deposited into the Protein Data Bank under the PDB codes 7OMK for GRF, and 7Z5A for NEI glycosylase domains of NEIL3.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Sebastian Zoll for his invaluable advice, and Tamara Jenkins for proofreading the manuscript.
FUNDING
European Regional Development Fund; OP RDE; Project: ‘Chemical biology for drugging undruggable targets (ChemBioDrug)’ [CZ.02.1.01/0.0/0.0/16_019/0000729]; Institute of Organic Chemistry and Biochemistry; Academy of Sciences Czech Republic [RVO: 61388963]. Funding for open access charge: Academy of Sciences Czech Republic [RVO: 61388963].
Conflicts of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments