





Abstract
PLEs (phage-inducible chromosomal island-like elements) are phage parasites integrated into the chromosome of epidemic Vibrio cholerae. In response to infection by its viral host ICP1, PLE excises, replicates and hijacks ICP1 structural components for transduction. Through an unknown mechanism, PLE prevents ICP1 from transitioning to rolling circle replication (RCR), a prerequisite for efficient packaging of the viral genome. Here, we characterize a PLE-encoded nuclease, NixI, that blocks phage development likely by nicking ICP1’s genome as it transitions to RCR. NixI-dependent cleavage sites appear in ICP1’s genome during infection of PLE(+) V. cholerae. Purified NixI demonstrates in vitro nuclease activity specifically for sites in ICP1’s genome and we identify a motif that is necessary for NixI-mediated cleavage. Importantly, NixI is sufficient to limit ICP1 genome replication and eliminate progeny production, representing the most inhibitory PLE-encoded mechanism revealed to date. We identify distant NixI homologs in an expanded family of putative phage parasites in vibrios that lack nucleotide homology to PLEs but nonetheless share genomic synteny with PLEs. More generally, our results reveal a previously unknown mechanism deployed by phage parasites to limit packaging of their viral hosts’ genome and highlight the prominent role of nuclease effectors as weapons in the arms race between antagonizing genomes.
Lay Summary
Parasites of viruses, often referred to as satellites, are found in all domains of life and have been co-opted for host defense across diverse virus-host systems multiple independent times. This study describes the mechanism by which such an element prevents a bacterial virus (a ‘phage’) from otherwise infecting Vibrio cholera and related bacteria. The study is of broad interest to investigators with interests in phage-host interactions and microbial genetics.
INTRODUCTION
Biological conflicts represent a major selective pressure shaping microbial diversity from the level of genes to communities (1,2). Nowhere are these conflicts more antagonistic than between bacteria and the lytic viruses (phages) that infect them. Bacterial genomes contain abundant anti-phage defense systems that function through diverse mechanisms to restrict phages. In addition to the selective pressure imposed by an evolving repertoire of host immunity, phages themselves are hosts to their own subcellular parasites. Phage parasites, often referred to as phage satellites, may appear to be defunct prophages, as they are integrated into specific attachment sites in their bacterial hosts’ genomes (3), yet phage parasites conspicuously lack the full suite of structural genes required for virion assembly (4). In response to proteins encoded by their viral hosts, phage parasites excise (5), replicate (6) and steal structural components from their viral hosts to package their own genome and, upon lysis of the cell, disseminate their genome to naïve neighboring cells in modified virions (7,8). To accomplish this, phage parasites encode mechanisms to inhibit packaging of their viral host’s genome and favor packaging of the parasite genome. These mechanisms must balance inhibition of their viral host’s life cycle with the phage parasites’ requirement for products encoded by the host virus. In this way, phage parasites employ finely tuned inhibitory mechanisms to redirect viral host resources for the parasites’ own use. Much of our understanding of the mechanisms that phage parasites use to subvert their viral hosts comes from the well-characterized phage-inducible chromosomal islands (4,9), of which Staphylococcus aureus pathogenicity islands (SaPIs) are the archetypical members (10). However, diverse families of unrelated phage parasites have likely evolved independently (11), and though often overlooked, such parasites are likely ubiquitous within bacterial genomes (12). Understanding diverse phage satellites can help define mechanistic paradigms of phage parasitism and uncover novel innovations not seen in model systems interrogated thus far.
PLEs (phage-inducible chromosomal island-like elements) are phage parasites unrelated to SaPIs that have proven lucrative for uncovering new mechanisms of parasitism (13–15). Based on current data, PLEs are restricted to toxigenic Vibrio cholerae, the causative agent of the diarrheal disease cholera. PLEs appear to exclusively parasitize the lytic phage ICP1, which is also frequently shed in cholera patient stool samples (16). PLEs have been identified in V. cholerae genomes dating back to the 1940s, and to date 10 distinct PLEs have been identified that share considerable nucleotide identity (17,18). All PLEs are ∼17 kb in size and encode ∼26 open reading frames (ORFs) that largely lack predicted functions or known PFAM domains. Surveillance efforts indicate that PLE1 is the predominant PLE in contemporary V. cholerae isolates from cholera patients in Bangladesh, where cholera is endemic (13,17). For this reason, PLE1 has been the subject of mechanistic studies aimed at understanding PLE activity (14,15,19,20) and will generally be referred to here as ‘PLE’ unless another variant is specified. Although PLE is known to completely abolish ICP1 production (17,21), the mechanisms PLE uses to inhibit ICP1 remain somewhat elusive. PLE employs multiple strategies to inhibit ICP1 progeny production, as no single ORF knockout allows ICP1 to overcome PLE (14). To antagonize PLE in nature, ICP1 isolates encode anti-PLE effectors: either a CRIPSR–Cas system that targets the PLE genome (21) or a site-specific nuclease directed to the PLE origin of replication (22). Such phage-encoded anti-PLE nucleases speak to the fitness costs ICP1 faces when PLE activity is left unimpeded.
Like other known phage parasites such as SaPIs, PLE modifies its viral host’s capsid, constraining ICP1’s capsid to ∼1/3 of its normal size to accommodate the smaller PLE genome and exclude ICP1’s larger ∼125 kb genome (15). Beyond redirecting virion morphogenesis proteins to package its own genome and restrict ICP1, recent work shows that PLE uses a multipronged approach to attack and compete with ICP1 DNA replication, both by reducing total ICP1 genome copy and by inhibiting a key step in ICP1’s genome replication program (19). PLE replication requires the PLE-encoded replication initiation factor RepA and hijacking of ICP1’s replication machinery to drive a nearly ∼1000-fold increase in genome copy during ICP1 infection (17,19). Concurrent with robust levels of PLE replication, PLE reduces ICP1’s DNA replication ∼4-fold, suggesting a link between PLE replication and inhibition of ICP1 replication (19). In addition to restricting ICP1’s genome replication, PLE also alters the mode of ICP1 replication (19). At 8 minutes post-infection (mpi) of a permissive PLE(−) host, ICP1 replication proceeds through bidirectional theta replication (19). Later in infection, between 12 and 16 mpi, ICP1 transitions to rolling circle replication (RCR) (19). RCR is a common mode of replication for viral genomes and plasmids (23–25). Continued rounds of RCR generate linear genome concatemers, an essential step for many viruses because linear concatemers are the DNA substrate recognized by the terminase complex that initiates DNA packaging into capsids (26). When ICP1 infects a PLE(+) V. cholerae host, early bidirectional theta replication proceeds normally; however, ICP1’s transition to RCR is inhibited by an unknown mechanism (19). Importantly, even when PLE is unable to replicate [as in PLE (+)ΔrepA] and total ICP1 replication is partially restored, ICP1’s transition to RCR is still inhibited, suggesting that an additional PLE-encoded factor impedes ICP1’s transition to RCR (19). To our knowledge, PLE is unique among phage parasites in targeting genome replication of its viral host, highlighting this stage of the viral life cycle as a ripe arena for uncovering new mechanisms of interference.
Here, we work to uncover the PLE-encoded mechanism that inhibits ICP1’s transition to RCR. We identify aberrant cut sites in ICP1’s genome that appear during infection of a PLE(+) host and identify the PLE-encoded nuclease necessary for generating these cut sites. This PLE-encoded nuclease, referred to as NixI, is expressed coincident with ICP1’s attempt to transition to RCR in a PLE(+) host. We show that NixI inhibits ICP1 genome replication, nearly abolishing virion production. We identify a sequence motif that is necessary for NixI-mediated cleavage and provide evidence that PLE not only is protected from NixI-mediated cleavage, but actually requires NixI-mediated cleavage of ICP1 to achieve wild-type levels of PLE replication. We further identify homologs of the PLE-encoded nuclease in what we consider to be an expanded family of putative phage parasites in nontoxigenic V. cholerae and other non-cholera Vibrio species.
MATERIALS AND METHODS
Strains and media
A complete list of strains used in this study is available in Supplementary Table S1. Vibrio cholerae strains used in this study are derivatives of E7946. ICP1_2006_E engineered to lack CRISPR–Cas (ΔCRISPR, Δcas2–3) was used throughout this study and is referred to as ICP1 for simplicity (20). Routine culturing of V. cholerae was done in lysogeny broth (LB) at 37°C with aeration. Cells grown in Terrific Broth (TB) were used for protein purification. Antibiotics were supplemented as appropriate at the following concentrations: 50 μg/ml kanamycin for maintenance of the pSUMO vector, 100 μg/ml spectinomycin, and 1.25 or 2.5 μg/ml chloramphenicol for broth or plate conditions, respectively.
Cloning
Plasmids to express nixI and nixIN95A are pMMB67EH derivative plasmids with a theophylline-inducible riboswitch (riboswitch E) as described previously (20), and were constructed using Gibson assembly. For expression of nixI homologs from V. cholerae YBA_A06 (WP_057552372) and Vibrio parahaemolyticus S042 (WP_029837635), the genes were commercially synthesized by GenScript and cloned into the pMMB67EH derivative. High-copy pUC19 derivative plasmids were used as DNA substrates for nuclease assays. Deletions in V. cholerae were generated through natural transformation as described previously (27). Naturally competent V. cholerae were transformed with PCR products generated by splicing by overlap extension PCR containing arms of homology flanking frt recombination sites as described previously (27). All deletions and constructs were verified by Sanger sequencing.
Protein purification
Protein purifications were carried out in Buffer A (150 mM Tris–Cl, pH 7.8, 50 mM NaCl, 2 mM BME, 10 mM NaCl) unless otherwise indicated or modified. His-SUMO tagged NixIN95A was purified from Escherichia coli strain BL21. Catalytically active His-SUMO tagged NixI was purified from E. coli BL21 pLysS background to reduce leaky expression of nixI, which is toxic to E. coli. For each E. coli expression strain, a 100 ml starter culture in TB was grown overnight at 37°C on a shaking incubator at 250 rpm with the appropriate antibiotic(s): kanamycin at 50 μg/ml to maintain the pSUMO plasmid and chloramphenicol at 25 μg/ml to maintain the pLysS plasmid. The next day, four 1L baffled flasks of TB with appropriate antibiotic(s) were each inoculated with 20 ml of overnight starter culture, and grown at 37°C with shaking. When the A600 reached 0.5–0.7, the temperature of the incubator was reduced to 18°C, 0.5 mM IPTG was added to induce protein expression and cultures were incubated for 20 h with shaking. Cells were harvested by centrifugation at 4000 × g for 15 min and then resuspended in 25 ml of lysis buffer: 150 mM Tris–HCl, pH 8, 50 mM NaCl, 2 mM BME, 1× Pierce phenylmethylsulfonyl fluoride protease inhibitors + 0.5% Triton X and 1% glycerol. The cell suspension was sonicated for a total of 5 min as cycles of 10 s ‘ON’ pulses with 20 s ‘OFF’. After sonication, cell debris was removed by centrifugation at 20 000 × g for 40 min, and the supernatant was filtered using GE 0.2 μm regenerated cellulose membranes. Lysate was bound to a GE His-Trap and pumped through using a peristaltic pump. A high-salt wash including 2 M NaCl was applied to remove DNA. The His-SUMO tagged proteins were eluted in 250 mM imidazole in 150 mM Tris–Cl, pH 7.8, 50 mM NaCl and 2 mM BME. The eluted protein was then dialyzed against Buffer A + 5% glycerol overnight using SnakeSkin (Thermo Scientific) at 10 kDa MWCO dialysis membrane. Dialysis was performed in concert with cleavage of the His-SUMO tag using the SUMO protease (Sigma-Aldrich) at 20 U/mg of protein. To remove the SUMO protease and the cleaved tag, the protein was reapplied to Ni-NTA resin and a batch purification was performed, where the cleaved protein of interest remained unbound in the buffer. Protein concentration was measured using a BioPhotometer D30 (Eppendorf). Additional purification of the Ni-affinity purified NixI and NixIN95A was carried out using a Heparin column, equilibrated with Buffer A except with a higher NaCl concentration of 150 mM. The column was washed with Buffer A + 150 mM NaCl until no protein was detected in the flow through, then eluted with 2 M NaCl in Buffer A and dialyzed against Buffer A overnight.
Electrophoretic mobility shift assays
NixIN95A was incubated with 100 bp probes amplified from ICP1, internal to gp156 and from pGp156* with the putative recognition motif mutated from GTAATCTT to TGTATAGT. Primer sequences are available in Supplementary Table S2. Fifteen nanomolar DNA probe was added for each 20 μl reaction, and incubated with varying concentrations of NixIN95A. Reactions were carried out for 30 min at 30°C and then chilled at 4°C. Reactions were run on an 8% polyacrylamide gel, pre-run at 120 V for 60 min. Electrophoretic mobility shift assay (EMSA) reactions were run at 140 V for 40 min at 4°C, and then visualized by staining with GelRed.
Nuclease assays
Nuclease assays with NixI and NixIN95A were carried out in 150 mM Tris–Cl, pH 7.8, 50 mM NaCl, 1 mM DTT and 10 mM MgCl2. Plasmid DNA substrates were added at 100 ng per 20 μl reaction, with NixI at varying concentrations as indicated. Reactions were incubated at 30°C for 15 min, and then 1 μg/ml of Proteinase K was added to each reaction to digest bound protein and incubated at 37°C for 30 min. The entire reaction volume was then run on a 0.8% agarose gel stained with GelRed. To generate cleaved controls, 100 ng of vector was digested for 1 h at 37°C with 10 U of BamHI-HF or EcoRI-HF (NEB) to create a single double-stranded cut or 10 U Nb.BtsI (NEB) to create a single nick, and then the enzymes heat inactivated at 80°C for 20 min.
Densitometry measurements were performed using ImageJ to quantify nuclease activity on plasmid substrates. Percent cleaved was calculated as the intensity of the nicked + linearized plasmid divided by the total band intensity in each lane. Densitometry measurements reported here are the average of three replicate nuclease assays conducted on different days.
Sample preparation for DNA sequencing and read mapping
ICP1-infected PLE(+)ΔnixI V. cholerae samples were prepared in parallel with previously analyzed PLE(+) and PLE(−) V. cholerae samples, as described previously (19). Briefly, 2 ml LB cultures of each V. cholerae strain were infected at A600 = 0.3 with ICP1 at a multiplicity of infection (MOI) of 1, and aliquots taken at given time points from which total DNA was extracted for sequencing. The pellets were washed with 1 ml ice-cold phosphate-buffered saline, and total DNA was extracted using the QIAGEN DNeasy Blood and Tissue Kit. Library preparation was done using the NEBNext Ultra II FS DNA Library Prep Kit, and sequenced using Illumina HiSeq4000 at University of California Berkeley QB3 Core Facility. Illumina sequencing reads for each sample and time point were mapped as before (19). Briefly, reads were mapped onto ICP1 reference genome using Bowtie 2 v2.3.4.1 using the default settings except for ‘-end-to-end’ and ‘-very-sensitive’. Read coverage was normalized by the total number of reads that mapped to the reference.
In vivo cleavage site identification
Illumina reads from samples at 16 mpi were analyzed for indications of DNA cleavage from PLE(−), PLE(+) and PLE(+)ΔnixI V. cholerae. Cleavage ratios in ICP1’s genome were calculated following an approach adapted from Culviner and Laub (28). To look for putative cleavage sites, the log2 ratio of read coverage averaged across three biological replicates at each nucleotide position in ICP1 was calculated for the following comparisons: PLE(+):PLE(−) and PLE(+)ΔnixI:PLE(−). The difference in log2 coverage was determined for each nucleotide position by subtracting the coverage at the nucleotide before and after each position to determine coverage decrease in both a 5′ to 3′ and a 3′ to 5′ orientation. These differences were then ranked, identifying 63 putative sites with coverage change >log2(0.026) (Supplementary Figure S1A).
Efficiency of center of infection assays
Cultures of V. cholerae were grown in LB supplemented with chloramphenicol to maintain the plasmid expressing NixI, NixIN95A or the empty vector control. At an A600 = 0.2, expression from the plasmid was induced with 1 mM IPTG + 1.5 mM theophylline. At A600 = 0.3, ∼15 min post-induction, 2 ml cultures were infected at MOI = 0.1 and incubated for 7 min to allow adsorption of ICP1. The infected culture was then diluted 1:2500, 1:25 000 and 1:250 000 into pre-warmed LB supplemented with chloramphenicol and inducer to a final volume of 2 ml. Immediately after dilution, 500 μl of 1:2500 diluted sample was removed to quantify the input phage. To quantify phage produced, 500 μl of each dilution was collected 40 min after the initial infection. To all samples, 20 μl of chloroform was added to each sample, and debris and chloroform were removed by centrifugation at 5000 × g for 15 min. Supernatant was used to quantify phage by a plaque assay on a permissive PLE(−) host. Efficiency of center of infection (ECOI) was quantified as the ratio between the number of phages produced per milliliter minus the number of input phage, from the strains of interest compared to the control culture: either an empty vector control when assessing restriction by nixI or wild-type PLE.
Quantification of genome replication by real-time quantitative PCR
ICP1 and PLE replication was quantified by qPCR as described previously, where strains used for quantifying ICP1 replication were infected at MOI = 0.1 and strains used for quantifying PLE replication infected at MOI = 2.5 (17). For strains expressing plasmid constructs, a 2 ml culture of interest grown with chloramphenicol was induced with 1 mM IPTG + 1.5 mM theophylline at A600 = 0.2, ∼15 min before infection when A600 = 0.3. A T = 0 aliquot was immediately boiled to stop DNA replication. Cultures were incubated for 20 min at 37°C with aeration, and then a 20 μl aliquot was boiled. DNA replication was determined by quantifying the fold change in replication at T = 20 compared to genome copy at T = 0. To determine the fold change, the Cq value was compared to that of a standard curve of known concentrations of ICP1 or PLE genomic DNA. ICP1’s genome was amplified with primers Zac68/69 and PLE replication with primers Zac14/15 using iQ SYBR Green Supermix (Bio-Rad) and the CFX Connect Real-Time PCR Detection System (Bio-Rad). Replication efficiency was compared to a PLE(−) permissive strain harboring an empty vector and similarly grown in the presence of chloramphenicol and inducer.
Identification of an expanded family of PLEs
An HHblits search with PLE1’s NixI protein sequence returned significant homology across the protein sequence to a protein found in non-O1 V. cholerae (WP_057552372). The identical protein was found in a total of 10 non-O1 V. cholerae isolates isolated in Massachusetts. Analysis of the 10 kb regions flanking the NixI homolog showed shared synteny with PLEs; specifically, these regions are characterized by a 5′ integrase, a total sequence length of ∼17 kb and an ORF-less region between divergently transcribed operons. To identify additional putative phage satellites, a PSI-BLAST search excluding V. cholerae was performed with NixI from V. cholerae’s PLE1 as well as the NixI homolog from nontoxigenic V. cholerae YB2_A06 (WP_057552367). We considered proteins potential homologs of NixI if they shared >25% amino acid identity across >85% of the protein length (Supplementary Table S3). To ensure that annotations were called correctly, the putative phage satellites from V. parahaemolyticus s042 and V. diazotrophicus 60.18M were verified with RAST.
Statistical and visual analyses
Statistics and graphs were generated using Prism, version 9.0.2. Gene maps were made using the gggenes package in R, version 4.0.0. Protein alignments and phylogenetic trees were made using MUSCLE within Jalview, version 2.11.0. Consensus motifs of NixI cleavage sites were produced by the WebLogo server (https://weblogo.berkeley.edu/logo.cgi).
RESULTS
ICP1’s genome is cleaved during infection of PLE(+) V. cholerae
While assessing ICP1 replication profiles by mapping Illumina read coverage for each nucleotide position (referred to from here on as DNA-seq) (19), we noted regions in ICP1’s genome where read coverage dropped precipitously during infection of PLE (+) but not PLE(−) V. cholerae. These coverage drops suggested potential sites of nuclease cleavage. In order to characterize these putative cut sites more systematically, we drew on a previously published method that identified cleaved sequences in transcripts from RNA-seq data in order to assess activity of an endoribonuclease toxin (28). To generate cleavage maps across ICP1’s genome, we calculated the coverage ratio of ICP1 reads 16 mpi of PLE(+) relative to PLE(−) V. cholerae. This time point is late in the infection cycle, at a time when ICP1 and PLE are both replicating (19) and before the onset of lysis ∼20 mpi of PLE(+) V. cholerae (14,17) (Figure 1A). We identified a total of 63 candidate cut sites in ICP1 whose appearance varied ± PLE with log2 coverage drops >0.026, with several coverage drops clustering in the same region of ICP1’s genome (see the ‘Materials and Methods’ section; Supplementary Figure S1A). We identified coverage drops across ICP1’s putative packaging (pac) site that were present during infection of a PLE(−) host but were reduced in a PLE(+) host (Supplementary Figure S1A and B). The pac site is recognized and cleaved by the terminase complex to initiate genome packaging, and a lack of cutting in a PLE(+) host is consistent with PLE-mediated inhibition of ICP1 genome packaging. The remaining putative cut sites were coverage drops that appeared in ICP1’s genome when infecting a PLE(+) host but not a PLE(−) host, suggesting ICP1’s genome is aberrantly cut when infecting a PLE(+) host. Two sites in ICP1’s genome, located in gp156 and gp121, showed the most striking drops in coverage, while additional sites showed more subtle drops in coverage, including a second site in gp121 located ∼60 bp 5′ of the dominant coverage drop (Figure 1B and C, and Supplementary Figure S1C). We therefore chose to focus on the sites in gp156 and gp121 in greater detail. We identified a shared motif based on the two sites in gp121 and single site in gp156 of GWAAWC^TT (W = A or T), where the ‘^’ symbol denotes the position of potential cleavage (Figure 1D). Where multiple coverage drops occurred within a 100 bp region (as occurred for many regions; Supplementary Figure S1A), the sequence at the most dominant coverage drop was used as input to generate the consensus motif for the PLE-dependent putative cut sites for a total of 17 sites. These sites generated a less stringent motif GNAANC^TT that occurs 99 times in ICP1, of which 16 sites displayed a coverage drop at the 16 min time point (Supplementary Figure S1D). The sites in ICP1’s genome that have the motif but were not cleaved may be protected due to protein occupancy or were perhaps missed because this assay was performed at just one time point. The analysis of read coverage across ICP1’s genome suggests a limited number of pronounced coverage drops that appear only when infecting a PLE(+) host, consistent with nucleolytic cleavage at these regions.
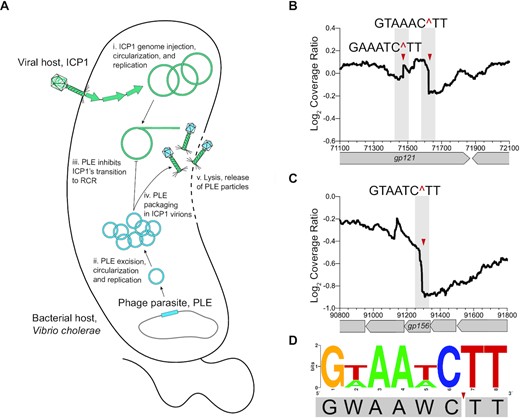
ICP1’s genome exhibits signs of cleavage when infecting PLE(+) V. cholerae. (A) Model of ICP1 infection in a PLE(+) V. cholerae: The viral host ICP1 injects its genome, triggering PLE excision and gene expression. Both ICP1 and PLE replicate their genomes initially, yet midway through infection PLE prevents a further increase in ICP1 genome copy and inhibits ICP1’s transition to RCR. PLE hijacks ICP1 structural proteins for packaging, and no ICP1 progeny is produced. Vibrio cholerae lyses ∼20 mpi releasing only PLE transducing particles. (B) Cleavage ratio PLE(+)/PLE(−) across ICP1’s genome position (x-axis) showing the most prominent putative cut site within ICP1’s gp121, with the two putative cut sites indicated by the red triangles and the nucleotides flanking the cut region above. The cleavage ratio is calculated as the average coverage of three biological replicates. (C) As in (B), but displaying the other most prominent cut region within ICP1’s gp156. Additional cut sites are summarized in Supplementary Figure S1A. (D) WebLogo displaying the consensus motif for cut sites in gp121 and gp156 with the cut site indicated by the red triangle.
ICP1’s genome is targeted by a PLE-encoded nicking endonuclease, NixI
We hypothesized that the putative cleavage sites in ICP1 apparent during infection of PLE(+) V. cholerae were caused by a PLE-encoded nuclease. However, we lacked a candidate nuclease, as the majority of PLE-encoded ORFs have no primary sequence similarity to any annotated genes. Using the more sensitive HHpred, which searches for remote homology based on predicted structural domains, we identified a single PLE-encoded ORF, NixI (WP_032468646), that shares similarity with the intron-encoded homing endonuclease I-HmuI (probability score = 97.1%, e-value = 0.0058) (29). Despite sharing only 10% amino acid identity across the protein, NixI shares an HNH/N catalytic motif with I-HmuI (Figure 2A). No other proteins shared significant homology with NixI by HHpred, which only compares proteins with solved structures. I-HmuI is the only crystallized protein from a family of closely related nicking HNH/N endonucleases (29,30), and a MUSCLE alignment including these proteins identified several conserved residues at the catalytic core, including an experimentally validated catalytic asparagine from I-HmuI at position 95 in NixI (29) (Figure 2A). To address whether nixI may encode for a nuclease, we cloned nixI or a mutated nixIN95A allele, which we postulated would be catalytically inactive, under an inducible promoter in V. cholerae and plated for cell viability with and without inducer. Consistent with nixI functioning as a nuclease, induction completely abolished V. cholerae colony formation, as the predicted recognition motif we identified in ICP1’s genome (GWAAWC^TT, Figure 1D) is found 638 times in V. cholerae’s chromosomes. In contrast, induction of NixIN95A had no impact on colony formation (Figure 2B), indicating that toxicity of NixI is associated with nucleolytic activity.
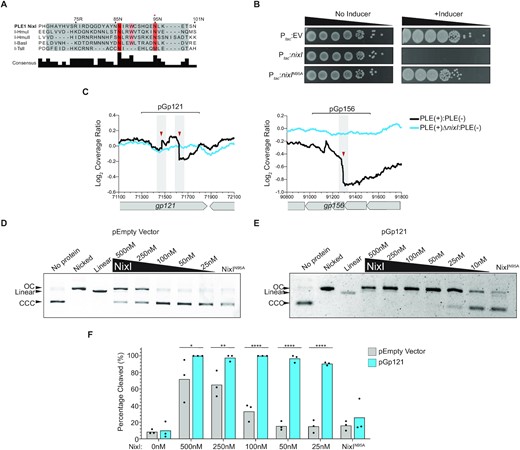
PLE encodes a nuclease, NixI, that cuts ICP1’s genome in vivo and in vitro. (A) MUSCLE alignment of the catalytic core of phage intron-encoded homing endonucleases including I-HmuI, where I-HmuI’s active site domain is highlighted in gray and residues that share >70% identity in orange/red. The red asterisk above 95N indicates the experimentally validated catalytic asparagine residue from I-HmuI (29). (B) Ten-fold dilution series of mid-log grown V. cholerae containing plasmids for inducible expression of nixI, nixI with the putative catalytic asparagine mutated to an alanine (N95A) or an empty vector control, spotted on plates +/− inducer. (C) Coverage ratios PLE(+)/PLE(−) and PLE(+)ΔnixI /PLE(−) across ICP1’s putative cleaved genes gp121 and gp156 as indicated in Figure 1. Brackets above indicate the regions cloned into plasmids and used as a DNA substrate for in vitro assays. (D) In vitro cutting by purified NixI at the concentrations indicated above the gel or 500 nM NixIN95A of empty vector plasmid, showing linearized or nicked plasmid controls. (E) As in (D), but showing cutting of pGp121. For (D) and (E), CCC = covalently closed circular DNA and OC = open circular DNA. (F) Densitometry measurements calculating the percent of substrate cleaved at different concentrations of NixI (concentrations indicated on the x-axis) or 500 nM NixIN95A of the empty vector plasmid or pGp121. Bar height is the mean and dots are measurements from independent assays. Asterisks indicate statistically significant differences between cleavage of the empty vector plasmid and pGp121 (*P < 0.0278, **P < 0.0079, ****P < 0.0001, where no asterisks are shown no difference was detected, two-way ANOVA, Sidak correction).
Having identified a putative PLE-encoded nuclease, we next asked whether nixI is required for the appearance of the putative cleavage sites in ICP1’s genome during infection of PLE(+) V. cholerae. We infected PLE ΔnixI V. cholerae with ICP1 and again performed DNA-seq to evaluate changes in cleavage ratios of PLE(+)ΔnixI relative to V. cholerae PLE(−). Strikingly, we observed that all coverage drops in ICP1’s genome seen during a PLE(+) infection were abolished during infection of a PLE(+)ΔnixI host (Figure 2C and Supplementary Figure S1C), indicating that nixI is necessary for cleavage of ICP1’s genome in vivo. However, cleavage at ICP1’s pac site was still inhibited in a PLE(+)ΔnixI host, indicating a PLE-encoded mechanism of inhibiting ICP1’s DNA packaging independent of nixI (Supplementary Figure S1B).
To directly evaluate NixI’s nuclease activity, we purified NixI and NixIN95A (Supplementary Figure S2) to conduct in vitro nuclease assays. Many nucleases require specific DNA topology such as negative supercoiling for activity, so we first assayed whether NixI had nucleolytic activity on plasmid substrates, allowing for visualization of both nicking, as evidenced by a loss of supercoiled plasmid, and double-stranded breaks. We observed nuclease activity on an empty vector plasmid at high protein concentrations of NixI (≥250 nM) after a 15-min incubation period (Figure 2D). We postulated that NixI exhibits a preference for cleavage of sites in ICP1’s genome that we identified coverage drops in during infection of a PLE(+) host (Figure 2C). To test this, a 500 bp region flanking the putative cut sites in gp121 was cloned onto the same plasmid and used as a substrate for NixI cleavage assays. NixI nucleolytic activity was dramatically enhanced for a plasmid template containing the region from gp121, allowing us to observe cleavage with only 25 nM NixI (Figure 2E). Quantification of results from independent assays showed that cleavage of the plasmid harboring the region from gp121 was significantly greater than cutting of an empty vector at all concentrations tested (Figure 2F). Importantly, nuclease activity was largely eliminated for NixIN95A (Figure 2D–F) even at a high protein concentration of 500 nM, supporting our prediction from remote homology that NixI possesses an HNH/N catalytic motif (Figure 2A). To determine whether NixI is a nickase or double-stranded cutter, we compared cut products to plasmid controls that had been nicked or linearized using commercially available endonucleases with known sequence specificity. NixI generated one or more nicks in the DNA substrate but did not cause double-stranded cuts (Figure 2D and E). We next assayed whether NixI cleaves PCR products. Even at high protein concentrations, we observed little nucleolytic activity of PCR products, as visualized on native or denaturing agarose gels (Supplementary Figure S3). Together, these data show that PLE encodes a nicking endonuclease, NixI, that is necessary for cleavage of ICP1’s genome in vivo and that shows in vitro specificity for sequences found in ICP1’s genome. We propose that NixI is a PLE-encoded nuclease effector that targets ICP1, functioning as part of PLE’s strategy to antagonize its viral host.
NixI’s nuclease activity is dependent on the consensus motif GWAAWCTT
Having established that PLE-encoded NixI is a nickase with enhanced activity for a region from ICP1 that was cleaved in vivo, we sought to address whether the consensus motif (Figure 1C) is necessary for NixI activity in vitro. We chose to focus on the region from gp156 because it contains a single instance of the consensus motif, while gp121 contains two motifs. We made nucleotide substitutions to alter the motif from GTAATCTT to TGTATAGT, creating the plasmid pGp156*, which is identical to the plasmid harboring the region from gp156 (pGp156) except for the changes to the 8 bp motif. We compared NixI’s ability to cleave wild-type pGp156 and pGp156* in side-by-side nuclease assays and quantified the amount of substrate cleaved (Figure 3A). NixI nuclease activity was significantly reduced for substrate with the mutated motif compared to the wild-type sequence at NixI concentrations of 25 and 50 nM (Figure 3A). We next asked whether this loss of nuclease activity could be explained by NixI’s hampered ability to bind sequences lacking the consensus motif. We performed EMSAs with probes internal to gp156 that have the wild-type motif or the mutant derivative as in pGp156*. We found that DNA binding by NixI was dramatically reduced for substrate with the mutated motif (Figure 3B). Quantification of the amount of bound probe across replicate EMSAs showed a significant reduction in NixI binding to the probe with the mutated motif compared to the wild-type probe at all protein concentrations tested (Figure 3C). Together, these data show that the motif GWAAWCTT is important for NixI DNA binding and cleavage.
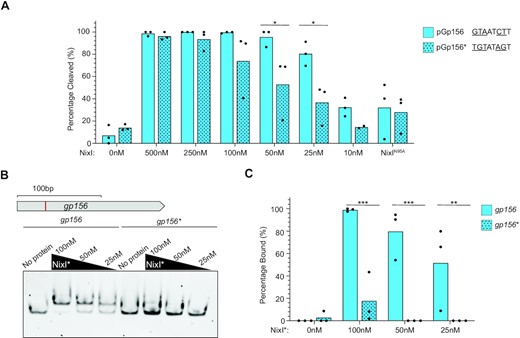
The consensus motif is required for NixI-mediated cutting and DNA binding. (A) Densitometry measurements calculating the percent of substrate cleaved from in vitro nuclease assays at different concentrations of NixI (concentrations indicated on the x-axis) or 500 nM NixIN95A of pGp156 (as shown in Figure 2C) or pGp156*, which are identical vectors except for the nucleotide substitutions made in the consensus motif indicated to the right. Bar height is the mean and dots are measurements from independent assays. Asterisks indicate statistically significant differences between cleavage of pGp156 and pGp156* (*P < 0.0332, where no asterisks are shown no difference was detected, two-way ANOVA, Sidak correction). (B) EMSAs using 100 bp probes internal to gp156 containing the GTAATCTT motif (top schematic, showing identified cut site in red) or the gp156* mutant derivative incubated with NixIN95A (NixI*) at the concentration indicated above the gel. (C) Densitometry measurements from three independent EMSAs showing NixIN95A (NixI*) binding to the wild-type or mutated gp156 probe. Bar height is the mean and dots are measurements from independent assays. Asterisks indicate statistically significant differences between binding of gp156 and gp156* (**P < 0.0021, ***P < 0.0002, where no asterisks are shown no difference was detected, two-tailed t-test, Sidak correction).
NixI inhibits ICP1 progeny production and replication
Having shown that NixI is necessary and sufficient to generate the cleavage patterns observed in ICP1’s genome during infection of a PLE(+) host, we next asked whether NixI activity inhibits ICP1’s life cycle. Previous work has identified PLE-encoded mechanisms that effect ICP1 gene expression (15) and that collapse ICP1-induced lysis inhibition (14); however, none of these mechanisms alone are sufficient to prevent ICP1 progeny production, showing that there are additional mechanisms of PLE-mediated ICP1 interference. We anticipated that NixI cleavage of ICP1’s genome would have negative consequences for ICP1’s capacity to produce phage progeny. Because of NixI’s toxicity to V. cholerae (Figure 2B), we were unable to assess plaque formation in a strain expressing NixI. Therefore, we assayed nixI-mediated inhibition with an ECOI assay, which measures ICP1 virions produced from a single round of infection of V. cholerae by collecting virions produced from an infected host of interest and quantifying the progeny phage produced on a permissive host using a plaque assay (Figure 4A). Vibrio cholerae expressing nixI, but not nixIN95A, reduced ICP1 progeny production 100-fold (Figure 4B). However, nixI was not required for PLE-mediated inhibition of ICP1, as PLE(+)ΔnixI was still inhibitory to ICP1 (Figure 4C). This corroborates previous work that showed no single PLE-encoded ORF, including nixI, is necessary for inhibition of ICP1 by the plaque assay (14). NixI, however, is sufficient to limit ICP1 progeny production in a single round of infection, the first PLE-encoded product shown to recapitulate the block in progeny production from wild-type PLE (Figure 4B and C).
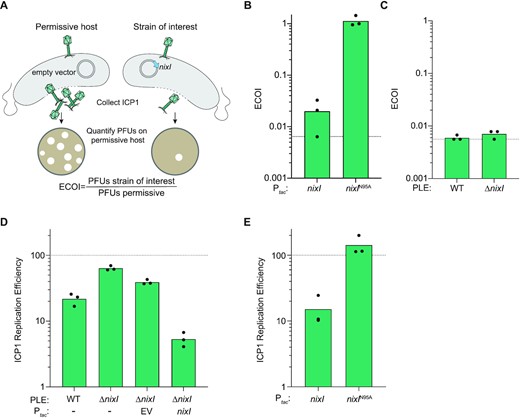
NixI is necessary and sufficient to inhibit ICP1 genome replication and sufficient to inhibit production of ICP1 progeny. (A) Schematic of the ECOI assay, where ICP1 produced by a single round of infection in a strain of interest (here, expressing nixI, on the right) is quantified as a ratio of the number of phages produced by a permissive host (left). (B) ECOI of ICP1 on PLE(−) V. cholerae expressing nixI or nixIN95A from a plasmid, relative to phage progeny produced from an induced empty vector control. The dashed line indicates where the progeny phage output is equal to the input phage. (C) ECOI of ICP1 in PLE(+) V. cholerae or PLE(+)ΔnixI relative to phage progeny produced from a permissive control. The dashed line indicates where the progeny phage output is equal to the input phage. (D) ICP1 replication efficiency in the PLE(+) strains indicated relative to infection of permissive PLE(−) V. cholerae harboring an empty vector control (dashed line) 20 mpi as assessed by qPCR. (E) ICP1 replication efficiency in PLE(−) V. cholerae pre-induced to express the gene indicated relative to infection of permissive PLE(−) V. cholerae harboring an empty vector control (dashed line) 20 mpi as assessed by qPCR. For (B)–(E), bar height displays mean and dots indicate independent biological replicates.
We anticipated that nixI inhibits ICP1 progeny production by impairing phage genome replication, which is a hallmark of PLE activity (19). We therefore measured ICP1 genome replication by qPCR in a wild-type PLE(+) host compared to a ΔnixI mutant derivative. As was previously observed (19), PLE reduced ICP1 genome copy to ∼20% of the levels observed in a permissive infection. Deletion of nixI restored ICP1 replication to ∼65% of the levels of replication observed in a permissive host (Figure 4D). Complementation of nixI in trans reduced ICP1 replication to a greater extent than wild-type PLE (Figure 4D). This is likely because endogenous nixI is not expressed until midway through infection, between 8 and 12 mpi (31). At this point in infection, ICP1’s has undergone several rounds of theta replication and is transitioning to RCR, while PLE replication begins to accelerate (19). Pre-induction of nixI as was done here (Figure 4D) would likely interfere with all stages of ICP1 replication and could therefore exceed PLE’s impact on ICP1 DNA replication. To assess whether NixI directly impacts ICP1’s genome replication in the absence of other PLE-encoded products, we also evaluated ICP1 genome replication in a PLE(−) background. As seen with nixI complementation in trans, ectopic expression of nixI was sufficient to inhibit ICP1 replication to 10% that of a permissive infection, again greater inhibition than is observed in the context of wild-type PLE (Figure 4E). Induction of the catalytically dead nixIN95A did not inhibit ICP1 replication (Figure 4E), demonstrating that catalytic activity of NixI is necessary to inhibit ICP1 replication. Together, these data demonstrate that nixI is a potent inhibitor of ICP1 replication, resulting in decreased virion production.
PLE is protected from cleavage through a sequence-independent mechanism
Organisms that use nuclease effectors as weapons for intergenome conflict must ensure that the nuclease can discriminate between self and non-self DNA. We have shown that PLE encodes a nickase that cleaves ICP1’s genome leading to a decrease in replication and phage progeny production. Despite NixI’s ability to restrict phage genome replication, PLE replicates to high copy number and transduces to new hosts, suggesting PLE is resistant to NixI activity. Indeed, genome integrity is important for PLE’s life cycle since ICP1 can antagonize PLE by employing its own nuclease effectors, namely Cas2–3 and Odn, which limit PLE replication and transduction (21,22). We therefore anticipated that PLE would be protected from NixI-mediated cleavage rather than sacrifice its own genome to thwart ICP1.
To address how PLE is protected from NixI activity, we first asked whether PLE contains the consensus motif, which we demonstrated is an important determinant of NixI activity (Figure 3). We found that PLE1, which has been the focus of our study, does not contain the consensus motif, suggesting that PLE may simply avoid NixI-mediated cleavage based on sequence alone. To corroborate this finding, we next looked at whether PLEs encoding identical NixI proteins also lack the consensus motif. Surprisingly, both PLE2 and PLE10, which have amino acid identical copies of PLE1’s NixI, contain three and four instances of the motif, respectively (Figure 5A). This prompted us to test whether the motif alone is sufficient to direct NixI-mediated cutting. We cloned the 8 bp motif GTAATCTT from gp156 into the plasmid (generating pMotif) and assessed cleavage of this substrate in vitro. Cutting of pMotif was greatly reduced compared to pGp156, which has additional sequence flanking the motif. For example, only 15% of pMotif was nicked compared to nearly 95% of pGp156 at 50 nM of NixI (Figures 3A and 5B). Overall, cleavage of pMotif was comparable to cutting of the empty vector at all concentrations tested (Figures 2F and 5B). This demonstrates that although the GWAAWCTT motif is necessary for NixI cleavage and binding (Figure 3), it is not sufficient to direct NixI cutting, suggesting that additional unidentified sequence features flanking the motif may be required.
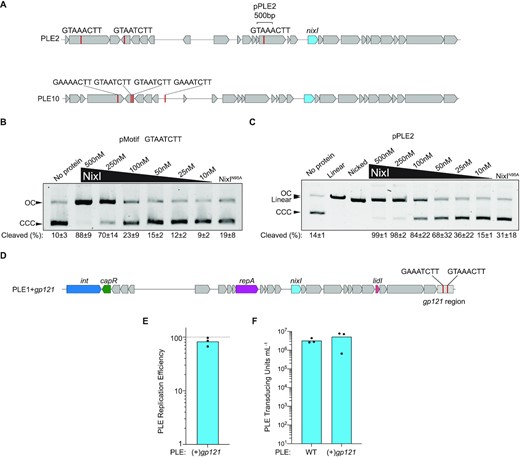
PLE is protected from NixI-mediated cleavage independent of sequence identity. (A) Gene maps of PLE2 and PLE10 showing instances of the consensus motif as red lines and the region cloned to create pPLE2, and the gene that encodes for identical amino acid identical NixI in blue. (B) In vitro cutting by NixI (concentrations indicated above the gel) or 500 nM NixIN95A of pMotif that contains only the 8 bp motif from gp156 (indicated above the gel) displaying the percent of cleaved substrate as the average of three independent assays and the standard deviation below the gel. CCC = covalently closed circular DNA; OC = open circular DNA. (C) As in (B), but showing cutting of pPLE2 and displaying linearized and nicked plasmid controls. (D) Gene map of engineered PLE1 where the region from ICP1’s gp121 shown to be cut in vivo and in vitro was cloned at the 3′ end of the integrated PLE. (E) Replication efficiency of PLE1 possessing ICP1’s gp121 region relative to the wild-type PLE (dashed line) 20 mpi as assessed by qPCR. (F) Transduction of wild-type PLE1 and PLE1 with the gp121 region. For (E) and (F), bar height is the mean and dots are measurements from independent assays.
We next asked whether regions from PLEs that contain the motif avoid NixI activity because they lack additional sequence features required for cutting. Toward this, we cloned a 500 bp region flanking one of the identified motifs from PLE2 into the plasmid (referred to as pPLE2, Figure 5A) and assayed NixI cutting in vitro (Figure 5C). Cleavage of pPLE2 approximated cleavage of pGp156 at protein concentrations ≥50 nM, suggesting that this region from PLE2 has the sequence features that are sufficient for NixI cutting in vitro.
Having determined that regions from PLE2 would not be protected from NixI activity due to sequence identity alone, we sought to reconcile this finding with previous studies showing that PLE2 achieves robust replication and transduction (17) and expresses NixI during ICP1 infection (31). We therefore asked whether we could make PLE1 sensitive to NixI activity by adding to it the region from gp121 that contains two motifs and exhibited robust cleavage in vivo and in vitro (Figure 2), predicting PLE1 with the gp121 region (‘PLE + gp121’, Figure 5D) would suffer a replication defect as we observed for ICP1. Strikingly, PLE harboring the cut region from gp121 was able to replicate as well as wild-type PLE, as there was no significant difference between PLE replication with and without the cut sites (P = 0.1767, paired t-test; Figure 5E). Given that qPCR to assess PLE replication amplifies only a small region of the genome that is 4 kb away from the introduced gp121 sites, we also evaluated whether introduction of the gp121 region manifested as a transduction defect. Consistent with our finding of no evidence of a replication defect, PLE + gp121 transduced as well as wild-type PLE (Figure 5F). These data show that PLE is protected from NixI cutting even when it harbors sequence that is robustly cut in vitro and cut in the context of ICP1’s genome in vivo. Collectively, these data show that PLEs have not evolved to avoid NixI activity on the basis of sequence alone, but suggest that PLEs are protected from NixI-mediated cleavage through a yet unidentified mechanism.
NixI promotes PLE replication
PLE replication relies on hijacking replication resources from ICP1, leading us to hypothesize that PLE may limit ICP1 replication to successfully co-opt replication machinery for its own replication. This suggested that NixI-mediated cleavage of ICP1’s genome and its subsequent limitation of ICP1 replication may be necessary for PLE to replicate efficiently. We therefore assayed PLE replication efficiency of PLE ΔnixI relative to wild-type PLE by qPCR (Figure 6A). During ICP1 infection, we observed that PLE replication is reduced to 25% of wild type in the absence of nixI (Figure 6A). Complementation of nixI in trans restored PLE replication to wild-type levels. Importantly, these data help us to contextualize the effect of nixI expression on ICP1 replication. Because nixI induction is toxic to V. cholerae (Figure 2B), we initially had concerns that the observed reduction in ICP1 replication in the presence of nixI (Figure 4E) was the result of indirect effects on V. cholerae, rendering the host unable to support phage infection and DNA replication in general. However, PLE replication is increased by nixI expression in trans (Figure 6A), demonstrating that nixI-mediated inhibition of ICP1 replication is specific and not a pleiotropic effect of cellular toxicity.
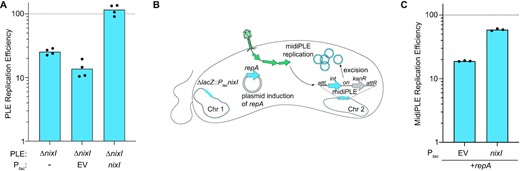
Expression of nixI is required for PLE replication and can augment replication of a minimal replicating PLE (midiPLE). (A) Replication efficiency of V. cholerae PLE mutant and complemented derivatives 20 mpi by ICP1 relative to replication of wild-type PLE (dashed line). (B) Model of midiPLE induction and replication during ICP1 infection. Plasmid-encoded repA, PLE’s essential replication protein (19) and chromosomally encoded nixI or an empty vector were pre-induced for 15 min prior to infection by ICP1. ICP1 infection triggers midiPLE excision from the V. cholerae chromosome and replication of midiPLE is detected by qPCR. (C) Replication efficiency of midiPLE in the presence and absence of nixI relative to wild-type PLE (dashed line) 20 mpi. For (A) and (C), bar height indicates mean and dots are independent biological replicates.
Previous work characterizing PLE replication identified PLE’s minimal unit of replication (referred to as midiPLE) comprised of PLE’s attachment sites, integrase and origin of replication integrated into PLE’s native locus in V. cholerae (Figure 6B). When ICP1 infects, midiPLE excises from the chromosome, and in the presence of PLE’s essential replication initiation factor, repA, midiPLE replicates to high levels; however, midiPLE replication does not reach the same robust levels of replication as is observed with the full-length wild-type PLE (19). As demonstrated earlier, NixI is required for wild-type levels of PLE replication (Figure 6A); therefore, we asked whether NixI could boost midiPLE replication to levels more comparable to wild-type PLE. Induction of nixI increased midiPLE replication to ∼60% of wild-type PLE replication, greater than repA-driven midiPLE replication alone (Figure 6C). The impact of NixI on midiPLE replication could be due to inhibition of ICP1, or NixI may have a direct role in PLE replication, as nicking enzymes are required factors for initiating RCR of plasmids and viral genomes (23). Previous work characterizing PLE’s replication profiles over the course of infection suggested that PLE also undergoes a transition to RCR (19), which we anticipate would be necessary for PLE to linearize its genome for packaging. We postulated that NixI may act as the nicking nuclease for initiating PLE’s RCR, which is often the same region where packaging is initiated from (the pac site). To test whether NixI can cleave PLE’s RCR ori, we identified a candidate region in PLE’s ORF-less space to clone into our plasmid as a substrate for nuclease assays. We reasoned that PLE’s RCR ori would be highly conserved among all known PLEs, and cloned a 500 bp region previously identified as highly conserved (22) (Supplementary Figure S4A). We found that even at high protein concentrations NixI exhibited minimal cleavage of this substrate suggesting NixI does not directly cut PLE’s RCR ori (Supplementary Figure S4B). Additionally, if NixI directly cuts midiPLE’s RCR origin, pre-induction of nixI would be detrimental to midiPLE replication, as it would nick the origin potentially before the expression of the remaining proteins required for RCR (such as single-stranded DNA-binding protein) and midiPLE would likely fail to replicate. Because NixI increases midiPLE replication (Figure 6C), even when ectopically expressed prior to infection, we anticipate NixI is not PLE’s RCR initiator nickase, but rather cleaves ICP1’s genome to liberate replication resources for midiPLE. The targeted nicking of ICP1 could liberate replication resources for PLE and hinder ICP1’s ability to package full-length genomes.
Homologs of NixI are a conserved feature of an expanded family of phage satellites in vibrios
To date, PLEs have only been found in toxigenic V. cholerae and have been shown to be dependent on ICP1. Accordingly, all PLE(+) V. cholerae that have been isolated possess the lipopolysaccharide O1 antigen that serves as the receptor for ICP1 (32). Bioinformatic searches for PLEs in sequenced genomes have searched for nucleotide similarity, as PLEs 1–10 all share homology at the nucleotide level. In an effort to predict NixI’s domain organization and function, we used HHblits (33) to look for homologs of NixI from PLE1 and found proteins that share ∼25% amino acid identity in nontoxigenic V. cholerae (e-value = 0.0000072). Notably, NixI homologs outside of those found in PLEs in toxigenic V. cholerae do not share detectable homology at the nucleotide level. Further investigation of the genomic context of these NixI homologs identified an expanded family of putative phage parasites similar to PLEs, as we elaborate on below. To determine whether the phage parasites from nontoxigenic V. cholerae were indicative of an even larger family of satellites, PSI-BLAST searches were performed with the NixI homologs from putative satellites in nontoxigenic V. cholerae and excluding V. cholerae as a subject. This search returned several homologs in other vibrios such as V. parahaemolyticus and V. casei. To expand this family of potential satellites, reciprocal searches were performed with the tyrosine recombinase/integrase from V. parahaemolyticus strain s042 and additional elements were gathered. In all cases, the NixI homologs were found on putative mobile genetic elements that possess key features of PLEs. First, they all encode a 5′ integrase/recombinase: either a large serine recombinase (which shares ∼50% amino acid identity with PLE-encoded large serine recombinases from toxigenic V. cholerae) or a tyrosine recombinase (Figure 7A). All putative phage parasites are ∼17 kb in length and, like PLEs from toxigenic V. cholerae, encode for almost exclusively hypothetical proteins. However, PFAM domain searches indicated that the satellites have ORFs with domains in previously characterized PLE-encoded ORFs (Figure 7A). Specifically, we searched for proteins with homology or shared domains with PLE1’s CapR (15), RepA (RepA_N domain) (19) and LidI (transmembrane domain) (14). All putative satellites encode a predicted replication initiation protein at the same location as PLEs’ RepA, but interestingly, we saw two different replication proteins: either a RepA_N domain or a DNA Pol I domain. The putative phage parasites have a small transmembrane domain protein similar to PLE’s LidI (14), suggesting that disruption of lysis inhibition may be a conserved function of phage parasites found in other Vibrio species. None of the parasites encoded for proteins with similarity to CapR. In addition, all of the putative satellites are syntenic with PLEs, possessing a large ORF-less space where PLEs’ origin of replication is located (19), as well as a smaller ORF-less space upstream of nixI, where PLEs encode a predicted small RNA (31). Although this analysis is not exhaustive, it shows for the first time that putative phage parasites similar to PLEs are found outside of toxigenic V. cholerae.
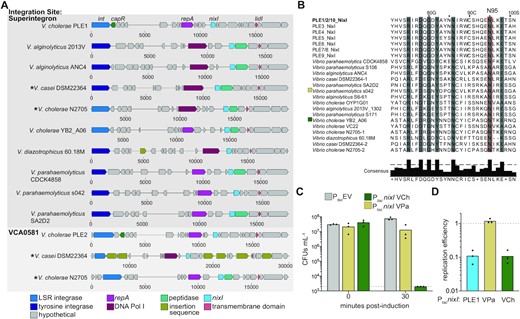
NixI homologs characterize an expanded family of putative phage parasites in Vibrio species. (A) Gene maps (scale below map displays relative nucleotide position) of putative phage parasites from vibrios, where homologs of functionally characterized genes from PLE1 (labeled top) are colored according to shared predicted function identified by PFAM domains or HHpred. Top: phage parasites integrated into the repeat regions of the superintegron; bottom: phage satellites integrated into vca0581, the integration site shared by PLEs 2, 7, 8 and 9. Asterisks are used to note where two similar phage satellites occur in different locations in the same genome. LSR = large serine recombinase. (B) MUSCLE alignment of NixI homologs, showing the experimentally determined catalytic asparagine from PLE1’s NixI (identical to NixI from PLEs 2 and 10) boxed in red (N95), and residues sharing >70% consensus shaded in gray. When more than one satellite was found in the same strain, they are differentiated by ‘-1’ or ‘-2’ after the strain name. (C) Cell viability as measured by CFU/ml of V. cholerae expressing nixI homologs from V. cholerae YB2_A06 (VCh) and V. parahaemolyticus s042 (VPa), plated prior to induction and 30 min post-induction. The dashed line indicates the limit of detection. (D) Same colors as in (C), showing ICP1 replication efficiency in strains pre-induced to express nixI homologs relative to V. cholerae harboring an empty vector control (dashed line) 20 mpi as assessed by qPCR. For (C) and (D), bar height displays mean and dots indicate independent biological replicates.
We next looked at the sequence flanking the putative satellites to determine whether they integrate into conserved attachment sites in their bacterial hosts. In the laboratory, PLEs 1, 3, 4, 5, 6 and 10 from toxigenic V. cholerae integrate into V. cholerae repeats that occur >100 times in the superintegron, each serving as a potential attachment site (17,18). Although long-read sequencing is required to resolve the repetitive nature of the superintegron to definitively state where these putative satellites integrate, the majority are integrated into the superintegron repeats even though some possess a tyrosine recombinase unrelated to the large serine recombinases found in PLEs from toxigenic V. cholerae. PLEs 2, 7, 8 and 9 integrate into the M48 metallopeptidase gene (vca0581) outside of the superintegron (17,18). Interestingly, two of the identified Vibrio genomes, V. cholerae N2705 and V. casei DSM22364, have multiple putative phage parasites: one integrated into a superintegron repeat and the other in the M48 metallopeptidase gene. Multiple PLEs in a single V. cholerae genome have not previously been described. The presence of multiple satellites similar to PLEs found in the same genome suggests the modular nature of PLEs that has been observed (17,18) is driven by recombination between phage parasites in the same host. The second putative satellite in V. casei (in the M48 metallopeptidase gene) is littered with insertion sequences, suggesting that there may be no fitness benefit to having multiple phage parasites and that they can be lost through degeneration (Figure 7A).
NixI homologs from PLEs in toxigenic V. cholerae are all highly similar, sharing >65% amino acid identity. In contrast, NixI homologs from the putative satellites outside of toxigenic V. cholerae are highly divergent, sharing only 20–25% amino acid identity (Supplementary Table 3). A MUSCLE alignment of all unique NixI homologs from Vibrio species indicates that the HNH/N catalytic asparagine identified in this study is conserved (Figure 7B). A neighbor-joining tree based on this alignment shows NixI encoded by PLEs from toxigenic V. cholerae are closely related, while the NixI homologs from nontoxigenic V. cholerae cluster together (Supplementary Figure S5). NixI from other vibrios did not group by species, suggesting the potential for genetic exchange between putative phage parasites found in the genomes of V. parahaemolyticus, V. alginolyticus and V. casei (Supplementary Figure S5). To determine whether NixI is functionally conserved among the newly identified putative phage parasites, we expressed nixI from V. cholerae YB2_A06 (an environmental nontoxigenic isolate) and from V. parahaemolyticus s042 in our lab strain of toxigenic V. cholerae, and the impact of NixI induction on cell viability was assayed. Induction of the homolog from V. cholerae YB2_A06 completely inhibited colony formation of our lab strain of V. cholerae, while the homolog from V. parahaemolyticus was only mildly toxic (Figure 7C). To determine whether these nixI homologs are inhibitory to ICP1, we quantified ICP1 replication in V. cholerae pre-induced to express either homolog, or nixI from PLE1, and compared replication efficiency to that of a permissive host with the empty vector. Interestingly, the homolog from V. cholerae YB2_A06 inhibited ICP1 replication, yet the homolog from V. parahaemolyticus did not (Figure 7D). Vibrio cholerae YB2_A06 is a non-O1 isolate that would not be susceptible to ICP1 infection. Induction of PLE’s genetic program requires infection by ICP1 (31), so specificity of NixI homologs for their cognate phages could be driven by expression only in response to infection by their viral host, rather than recognition of a motif unique to their host phage’s genome. Together, we show that PLEs are not restricted to toxigenic V. cholerae, but functionally analogous elements also reside in nontoxigenic V. cholerae as well as other vibrios. Although members of this expanded family of phage satellites remain to be experimentally validated, these data suggest that PLE-encoded strategies that restrict ICP1, such as nixI-mediated cleavage, may be conserved among divergent satellites and represent a mechanism to parasitize a broader class of inducing phage.
DISCUSSION
Phage parasites can be considered host defense systems, as the parasite inhibits production of its viral host, benefiting the bacterial population (4,17). However, phage parasites are fundamentally selfish elements, promoting both their vertical transmission by protecting the bacterial host population from viral takeover and their own horizontal transmission through parasitism of their viral host’s structural components (7,15,34). Phage parasites employ finely tuned mechanisms to prevent packaging of the viral host’s genome, but allow for progression of the viral host’s gene expression program for production of structural proteins that are hijacked by the phage parasite. For this reason, it is perhaps unsurprising that all phage parasites studied outside of V. cholerae reduce but do not eliminate production of their viral host (7,9,35,36). Vibrio cholerae’s PLEs are unique in their capacity to completely restrict their viral host ICP1 (17), yet the PLE-encoded mechanisms responsible for ICP1 restriction have remained mysterious. In this work, we show that NixI from PLE1 functions as a nuclease effector, adopting a strategy that is widely used by bacterial defense systems to restrict phage production (37–39). We propose a model where NixI functions as a nicking endonuclease, introducing site-specific nicks into ICP1’s genome. Anti-phage defense systems that encode nicking endonucleases have recently been characterized, suggesting nicking is an effective strategy to destroy invading viral DNA (40,41). Although we did not detect any exonuclease activity from NixI in vitro, the coverage profiles across ICP1 suggest that additional exonuclease(s) may act 3′ of the cleaved regions, perhaps as an effort by ICP1 to repair DNA damage. While nicking may directly cause replisome disassembly (42), attempts to repair nicks may also recruit DNA repair and recombination factors to these sites, stealing them away from collapsed replication forks leading to further defects in replication (43). We show here that NixI is the most inhibitory PLE-encoded product characterized thus far, recapitulating PLE’s inhibition of ICP1 progeny production and reducing ICP1 replication by an order of magnitude. Targeting viral host replication with a nuclease effector is a strategy of viral host interference that, to our knowledge, has not been previously reported for phage parasites. Though as we discuss below, PLE-mediated restriction of ICP1 replication through NixI does not completely prevent ICP1 replication, as PLE relies on the maintenance of ICP1’s genome for expression of replication (44) and structural (15) proteins that PLE parasitizes. NixI is not necessary for PLE-mediated inhibition of ICP1, consistent with there being multiple PLE-encoded mechanisms to restrict ICP1 (14). While the remaining mechanisms of ICP1 restriction remain to be determined, they may target nodes in ICP1’s life cycle to divert structural components toward PLE packaging, a strategy used by all known phage parasites studied to date (11,45,46).
Several lines of evidence implicate NixI as the PLE-encoded factor that inhibits ICP1’s transition to RCR. First, bioinformatic analyses identified NixI as the only putative nuclease encoded by PLE. Additionally, transcription of the nixI operon peaks between 12 and 16 mpi, at a time when ICP1 is transitioning to RCR (19,31). NixI is necessary for the appearance of cut sites in ICP1’s genome, which are evident at 16 mpi of PLE(+) V. cholerae. Inhibiting ICP1 replication may liberate ICP1- and/or V. cholerae-encoded replication resources for PLE. The components of ICP1’s and PLE’s replisomes remain to be experimentally validated, though ICP1 is predicted to have a T7-like replisome comprised of DNA Pol I, a primase–helicase, an RNaseH homolog and a yet unidentified single-stranded DNA-binding protein (47,48). PLE lacks homologs of these key replisome components, suggesting that it hijacks ICP1’s replisome, which is supported by experimental evidence showing that PLE replication depends on an ICP1-encoded helicase (44). PLE-mediated inhibition of ICP1’s RCR would prevent ICP1 from linearizing its genome, which is necessary for efficient packaging. In this way, inhibition of RCR may be functionally analogous to a strategy used by phage parasites in S. aureus (known as SaPIs) to prevent packaging of their viral host’s genome. SaPIs inhibit their viral hosts’ small terminase (TerS) subunit and encode their own replacement (49). TerS is the component of the packaging machinery that specifically recognizes the viral DNA to initiate translocation of the viral genome into virions (26). By blocking TerS and encoding their own, SaPIs ensure that their own genome is preferentially packaged. Adopting a similar strategy, divergent satellites found in E. coli encode a protein that redirects the viral host’s TerS to specifically package the parasites’ genome (50). Our analyses suggest that ICP1 packaging is still inhibited by PLEΔnixI, as cleavage of ICP1’s pac site is still blocked (Supplementary Figure S1B). This suggests that PLE encodes additional inhibitory mechanism(s) that may block ICP1’s terminase complex, and may act synergistically with NixI to ensure that ICP1’s genome is not packaged.
An essential component of nuclease effectors is the ability to specifically target foreign DNA. We therefore anticipated that PLE would have mechanisms to protect itself from self-targeting by NixI. Based on the work presented here, we hypothesize that PLE is protected through other mechanisms that are more nuanced than distinguishing self from non-self based on sequence alone. Recent work has identified an anti-CRISPR protein that counters CRISPR–Cas immunity by nicking supercoiled DNA, which is the preferred substrate for the SpyCas9 nuclease effector, allowing for phage escape from CRISPR targeting (51). Here, we have shown that negative supercoiling, which is the state of plasmid substrates used for in vitro cutting assays, is robustly cleaved by NixI, while we saw no evidence that linear PCR products are nicked, suggesting NixI activity depends on DNA topology. It is therefore possible that PLE possesses alternative DNA topology to avoid NixI cleavage, perhaps by alleviating its own supercoiling, while NixI may preferentially cleave supercoiled regions of ICP1’s genome. We cannot rule out the possibility that PLE might not be protected from NixI cleavage, but rather is able to overcome NixI targeting through robust genome replication. PLE replicates to ∼1000 genome copies per cell, exceeding the copy number increase of ICP1. PLE possesses multiple mechanisms to hinder ICP1’s genome replication, as ICP1 replication is still reduced in a PLE(+)ΔnixI background. This defect in replication may render ICP1 more susceptible to NixI activity, while robust PLE replication may outpace the rare nicks introduced by NixI on a self-targeting PLE. We further observe many instances of NixI-dependent cleavage drops in ICP1’s genome in vivo during infection of a PLE(+) host, suggesting that cutting at many regions is required for NixI to hamper ICP1 replication. However, if PLE is not protected from NixI activity, PLEΔnixI should show elevated replication. Rather, we observe a replication defect in the absence of nixI. However, it is possible that there may be counteracting forces at play: NixI cleaves ICP1’s genome limiting competition with its viral host and liberating replication resources for PLE, yet NixI can nick PLE hampering its replication. These additive effects may result in the intermediate PLE replication defect seen with PLEΔnixI. Determining how PLE is protected from self-targeting may reveal new ways of self–non-self discrimination by nuclease effectors.
Established and novel phage defense systems can be predicted in silico through the presence of conserved domains or by their genomic proximity to known defense systems (43,44). Our work complements recent studies showing that phage parasites are likely ubiquitous in bacterial genomes (11,12) and highly similar parasites can be found in divergent taxa, such as the parasite P4 that was previously thought to be restricted to E. coli but was recently found across several genera of Enterobacterales (52). Phage parasites lack hallmark genes allowing for easy identification but can be identified as mobile genetic elements carrying an integrase/recombinase. Additional gene cargo carried by phage parasites can influence the fitness of their bacterial hosts, beyond their established role in restricting viral host propagation, as phage parasites have recently been shown to encode defense systems that function more broadly against infecting phages (53). Demonstrating bona fide phage parasitism is challenging given that a compatible tripartite system must be identified. While the cellular host is known from the integrated parasite genome, the cognate viral host may not be as obvious, especially if the viral host is not temperate. Interestingly, phages similar to ICP1 (48) have been isolated and shown to infect non-cholera vibrios (54) that may be candidate viral hosts for the new parasites identified here. Complicating the identification of viral hosts is the arms race that can occur between viral hosts and their parasites, as occurs with ICP1 and PLEs, which may obscure inhibitory activity if the phage being tested has evolved to antagonize the phage parasite in question (13,22). The outcome of phage infection can be hard to predict based solely on the genome sequences of bacterial hosts and their phages, influencing microbial ecology and complicating phage therapy efforts. Vibrios are responsible for diseases in aquaculture, prompting research into the use of phage therapy to control outbreaks (55). Phage parasites may hinder these efforts, and research into the viral host range and prevalence of putative parasites in aquaculture settings should be considered before application of phages.
Together, we have shown that PLE encodes a nuclease that specifically targets ICP1’s genome for cleavage, culminating in decreased ICP1 replication and inhibition of ICP1 progeny production. NixI homologs are a feature of an expanded family of putative phage satellites, suggesting that nuclease effectors are a widespread approach used by phage parasites to limit viral host replication. NixI is expressed specifically in response to ICP1 infection, aiding in PLE’s efficient co-option of ICP1 components for its own life cycle. This work furthers our understanding of the tight co-evolutionary relationship between ICP1 and PLE, where adaptations and counteradaptations between these two genomes have been ongoing for nearly 100 years, shaping phage infection dynamics within cholera-infected patients.
DATA AVAILABILITY
The sequencing data from phage-infected PLE(+)ΔnixI V. cholerae generated in this study have been deposited in the Sequence Read Archive database under BioProject accession code PRJNA577694.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank all current and former members of the Seed lab for their many helpful conversations and feedback. Specifically, we would like to thank Dr Angus Angermeyer for his help analyzing ICP1’s coverage ratios. We would also like to thank the Komeili lab for use of their AKTA for protein purification and the University of California, Berkeley QB3 Core Facility for assistance with whole genome sequencing.
FUNDING
National Institute of Allergy and Infectious Diseases [R01AI127652 and R01AI153303 to K.D.S.]. The contents of this project are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or NIH. Funding for open access charge: University of California, Berkeley Startup Funds. K.D.S. is a Chan Zuckerberg Biohub Investigator and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. K.N.L. was supported by an NSF Graduate Research Fellowship (Fellow ID no. 2017242013). P.D. was supported by a Rose Hills Summer Undergraduate Research Fellowship.
Conflict of interest statement. K.D.S. is a scientific advisor for Nextbiotics, Inc.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments