





Abstract
Mobile genetic elements such as phages and plasmids have evolved anti-CRISPR proteins (Acrs) to suppress CRISPR-Cas adaptive immune systems. Recently, several phage and non-phage derived Acrs including AcrIIA17 and AcrIIA18 have been reported to inhibit Cas9 through modulation of sgRNA. Here, we show that AcrIIA17 and AcrIIA18 inactivate Cas9 through distinct mechanisms. AcrIIA17 inhibits Cas9 activity through interference with Cas9-sgRNA binary complex formation. In contrast, AcrIIA18 induces the truncation of sgRNA in a Cas9-dependent manner, generating a shortened sgRNA incapable of triggering Cas9 activity. The crystal structure of AcrIIA18, combined with mutagenesis studies, reveals a crucial role of the N-terminal β-hairpin in AcrIIA18 for sgRNA cleavage. The enzymatic inhibition mechanism of AcrIIA18 is different from those of the other reported type II Acrs. Our results add new insights into the mechanistic understanding of CRISPR-Cas9 inhibition by Acrs, and also provide valuable information in the designs of tools for conditional manipulation of CRISPR-Cas9.
INTRODUCTION
CRISPR-Cas systems, consisting of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins, are RNA-guided adaptive immune systems for bacteria and archaea to resist mobile genetic elements (MGEs) such as phages and plasmids (1–3). The identified CRISPR-Cas systems are broadly classified into the multi-effector systems (class 1) and the single-effector systems (class 2), covering six types (I–VI) and various subtypes (4,5). Among them, the type II CRISPR-Cas9 system has been widely used in genome editing applications. During primary phage infection, the Cas protein complex such as Cas1-2 acquires short segments of the exogenous DNA into its own CRISPR array in the adaptation stage. Subsequently, the CRISPR array serving as a memory reservoir is usually transcribed into a single pre-CRISPR RNA (pre-crRNA), which would be further processed into a mature crRNA. Upon phage reinfection, Cas effectors guided by crRNAs recognize and cleave the invasive nucleic acid complementary to crRNA (6). Additionally, a short stretch of nucleic acids, called protospacer adjacent motif (PAM) sequences of target DNA, is often crucial for recognition and degradation (7,8).
Competition between bacterium and phages drives their co-evolution. Not surprisingly, some MGEs especially phages have evolved fight back strategies including anti-CRISPR proteins (Acrs) to block CRISPR defense response (9). Dozens of Acrs have been identified since the first discovery in the phages of Pseudomonas aeruginosa (10–12). To date, several type II Acrs have been structurally and biochemically characterized. One common blockage mechanism of type II Acrs is to bind and restrict the conformational changes required for Cas9 endonuclease activity. For example, AcrIIA14, AcrIIC1 and AcrIIC3 bind to the HNH nuclease domain to trap Cas9 in a catalytically incompetent conformation (13–16). AcrIIA6 mainly interacts with WED and PI domain to allosterically alter conformational dynamics of Cas9 (17). Another common inhibitory strategy is to prevent the target DNA binding by mimicking PAM, which is shared by type II Acrs including AcrIIA2 and AcrIIA4 (18–23). Recent studies also reveal some different inhibition strategies, such as relieving DNA torsion and trapping of Cas9 at decoy sites (24,25).
Several new type II Acrs (AcrIIA16–19) have recently been unveiled to inactivate CRISPR-Cas9 systems (26). AcrIIA16–19 exhibit similar inhibition effects on Cas9 compared with AcrIIA4 in a CRISPRi assay, suggesting AcrIIA16–19 as potent inhibitors of Cas9. The co-immunoprecipitation experiment reveals direct interactions between Cas9 and AcrIIA16–19. Moreover, the Cas9 protein that co-expressed and co-purified with AcrIIA17 or AcrIIA18 is inactive for target DNA cleavage in vitro. Northern blot analysis shows that sgRNA falls to an undetected level in the presence of AcrIIA17 in vivo, indicative of inhibition of sgRNA loading or biogenesis. By contrast, the sgRNA is truncated by AcrIIA18 in vivo.
In the present work, we biochemically and structurally characterized the inhibitory roles of AcrIIA17 and AcrIIA18. AcrIIA17 engages with the bridge helix (BH) domain to inhibit Cas9-sgRNA ribonucleoprotein (RNP) complex assembly. Strikingly, AcrIIA18 cleaves sgRNA to a length inadequate to trigger the Cas9 endonuclease activity. This enzymatic mechanism is different from the aforementioned type II Acrs mechanisms. These results not only provide new insights into the Cas9 suppression mechanisms of type II Acrs, but may also facilitate the development of off-switch tools for CRISPR-Cas9 systems.
MATERIALS AND METHODS
Protein expression and purification
The plasmid encoding full-length SpyCas9 was obtained from Addgene (27). SpyCas9 was fused with an N-terminal His tag followed by a maltose-binding protein (MBP) tag. The coding sequences for AcrIIA17 and AcrIIA18 were synthesized (Genewiz) and cloned into the pET28-MHL vector. All the resultant plasmids were independently transformed into BL21(DE3) cells. 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) was added for induction of protein overexpression at 16°C. Cells were then harvested and resuspended in the binding buffer (25 mM Tris–HCl pH 7.5, 500 mM NaCl, 2 mM β-mercaptoethanol), and lysed by sonication. Target proteins were initially purified with Ni-NTA resin (QIAGEN). After extensive wash using the binding buffer supplemented with 10 mM imidazole, target proteins were eluted with an elution buffer (25 mM Tris–HCl pH 7.5, 500 mM NaCl, 300 mM imidazole, 2 mM β-mercaptoethanol). TEV was added to the eluates to remove the His or His-MBP tag. Ion-exchange chromatography was used for the further purification of target proteins (HiTrap SP for Cas9, HiTrap Q for AcrIIA17 and AcrIIA18). The protein samples were concentrated and loaded onto the Superdex 200 column (Cytiva) equilibrated with the gel-filtration buffer (25 mM Tris-HCl pH 7.5, 300 mM NaCl, 2 mM DTT). Peak fractions were collected and concentrated for further applications. SDS-PAGE was used to analyze the purity of protein samples in all steps. Full-length and truncated NmeCas9 were expressed and purified using the same procedure.
The SeMet-labeled AcrIIA18 proteins were obtained by overexpression using the SelenoMethionine Medium kits (Molecular Dimensions) following manufacturer's instructions, and were purified by the same protocol as above.
Crystallization and structure determination
Purified Se-Met labeled and native AcrIIA18 was crystallized at 18°C by the sitting-drop method. 1 μl protein solution (14 mg/ml) was mixed with 1 μl crystallization buffer containing 0.3 M calcium acetate and 15% (w/v) PEG 3350. All the crystals were cryo-protected in mother liquor supplemented with 20% glycerol and flash frozen in liquid nitrogen for data collection. Diffraction data were collected at beamline BL19U1 of Shanghai Synchrotron Radiation Facility (SSRF) (28). All diffraction data sets of AcrIIA18 were processed by HKL3000 (29). Seleniumpositions determination, density modification, and automated model building were performed by PHENIX (30). Improvement of the initial automated model was conducted iteratively with COOT (31) and phenix.refine (32). Statistics of data processing and structure refinement are listed in Table 1.
X-ray diffraction and refinement statistics
| AcrIIA18 | |
|---|---|
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 105.371, 32.405, 62.594 |
| α, β, γ (°) | 90, 120.366, 90 |
| Resolution | 50–1.38 (1.43–1.38) |
| Completeness | 98.45 (98.27) |
| R-merge | 0.06649 (0.1984) |
| I/sigma (I) | 16.24 (4.27) |
| CC 1/2 | 0.991 (0.905) |
| Redundancy | 3.7 (3.4) |
| Wavelength (Å) | 0.97853 |
| Refinement | |
| Resolution (Å) | 30.52–1.38 |
| Reflections used in refinement | 30 375 (3693) |
| R-work/R-free | 0.1842/0.2083 |
| Number of non-hydrogen atoms | 1694 |
| macromolecules | 1442 |
| ligands | 67 |
| solvent | 221 |
| B-factors | 21.13 |
| macromolecules | 19.49 |
| ligands | 45.80 |
| solvent | 29.76 |
| Ramachandran (%) | |
| favored | 99.42 |
| allowed | 0.58 |
| outliers | 0 |
| R.m.s.deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.11 |
| AcrIIA18 | |
|---|---|
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 105.371, 32.405, 62.594 |
| α, β, γ (°) | 90, 120.366, 90 |
| Resolution | 50–1.38 (1.43–1.38) |
| Completeness | 98.45 (98.27) |
| R-merge | 0.06649 (0.1984) |
| I/sigma (I) | 16.24 (4.27) |
| CC 1/2 | 0.991 (0.905) |
| Redundancy | 3.7 (3.4) |
| Wavelength (Å) | 0.97853 |
| Refinement | |
| Resolution (Å) | 30.52–1.38 |
| Reflections used in refinement | 30 375 (3693) |
| R-work/R-free | 0.1842/0.2083 |
| Number of non-hydrogen atoms | 1694 |
| macromolecules | 1442 |
| ligands | 67 |
| solvent | 221 |
| B-factors | 21.13 |
| macromolecules | 19.49 |
| ligands | 45.80 |
| solvent | 29.76 |
| Ramachandran (%) | |
| favored | 99.42 |
| allowed | 0.58 |
| outliers | 0 |
| R.m.s.deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.11 |
Values in parentheses are for highest resolution shell.
X-ray diffraction and refinement statistics
| AcrIIA18 | |
|---|---|
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 105.371, 32.405, 62.594 |
| α, β, γ (°) | 90, 120.366, 90 |
| Resolution | 50–1.38 (1.43–1.38) |
| Completeness | 98.45 (98.27) |
| R-merge | 0.06649 (0.1984) |
| I/sigma (I) | 16.24 (4.27) |
| CC 1/2 | 0.991 (0.905) |
| Redundancy | 3.7 (3.4) |
| Wavelength (Å) | 0.97853 |
| Refinement | |
| Resolution (Å) | 30.52–1.38 |
| Reflections used in refinement | 30 375 (3693) |
| R-work/R-free | 0.1842/0.2083 |
| Number of non-hydrogen atoms | 1694 |
| macromolecules | 1442 |
| ligands | 67 |
| solvent | 221 |
| B-factors | 21.13 |
| macromolecules | 19.49 |
| ligands | 45.80 |
| solvent | 29.76 |
| Ramachandran (%) | |
| favored | 99.42 |
| allowed | 0.58 |
| outliers | 0 |
| R.m.s.deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.11 |
| AcrIIA18 | |
|---|---|
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 105.371, 32.405, 62.594 |
| α, β, γ (°) | 90, 120.366, 90 |
| Resolution | 50–1.38 (1.43–1.38) |
| Completeness | 98.45 (98.27) |
| R-merge | 0.06649 (0.1984) |
| I/sigma (I) | 16.24 (4.27) |
| CC 1/2 | 0.991 (0.905) |
| Redundancy | 3.7 (3.4) |
| Wavelength (Å) | 0.97853 |
| Refinement | |
| Resolution (Å) | 30.52–1.38 |
| Reflections used in refinement | 30 375 (3693) |
| R-work/R-free | 0.1842/0.2083 |
| Number of non-hydrogen atoms | 1694 |
| macromolecules | 1442 |
| ligands | 67 |
| solvent | 221 |
| B-factors | 21.13 |
| macromolecules | 19.49 |
| ligands | 45.80 |
| solvent | 29.76 |
| Ramachandran (%) | |
| favored | 99.42 |
| allowed | 0.58 |
| outliers | 0 |
| R.m.s.deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.11 |
Values in parentheses are for highest resolution shell.
In vitro transcription and purification of sgRNAs
All sgRNAs were transcribed in vitro using home-made T7 RNA polymerase. Template dsDNA for transcription was generated by PCR. The reaction was processed in a buffer containing 0.1 M HEPES-K pH 7.9, 12 mM MgCl2, 30 mM DTT, 2 mM spermidine, 2 mM NTPs, 80 μg/ml T7 polymerase and 250 nM transcription template at 37°C overnight. Synthesized RNA was separated and gel-extracted by 12% denaturing TBE-urea PAGE gel and subsequently resolved overnight in elution buffer (200 mM NaCl, 1 mM EDTA). Extracted RNA was precipitated by 8 M LiCl and resuspended with diethylpyrocarbonate (DEPC) water for storage at −80°C.
In vitro DNA cleavage assay
Target DNA sequence containing a 5′-TGG-3′ PAM motif was cloned into the pET28-MKH8SUMO vector. The resultant plasmids were linearized by FspI (NEB) for the cleavage reaction. Cas9 and Acr proteins were pre-diluted by the cleavage buffer (25 mM Tris–HCl pH 7.5, 200 mM KCl, 5 mM MgCl2, 5 mM DTT, 5% glycerol). To test the effects of Acr proteins, the reaction components (300 ng of Cas9, Acr proteins and 40 ng of sgRNA) were pre-mixed in order as indicated in Figure 1A and B (lower inset), followed by addition of 100 ng target DNA. Molar ratios of Cas9-Acr ranging from 1:50 to 1:100 were used. The reaction mixtures were incubated at 37°C for 15 min at each step. 40 mM EDTA and 1 mg/ml proteinase K were then added to stop the reaction. The cleavage products were separated and visualized by 0.5% TBE agarose gel pertained with StarStain Red Nucleic Acid Dye (GenStar).
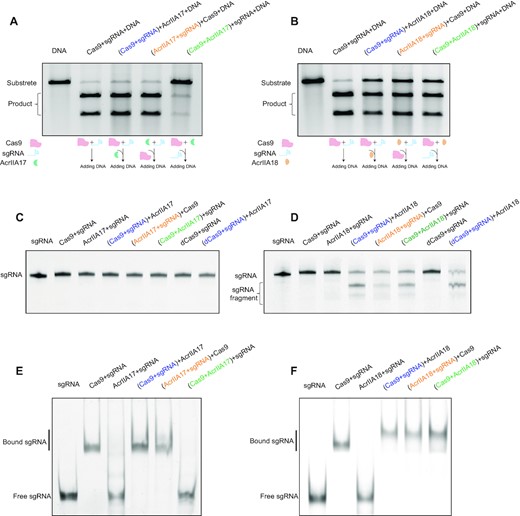
Biochemical and functional characterizations of AcrIIA17 and AcrIIA18. (A, B) In vitro DNA cleavage assay of Cas9 in the presence of AcrIIA17 (A) or AcrIIA18 (B). The assay was performed with different orders of addition as described in the schematic (lower inset). The pre-incubated components are shown in parentheses. These gels are representative of three replicate experiments. (C, D) A TBE-urea denaturing gel showing AcrIIA17- (C) or AcrIIA18-mediated (D) cleavage of sgRNA. The assay was performed with different orders of addition. dCas9: dead mutant Cas9. These gels are representative of three replicate experiments. (E, F) The gel electrophoresis mobility shift assay (EMSA) on sgRNA in the presence of AcrIIA17 (E) or AcrIIA18 (F). The assay was performed with different orders of addition. These gels are representative of three replicate experiments.
In vitro sgRNA cleavage assay
All sgRNA cleavage assays were performed in the cleavage buffer. 200 ng sgRNA was used for the cleavage assay. The reaction components (Cas9, Acr proteins and sgRNA) were mixed in three ways as described in Figure 1A and B. Two of the components were pre-incubated at 37°C for 15 min, and then the third component was added to the reaction system for another 15 min. The reaction products were denatured by adding TBE–urea loading buffer, and then separated on a 12% TBE–urea PAGE gel and visualized using StarStain Red Nucleic Acid Dye (GenStar).
Electrophoretic mobility shift assay
EMSA were performed in the cleavage buffer. For RNP assembly, the purified Cas9 proteins were incubated with sgRNA at a ratio of 2:1. For DNA EMSA, the non-target stand was labeled at the 5′ end with 6-carboxyfluorescein (FAM). The sequences used for EMSA are ATTGCCGTCAGGAAATTAGGTGCGCTTAGCTGGTATTG for non-target stand and CAATACCAGCTAAGCGCACCTAATTTCCTGACGGCAAT for target stand. The reaction components were incubated as described in sgRNA and DNA cleavage assay. The sample was loaded onto a 6% native PAGE and the gel run for 1 hour at 120 V at 4°C. For sgRNA EMSA, the gels were visualized by staining with StarStain Red Nucleic Acid Dye stain. For DNA EMSA, the fluorescence signal was analyzed using a Tanon 5200 imaging system.
Isothermal titration calorimetry
All proteins were in a buffer containing 25 mM Tris–HCl pH 7.5, 300 mM NaCl unless otherwise indicated. The isothermal titration calorimetry experiments were performed at 25°C using a MicroCal PEAQ-ITC system (Malvern). A typical titration experiment involved 19 injections of protein (200–300 μM) solution into the cell containing protein (20–30 μM) with a spacing of 120 s. MicroCal PEAQ-ITC analysis software was used for processing the ITC titration data.
Analytical ultracentrifugation
The sedimentation velocity measurements were performed using a Beckman Optima XL-I analytical ultracentrifugation. AcrIIA18 proteins were diluted to ∼0.5 mg/ml in buffer containing 25 mM Tris–HCl pH 7.5, 150 mM NaCl. The Sedfit and Sedphat programs (33) were used to analyze the sedimentation coefficient.
Gel-filtration assay
The gel-filtration assay was carried out using the Superdex 200 Increase 10/300 (Cytiva). AcrIIA17 was incubated with the Cas9 or BH-REC1 fragment at a 3:1 molar ratio on ice for 30 mins, and then the mixture was loaded onto the column pre-equilibrated with the gel-filtration buffer. The peak fractions were analyzed by SDS–PAGE.
His pull-down assay
The His-tagged full-length and truncated NmeCas9 proteins were first incubated with Ni-NTA resin for 1 h at 4°C in gel-filtration buffer. Tag-free AcrIIA17 proteins were then incubated with the beads for 1 h at 4°C. The samples were washed four times with 1 ml gel-filtration buffer supplemented with 20 mM imidazole to remove excess unbound protein, and then eluted with buffer containing 25 mM Tris–HCl pH 7.5, 300 mM NaCl and 500 mM imidazole. The eluate fractions were analyzed by SDS-PAGE.
RESULTS
AcrIIA17 and AcrIIA18 inhibit the Cas9-mediated DNA cleavage
The newly identified anti-CRISPR proteins AcrIIA17 and AcrIIA18 have been reported to inactivate the CRISPR-Cas9 system (26). We carried out in vitro DNA cleavage experiments to further characterize the AcrIIA17- and AcrIIA18-mediated inhibition of Cas9. Cas9 from Streptococcus pyogenes (SpyCas9), the most widely used nuclease for genome editing (34,35), can be blocked by AcrIIA17 and AcrIIA18, and thus was employed in this study unless otherwise noted. The reaction components were added in different assembly orders as indicated in Figure 1A, B.
Consistent with the previous report (26), neither mixing of AcrIIA17 with sgRNA before Cas9 (Figure 1A, lane 4) nor Cas9-sgRNA RNP complex formation before AcrIIA17 had effects on DNA cleavage (Figure 1A, lane 3). By contrast, the DNA cleavage was impaired when pre-incubating AcrIIA17 with Cas9 before sgRNA (Figure 1A, lane 5), which suggests that AcrIIA17 inhibits Cas9-mediated DNA cleavage through modulation of Cas9. Intriguingly, as opposed to AcrIIA17, AcrIIA18 showed inhibitory effects regardless of the mixing orders (Figure 1B). These data suggest that AcrIIA17 and AcrIIA18 inactivate Cas9 in distinct fashions.
AcrIIA18 cleaves sgRNA in a Cas9-dependent manner
Northern blotting shows that AcrIIA17 and AcrIIA18 might block Cas9 through manipulating the sgRNA (26). To further define the function of AcrIIA17 and AcrIIA18, we performed in vitro sgRNA cleavage assays. Interestingly, we did not observe sgRNA degradation by AcrIIA17 neither in the presence nor absence of Cas9 (Figure 1C). In contrast, sgRNA was degraded when incubated with both AcrIIA18 and Cas9 regardless of the mixing orders (Figure 1D). Furthermore, we performed the sgRNA cleavage assay with the catalytically dead Cas9 (dCas9), and found similar sgRNA cleavage pattern as that with wild-type Cas9 (Figure 1D), which therefore exclude the possibility that the ribonuclease activity on sgRNA originated from endonuclease active site of Cas9. However, AcrIIA18 alone showed no obvious RNA degradation activity (Figure 1D, lane 3). These data demonstrate that AcrIIA18 could cleave sgRNA in a Cas9-dependent manner.
AcrIIA17 prevents the RNP formation
Next, we used the gel electrophoresis mobility shift assay (EMSA) to further define the effects of AcrIIA17 and AcrIIA18 on RNP assembly. RNP formation resulted in the slow-migrating band in EMSA. Pre-incubation of AcrIIA17 with Cas9 before sgRNA led to disappearance of this retarded band (Figure 1E). Nevertheless, this RNP band is maintained when mixing AcrIIA17 with Cas9-sgRNA complex or pre-incubating AcrIIA17 with sgRNA before Cas9. The effects of AcrIIA17 in RNP assembly correlate with that of Cas9-mediated DNA cleavage (Figure 1A and E). These results indicate that AcrIIA17 may impede RNP assembly by competing with sgRNA (potentially with a lower binding affinity), thereby inhibiting Cas9 activity. By contrast, AcrIIA18 had no obvious effects on RNP complex assembly (Figure 1F). Taken together, these data demonstrate that AcrIIA17 and AcrIIA18 block Cas9 through modulating sgRNA but in distinct manners.
AcrIIA17 associates with the BH domain
As AcrIIA17 impairs the RNP assembly, it is therefore reasonable to assume that AcrIIA17 is involved in a direct physical association with Cas9. However, AcrIIA17 did not co-migrate with SpyCas9 in gel-filtration assay (Supplementary Figure S1A), indicating a weak or unstable binding between them. AcrIIA17 has been reported to efficiently block the type II-C Cas9 from Neisseria meningitides (NmeCas9) in human cells (26). Not surprisingly, the target DNA cleavage was efficiently blocked when pre-incubating NmeCas9 with AcrIIA17 before sgRNA (Supplementary Figure S2). We thus tested the binding between AcrIIA17 and NmeCas9. Indeed, AcrIIA17 and NmeCas9 eluted in the same fractions in gel filtration, indicative of a direct, stable association (Supplementary Figure S1B). A pull-down assay was performed to map the key regions of NmeCas9 for binding of AcrIIA17. Only the BH-REC1-containing fragments were found able to interact with AcrIIA17 (Supplementary Figure S3A, B). As expected, the BH-REC1 fragment of NmeCas9 co-migrated with AcrIIA17 at a stoichiometric ratio (Supplementary Figure S3C). Additionally, the ITC measurements showed that the BH-REC1 fragment and full-length NmeCas9 exhibit similar binding affinities toward AcrIIA17 (Figure 2A and Supplementary Figure S1C). However, the binding affinity was reduced by about 10-fold in the presence of high salt, indicative of dominant polar contacts between AcrIIA17 and the BH-REC1 segment (Figure 2A). Given the acidic nature of AcrIIA17 (a theoretical isoelectric point (pI) of ∼ 4), the highly basic BH domain may be the target of AcrIIA17. As expected, mutation of the basic residues on BH domain significantly impaired AcrIIA17-binding (Figure 2A), supporting the importance of the BH domain in the binding of AcrIIA17.
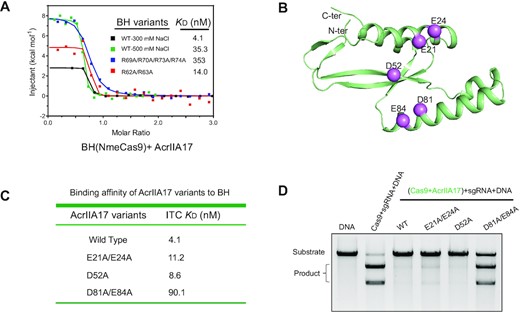
AcrIIA17 binds to the BH domain. (A) ITC measurements of AcrIIA17 and BH-REC1 of NmeCas9. Both wild-type (WT) and variants of BH-REC1 were examined. All the experiments are carried out in the presence of 300 mM salt unless otherwise indicated. (B) The predicted model of AcrIIA17 using AlphaFold2. The residues selected for mutagenesis are shown in sphere representations. (C) Quantification of the binding affinity between indicated AcrIIA17 variants and BH-REC1 fragment of NmeCas9 by ITC. (D) In vitro DNA cleavage assay in the presence of the wild-type or mutated AcrIIA17. This gel is representative of three replicate experiments.
The full-length AcrIIA17 protein eluted at a volume equivalent to a molecular weight of ∼17 kDa on a gel-filtration column, matching the theoretical molecular weight of monomeric AcrIIA17 (∼13 kDa) (Supplementary Figure S4A). Crystallization of AcrIIA17 failed after extensive trials. We then predicted the AcrIIA17 model using AlphaFold (36,37). Consistent with the acidic pI value, AcrIIA17 has a negatively charged concave surface (Supplementary Figure S4B, C), which may accommodate the positively charged BH domain. Alanine substitution mutations were introduced into the acidic patch of AcrIIA17 (Figure 2B), and the D81A/E84A mutant showed a 20-fold reduction in binding affinity (Figure 2C). More importantly, the D81A/E84A mutant also profoundly decreased the inhibition of Cas9 (Figure 2D). Given that the BH domain is a requisite for sgRNA loading (38–40), it is therefore likely that AcrIIA17 engages with BH domain, which precludes Cas9 from binding to sgRNA and thereby impairs RNP formation.
Crystal structure of AcrIIA18
We solved the crystal structure of AcrIIA18 at a resolution of 1.4Å (Table 1). Almost all the residues except for 40–44 could be clearly traced in the electron density map. The crystal asymmetric unit contains one AcrIIA18 molecule, which is in agreement with the gel-filtration assay and analytical ultracentrifugation measurements demonstrating monomeric AcrIIA18 in solution (Supplementary Figure S5). AcrIIA18 folds into a crab-like architecture, with a N-terminal domain (NTD) comprising residues 1–98 and a C-terminal domain (CTD) spanning residues 99–182 (Figure 3A). The NTD domain adopts a two-layer αβ sandwich fold where the β3–β6 strands assemble into a mixed parallel–antiparallel β-sheet, and together with the β1-β2 hairpin pack against the helices α1–α2 (Figure 3A, B). The CTD domain consists of an antiparallel β-sheet (β7–β11) flanked by a helix α3 (Figure 3A, B). The two domains spatially arrange in a linear manner. Neither the full-length protein nor the individual domains, has structural homolog in the PDB database according to the Dali search results (Z score < 5) (41), implying a novel fold for AcrIIA18. The β1–β2 hairpin projects out from the NTD, packing against helix α1 and β3–β5 sheet and thus forming a V-shaped pocket (Figure 3A, B). Although the surface electrostatic potential of AcrIIA18 is largely negatively charged, the V-shaped groove displays a positive electrostatic potential (Figure 3C), possibly playing a role in modulation of sgRNA. Moreover, the V-shaped pocket is ∼14–16 Å deep and 17–19 Å wide, big enough to accommodate the sgRNA. This structural feature is reminiscent of AcrVA1 (42,43), which employs two basic α-helices packing against each other in a V-shaped fashion to bind and cut crRNA.
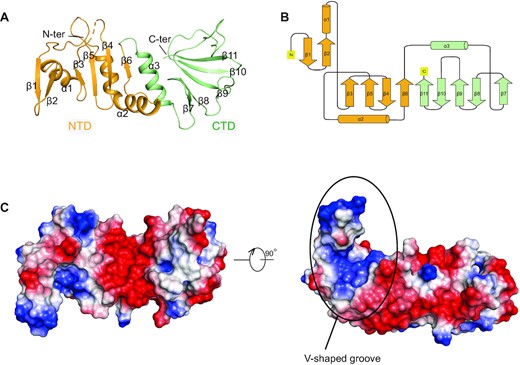
Crystal Structure of AcrIIA18. (A) Overall structure of AcrIIA18. The NTD and CTD are shown in orange and green, respectively. (B) Topology diagram of AcrIIA18. The color scheme is the same as in (A). (C) Electrostatic surface representations of AcrIIA18. The V-shaped groove is positively charged.
The β1–β2 hairpin in NTD is critical for sgRNA cleavage
To elucidate the structural basis of Cas9 inhibition by AcrIIA18, we designed a series of mutations within AcrIIA18. As the charged residues exposed to the solvent are expected to have functional implications in substrate binding and catalysis, 13 charged residues throughout both NTD and CTD domains are selected for point mutagenesis and functional assessment (Figure 4A and Supplementary Figure S6). Alanine replacement on NTD disrupted the sgRNA cleavage (Figure 4B). Consistently, Cas9-mediated DNA cleavage was largely restored by incubation of Cas9 with these AcrIIA18 mutants (Figure 4C). However, all the mutations except E124A on CTD showed little or no impact on AcrIIA18-mediated inhibition (Figure 4B). In line with this observation, these mutations exhibited a similar level of sgRNA degradation compared to wild-type AcrIIA18 (Figure 4C). These results show that NTD may contribute remarkably to AcrIIA18 activity.
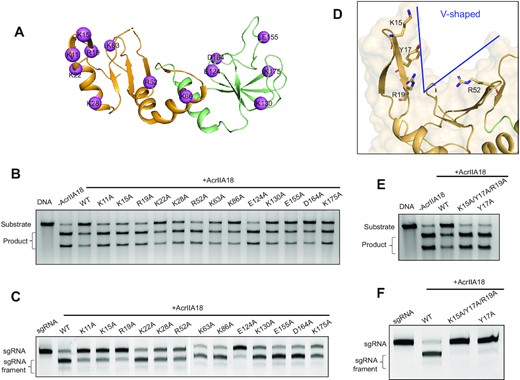
The N-terminal β-hairpin in NTD is critical for AcrIIA18 function. (A) The charged residues of AcrIIA18 selected for mutagenesis are shown in pink sphere representations (B) In vitro DNA cleavage assay in the presence of the wild-type or mutated AcrIIA18. This gel is representative of three replicate experiments. (C) Representative TBE-urea denaturing gels showing the sgRNA cleavage by wild-type and mutated AcrIIA18 in the presence of Cas9. This gel is representative of three replicate experiments. (D) The V-shaped groove of AcrIIA18. Charged residues Lys15, Arg19 and Arg52 are located in the V-shaped groove. Tyr17 stacks with Lys15 and Arg19. (E) Y17A mutant almost failed to inhibit Cas9-mediated DNA cleavage. This gel is representative of three replicate experiments. (F) Y17A mutant significantly impaired the AcrIIA18-induced sgRNA cleavage. This gel is representative of three replicate experiments.
The identified key residues for AcrIIA18-mediated sgRNA cleavage were found to cluster within the basic patch of the V-shaped cavity (Figures 3C and 4A). Particularly, Lys15 and Arg19 from β1–β2 hairpin are located on the inward face of the V-shaped cavity, and pack roughly perpendicular to another important residue Arg52 on the other side of the pocket (Figure 4D). These observations prompt us to speculate that the V-shaped cavity may be the RNase active site of AcrIIA18. To test this hypothesis, we mutated another potential essential residue within the cavity, Tyr17, which is sandwiched by Lys15 and Arg19 and also in close proximity to Arg52. Indeed, Y17A mutant almost failed to truncate sgRNA, as well as having very little effect on Cas9-mediated DNA cleavage (Figure 4E-F).
AcrIIA18 truncates sgRNA to inactivate Cas9
We next explored how the sgRNA truncation by AcrIIA18 affects Cas9 activity. We examined DNA binding by EMSA using Fam-labeled DNA. One retarded band corresponding to the Cas9–sgRNA–DNA ternary complex appeared when incubation of DNA with RNP (Figure 5A). Similar migration patterns were observed when including AcrIIA18 in this ternary complex, suggesting that the AcrIIA18 does not affect DNA binding. These data, coupled with our aforementioned sgRNA cleavage results (Figure 1D), demonstrate that the truncated sgRNA by AcrIIA18 are sufficient to bind target DNA, but could not trigger the endonuclease activity of Cas9. As shown in the urea-denaturing PAGE gel, the cleaved sgRNA products migrated at a position equivalent to the truncated sgRNA with a 15 nt spacer (denoted 15 nt sgRNA) (Figure 5B, C). Spacers with various lengths are truncated to the same position, while no cleavage was seen when incubation with 15 nt sgRNA, indicating that the cleavage site may occur around 15th nt (Figure 5D). Moreover, the endonuclease activity of Cas9 was nearly abolished when incubation with the sgRNA bearing 15 nt of sequence complementary to target DNA (Figure 5E). These data suggest that AcrIIA18 cuts the sgRNA to a length inadequate to generate a catalytically competent Cas9, thereby leading to Cas9 inhibition. Consistently, previous studies have identified the minimal length of 17 nt for the guide segment to trigger Cas9 endonuclease activity both in vivo and in vitro (44,45).

AcrIIA18 triggers sgRNA truncation, and the product is incapable of inducing Cas9-mediated DNA cleavage. (A) EMSA was performed using the carboxyfluorescein (FAM)-labeled target DNA as a probe. EMSA showing Cas9 binding to DNA in the presence of either AcrIIA18 or 15 nt sgRNA. 15 nt sgRNA: sgRNA with 15 nt spacer. This gel is representative of three replicate experiments. (B) Schematic diagrams of sgRNA. The proposed cleavage site of spacer is indicated by a scissors. (C) AcrIIA18 induces the truncation of sgRNA to 15 nt spacer. 15 nt sgRNA and 14 nt sgRNA were used as marker. This gel is representative of three replicate experiments. (D) Representative TBE–urea denaturing gels showing sgRNAs of 30, 20 and 15-nt, respectively, cleavage by AcrIIA18. This gel is representative of three replicate experiments. (E) In vitro DNA cleavage assay for Cas9 complexed with full-length (20 nt) or 15 nt sgRNA. This gel is representative of three replicate experiments.
DISCUSSION
In the constant arms race between host and viral parasite, bacteria have evolved CRISPR-Cas system to silence or destroy foreign mobile genetic elements (MGEs). MGEs also develop ani-CRISPR mechanisms to evade the CRISPR immune response. Phage-derived anti-CRISPR proteins have been extensively studied. Recently, several new anti-CRISPR proteins, including AcrIIA17 and AcrIIA18, have been reported to block the CRISPR-Cas9 system with distinct mechanisms. In this work, we biochemically and structurally characterized AcrIIA17 and AcrIIA18, revealing their inhibition mechanisms.
The inhibition mechanism of AcrIIA17
Our results suggest that AcrIIA17 may associate with the positively charged BH domain of Cas9 via an acidic patch. This interaction thus impairs sgRNA binding to Cas9 and the subsequent DNA recognition. Structural analysis of the RNP complex reveals that the BH domain is involved in the sgRNA recognition through polar contacts (Supplementary Figure S7) (16,38–40). Therefore, it is likely that AcrIIA17 may function as an RNA mimic to bind BH domain. The complex structure of AcrIIA17-Cas9 is needed to further elucidate the inhibition mechanism. Such a blockage mechanism is reminiscent of AcrIIC2, which competes with sgRNA for binding of BH domain (15,46). Intriguingly, sequence analysis reveals no homologue with known structures, and the predicted AcrIIA17 structure model is also distinct from AcrIIC2. AcrIIC2 dimerization is required for BH engagement, whereas AcrIIA17 was found as a monomer in solution (Supplementary Figure S4A) and binds to Cas9 with a ∼1:1 stoichiometric ratio (Supplementary Figure S1C). This supports the emerging notion that structurally diverse Acrs may share similar inhibition strategies. AcrIIA17 seems to be a more potent inhibitor for NmeCas9 compared with AcrIIC2, likely due to the high binding affinity (∼5 nM) between AcrIIA17 and NmeCas9 (∼200 nM for AcrIIC2-NmeCas9) (46).
Blocking of the RNP formation by AcrIIA17 would leave the sgRNA exposed to cellular ribonucleases that cleave the ribonucleotides. Consistently, Northern blotting reveals that the sgRNA falls to an undetected level in the presence of AcrIIA17 in vivo (26). The genes encoding AcrIIA17 are present predominantly in Streptococcus and Lactococcus genomes, which almost exclusively encode type II-A CRISPR-Cas systems. However, AcrIIA17 has more potent inhibitory effects on the non-cognate type II-C subtype in our in vitro assays and in the context of human cells (26). It would be interesting to further characterize the role of AcrIIA17 in bacteria.
The inhibition mechanism of AcrIIA18
In contrast to AcrIIA17 that hinders RNP formation, Cas9 is able to assemble with sgRNA and target DNA in the presence of AcrIIA18. Instead, AcrIIA18 inhibits Cas9 activity through cleaving the sgRNA. The inhibitory effect of AcrIIA18 seems to be less robust than that of AcrIIA17, which is possibly due to the incomplete sgRNA cleavage. As reported in a recent study, Cas9 guided with sgRNA containing a 16 nt spacer sequence still adopts a catalytically incompetent conformation (47). In our study, we found that AcrIIA18 cut the spacer to generate a shortened sgRNA with a 15 nt spacer, thereby failing to activate Cas9. AcrIIA18 functions in a manner akin to AcrVA1 that truncates the crRNA spacer to seed region (42,43). However, AcrIIA18 displays a distinct sgRNA cleavage profile. Specifically, AcrIIA18 appears to catalyze a single-turnover sgRNA cleavage at the 5′ end, while AcrVA1 efficiently acts on the 3′ end of crRNA with multiple-turnover. AcrVA1 binds to the PI domain of Cas12a through PAM mimic (42). Interestingly, AcrIIA18 was suggested to associate with Cas9 through a different interface (26). Considering that the 5′ end of sgRNA would be surrounded by REC, HNH and RuvC domains of Cas9 in RNP complex (38,40,48), we propose that AcrIIA18 may interact with these segments rather than PI domain. It also raises another interesting question of whether the catalytic inactive AcrIIA18 mutants may still be able to work as inhibitors through direct binding. Yet, more studies will be performed to determine the binding mode between AcrIIA18 and Cas9.
AcrIIA18 displays an elongated conformation with a novel two-domain architecture arranged side-by-side, which is different from the single domain (helical bundle) protein AcrVA1. It is likely that a V-shaped groove formed by β1–β5 strands and helix α1 of NTD would facilitate the accommodation of sgRNA. As expected, mutation of multiple residues inside the V-shaped groove impairs sgRNA cleavage by AcrIIA18. Surprisingly, mutation of Glu124 from CTD damaged the abilities of sgRNA cleavage and Cas9 inhibition (Figure 4A-C). Glu124 is involved in bifurcated interactions with Tyr84 and Tyr102 from NTD (Supplementary Figure S8), which possibly stabilizes the NTD-CTD interface. The E124A mutation may lead to an inappropriate interdomain orientation, which prevents AcrIIA18 from proper functioning. Supporting the importance of AcrIIA18 structural integrity, truncation of either NTD or CTD reduced protein stability and caused precipitation. Notably, full-length AcrIIA18 and NTD are nearly exclusively found in Streptococcus and Staphylococcus prophages, while CTD is widely distributed in other MGEs such as plasmids and other phylum like plylum Firmicutes (26), implying a more general unknown function of CTD.
In summary, AcrIIA17 and AcrIIA18 function at different stages of immunity. AcrIIA17 interacts BH domain to prevent the RNP formation. AcrIIA18 disables Cas9 through an enzymatic mechanism distinct from the steric or conformational constraining inhibition modes by other identified type II Acr proteins. Our results highlight the mechanistic versatility of anti-CRISPR employed by MGEs to subvert the CRISPR-mediated adaptive immunity. These findings will also provide clues for the development of tools for CRISPR-Cas9 modulation.
DATA AVAILABILITY
Coordinates and structure factor for AcrIIA18 have been deposited into the Protein Data Bank under the accession code 7VLM.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank all the members of Zhang Lab for their kind help. We thank the staffs from BL17B/BL18U1/BL19U1/BL19U2/BL01B beamlines of the National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.
FUNDING
National Natural Science Foundation of China [32071218]. Funding for open access charge: National Natural Science Foundation of China [32071218].
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments