





Abstract
The widespread and versatile prokaryotic CRISPR–Cas systems (clustered regularly interspaced short palindromic repeats and associated Cas proteins) constitute powerful weapons against foreign nucleic acids. Recently, the single-effector nuclease Cas12a that belongs to the type V CRISPR–Cas system was added to the Cas enzymes repertoire employed for gene editing purposes. Cas12a is a bilobal enzyme composed of the REC and Nuc lobe connected by the wedge, REC1 domain and bridge helix (BH). We generated BH variants and integrated biochemical and single-molecule FRET (smFRET) studies to elucidate the role of the BH for the enzymatic activity and conformational flexibility of Francisella novicida Cas12a. We demonstrate that the BH impacts the trimming activity and mismatch sensitivity of Cas12a resulting in Cas12a variants with improved cleavage accuracy. smFRET measurements reveal the hitherto unknown open and closed state of apo Cas12a. BH variants preferentially adopt the open state. Transition to the closed state of the Cas12a-crRNA complex is inefficient in BH variants but the semi-closed state of the ternary complex can be adopted even if the BH is deleted in its entirety. Taken together, these insights reveal that the BH is a structural element that influences the catalytic activity and impacts conformational transitions of FnCas12a.
INTRODUCTION
Many bacteria and almost all archaea possess the adaptive immune system termed CRISPR–Cas (clustered regularly interspaced short palindromic repeats–CRISPR associated proteins) as part of their defense repertoire against viruses and mobile genetic elements (1–3). The CRISPR–Cas immune response is divided into three stages: (i) foreign nucleic acids (protospacers) are integrated as spacers between repeat sequences into the CRISPR locus during the initial adaptation phase (4–6). (ii) In the expression and maturation phase, the CRISPR locus is transcribed and the resulting precursor CRISPR RNA (pre-crRNA) is further processed to yield the mature CRISPR RNA (crRNA) (7). (iii) In the final interference stage, the binary complex consisting of Cas protein(s) and crRNA binds invading DNA or RNA with sequence complementarity to the crRNA. This leads ultimately to the inactivation of the target nucleic acid most often as a result of endonucleolytic cleavage of the target DNA or RNA (1,8).
The single effector nucleases Cas9 and Cas12a garnered widespread attention as genome editing tools (9–13) because (i) Cas9 as well as Cas12a can be easily programmed to target a specific DNA sequence by complementary crRNAs and (ii) single effector nucleases act independently during the interference stage. Cas12a (formerly Cpf1) is an RNA-guided DNA endonuclease classified as class 2 type V-A Cas protein (3,14,15). Cas12a, in contrast to other Cas systems, is able to self-process the pre-crRNA (16). Programmed with a crRNA, Cas12a recognizes and binds a protospacer adjacent motif (PAM) in the target DNA with the sequence 5′-TTTV-3′ (V = G/C/A) (17,18). The single active site for DNA cleavage resides in the RuvC domain, which cleaves the non-target strand (NTS) faster than the target strand (TS) resulting in a sequential cut thereby creating a double strand break with staggered ends (8,19–22). The NTS is further shortened due to the trimming activity of Cas12a. After cleavage, Cas12a releases the PAM-distal part of the target DNA but remains bound to the PAM-proximal product (23,24). This cleavage event is termed ‘cis-cleavage’ of double-stranded DNA (dsDNA). However, Cas12a possesses also a trans-cleavage activity that is activated upon dsDNA cleavage. Here, a Cas12a-crRNA complex unspecifically binds to single-stranded DNA (ssDNA) and degrades it (25). The trans-cleavage activity of Cas12a has been repurposed to establish quantitative platforms for nucleic-acid detection (26–29).
Cas12a comprises a bilobal structure formed by the nuclease (Nuc) lobe (harboring the RuvC I, RuvC II, RuvC III, BH, and Nuc domains) and the recognition (REC) lobe (composed of the REC1 and REC2 domains). The wedge domain (WED I, WED II, WED III, PI (PAM interacting)) operates as hinge between the two lobes (Figure 1A) (2,17,30,31). At the 5′-end, the crRNA forms a pseudoknot structure that is anchored in the WED domain and additionally forms an extensive network of interactions with the RuvC and REC2 domains (2,30–32). The 3′-end of the crRNA encodes the sequence complementary to the target strand and interacts with the target DNA thereby forming an R-loop. Structural studies showed that Nuc and REC lobes undergo a closed-to-open transition upon loading of the DNA target in the crRNA-Cas12a complex (2,17). The bridge helix (BH) is a central helix in the protein that structurally links the REC and the Nuc lobe. For Cas12a from Acidaminococcus sp. (AsCas12a) it has been shown that amino acids in the BH interact with the sugar-phosphate backbone of the target DNA strand and play a role in the heteroduplex recognition of crRNA and target DNA (31). A tryptophan residue at the end of the BH (W958 in AsCas12a, W971 in Cas12a from Francisella novicida (FnCas12a)) is anchored in a hydrophobic pocket formed by the REC2 domain (Figure 1B/C). This interaction stabilizes the closed and active conformation of Cas12a (31,32). The crRNA has a parallel orientation to the BH and also connects the Nuc and the REC lobe (Figure 1A). In this context, the question remains if and how the BH influences the catalytic activity of Cas12a and whether the integrity of the BH as well as the anchoring in the REC lobe is required to ensure efficient cleavage activity of Cas12a. Furthermore, given the central position of the BH, the question arises whether the BH influences the conformational transitions of the enzyme.
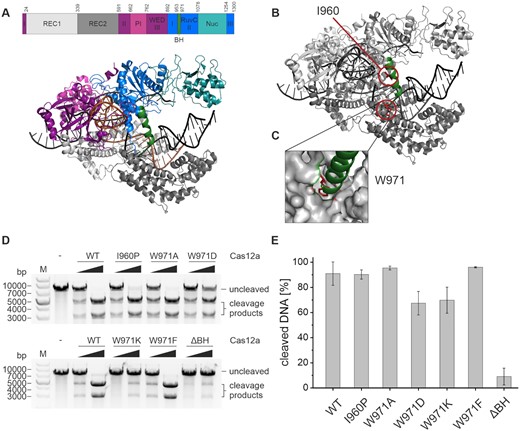
Structural organization of Francisella novicida Cas12a (FnCas12a) and influence of bridge helix mutations on Cas12a cleavage activity. (A) Domain organization and structure of FnCas12a in complex with crRNA (orange) and a double-stranded target DNA (black) (PDB: 6I1K). The bridge helix (BH) is highlighted in green. (B) Structure of Cas12a with residues I960 and W971 highlighted in red and the BH shown in green. (C) Close-up of the anchoring of W971 at the end of the bridge helix in a hydrophobic pocket in the REC2 domain. (D) Plasmid cleavage assay using WT Cas12a and Cas12a BH variants with increasing concentrations of Cas12a (5 and 37.5 nM), crRNA (5 and 37.5 nM) and target DNA at 5 nM. The reaction was incubated at 37°C for 1 h. The linearized target DNA plasmid (7300 bp) is cleaved into two products (4400 and 2900 bp). M, 1 kb plus DNA ladder; –, linearized target DNA without protein and crRNA. (E) Quantification of cleavage efficiency for the WT and Cas12a variants for reactions containing 37.5 nM protein and crRNA. Shown is the average, error bars show the standard deviation of three independent assays.
Here, we performed a mutational analysis of the BH and evaluated the cleavage activity and cleavage accuracy of a variety of BH variants. Cas12a is still catalytically active when the alpha-helical nature of the BH is disrupted or the anchoring of the BH in the REC lobe is prevented. However, mutations in the BH affect the trimming activity and decrease the mismatch tolerance of Cas12a. We employed single-molecule Förster resonance energy transfer (smFRET) measurements on diffusing molecules to follow the conformational transitions of FnCas12a and the BH variants during their catalytic cycle making use of site-specifically labeled Cas12a. We show that Cas12a in its apo state exists in both an open and closed conformation. The open conformation is preferred if the BH is not accurately anchored in the REC domain. Nevertheless, the crRNA induces the closure of the enzyme even if the anchoring of the BH is prevented or the BH is removed from the enzyme most likely because the adjacent helix 1 of the RuvC domain acts in concert with the BH and can ensure structural integrity. We demonstrate furthermore that the crRNA (independent of the BH) plays an essential role to direct the conformational transitions to reach a cleavage-competent state. A third, semi-closed conformation has to be adopted to allow the binding of target DNA. However, the stabilizing effect of the crRNA is sufficient to adopt the semi-closed state in all BH variants. Taken together, these data indicate that the BH is a structural element that influences the cleavage accuracy and trimming activity, increases the specificity of Cas12a and enables the apo enzyme to adopt the closed state thereby promoting efficient crRNA loading.
MATERIALS AND METHODS
Protein expression and purification
The Cas12a gene from Francisella novicida was amplified from the plasmid pFnCpf1_min (Addgene #69975) (1) and cloned into pGEX-2TK via the BamHI and EcoRI restriction sites. This resulted in an expression plasmid that encoded the wildtype Cas12a sequence with an N-terminal GST-tag and Thrombin cleavage site. Cas12a variants (E1006A (dCas12a), I960P, W971A, W971D, W971K, W971F, ΔY953-K969 (ΔBH), ΔY953-W971 (ΔΔBH)) were generated via site-directed mutagenesis (Supplementary Table S1). Protein expression was performed in Escherichia coli BL21(DE3) cells. The cells were grown at 37°C. At an optical density (600 nm) of 0.4, the cells were transferred to 18°C. Protein expression was induced with IPTG (1 mg/l) at an OD600 = 0.6. Cells were further incubated at 18°C overnight. After harvesting the cells (10 min, 6000 g, 4°C), they were resuspended in 20 ml Tris-Cas-A buffer (20 mM Tris–HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5% (v/v) glycerol). 3 μl Universal Nuclease Mix (Pierce™) and 1 tablet of cOmplete™ Protease Inhibitor Cocktail (Roche) were added and the cells were lysed via sonication. The lysate was centrifuged (1 h, 30 000 g, 4°C) and the supernatant applied to a GSTrap 5 ml FF affinity purification column (GE Healthcare) at 4°C equilibrated with Tris-Cas-A buffer. After binding of the protein, the column was washed with a high salt washing buffer (Tris-Cas-A buffer containing 1 M NaCl) and eluted with 100% Tris-Cas-B buffer (20 mM Tris–HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5% (v/v) glycerol, 10 mM glutathione). The eluted protein was incubated overnight with Thrombin protease (100 U/ml, 50:1 (v:v)) at 4°C to allow the cleavage of the GST-tag. The GST-tag and Thrombin protease were removed via heparin affinity chromatography (HiTrap Heparin 5 ml FF, GE Healthcare). Cas12a was eluted via an elution gradient with 1 M NaCl (20 mM Tris–HCl pH 7.5, 1 M NaCl, 5 mM MgCl2, 5% (v/v) glycerol). Cas12a typically eluted at a NaCl concentration of 760 mM. Protein concentrations were calculated using an extinction coefficient at 280 nm of 143 830 M−1cm−1 (ProtParam tool from www.expasy.org). Protein aliquots were flash frozen and the protein stored at −80°C until further use.
Site-specific incorporation of the unnatural amino acid para-azido-l-phenylalanine into Cas12a
To produce fluorescently labeled protein variants of Cas12a for single-molecule FRET measurements, the Amber-suppressor strategy was employed to site-specifically incorporate the unnatural amino acid para-azido-l-phenylalanine (AzF) (33,34). The Amber-stop-codon (TAG) was introduced either into the wildtype pGEX-2TK-Cas12a plasmid or the Cas12a plasmids already carrying mutations in the BH via site-directed mutagenesis at positions D470, K647 and/or T1222 (Supplementary Table S1). The plasmids with Amber-codons at the required positions were expressed in BL21(DE3), which additionally harbored the pEVOL-pAzF plasmid (Addgene #31186, (35)). This plasmid encodes an additional arabinose-inducible promoter that governs the expression of an amber-suppressor tRNA (tRNACUA) and a biorthogonal tRNA synthetase. Expression and protein purification were carried out as described above with the following modification: additionally, 200 mg/l AzF were added to the expression culture at OD600 = 0.3 and l-arabinose at a final concentration of 20 mg/l at OD600 = 0.4. Aliquots of the purified protein were flash frozen and stored at −80°C.
Protein labeling via unnatural amino acids incorporated into Cas12a
Cas12a variants with AzF at positions D470, K647 and/or T1222 were stochastically labeled via the Staudinger-Bertozzi ligation between a triphenylphosphine (at the DyLight fluorophores) and an azide moiety at AzF (36). The purified protein was incubated with a final concentration of 50 μM of DyLight550 and DyLight650 for 90 min at room temperature in darkness. The labeling reaction was centrifuged for 10 min at 21 000 g. The supernatant was purified via size exclusion chromatography using a Sephadex G-50 illustra NICK column and Cas batch buffer (50 mM Tris–HCl pH 7.5, 200 mM NaCl, 5 mM MgCl2, 2% (v/v) glycerol, 0.1% (v/v) Tween20) to remove the excess of dye. The labeled protein was directly used for single-molecule measurements.
Synthetic oligonucleotides
Oligodeoxyribonucleotides were purchased from Eurofins-MWG (Ebersberg, Germany). The target DNA sequence was derived from Swarts et al. (TS 5′-ACTCAATTTTGACAGCCCACATGGCATTCCAC TTATCACTAAAGGCATCCTTCCACGT-3′, NTS 5′-ACGTGGAAGGATGCCTTTAGTGATAAGTGGAATGCCATGTG GGCTGTCAAAATTGAGT-3′) (21). For cleavage assays, the target strand carried a Cy5 label and the non-target strand a Cy3 label either on the 5′-end or the 3′-end. Target and non-target strand were annealed by incubation in annealing buffer (final concentration: 3 mM HEPES pH 7.4, 10 mM potassium acetate, 0.2 mM magnesium acetate) at 95°C for 3 min and passive cool-down to room temperature.
In vitro transcription
crRNAs (crRNA 5′-UAAUUUCUACUGUUGUAGAUGUGAUAAGUGGAAUGCCAUGUGGG-3′, pre-crRNA 5′- GCUGAUUUAGGCAAAAACGGGUCUAAGAACUUUAAAUAAUUUCUACUGUUGUAGAUGUGAUAAGUGGAAUGCCAUGUGGG-3′ derived from Creutzburg et al. (37)) were prepared and purified using the T7 RiboMAX™ Express Large Scale RNA Production System (Promega). The DNA templates (T7crRNA 5′-CCCACATGGCATTCCACTTATCACATCTACAACAGTAGAAATTACCCTATAGTGAGTCGTATTATCGATC-3′, T7pre-crRNA 5′-CCCACATGGCATTCCACTTATCACATCTACAACAGTAGAAATTATTTAAAGTTCTTAGACCCGT TTTTGCCTAAATCAGCCCCTATAGTGAGTCGTATTATCGATC-3′) were purchased from Eurofins-MWG (Ebersberg, Germany). The concentration was photometrically determined and the RNA stored at −20°C.
Electrophoretic mobility shift assay
The binary complex composed of 200 nM Cas12a protein and 200 nM crRNA was pre-formed in 1x Cas buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 2% (v/v) glycerol, 1 mM DTT, 0.05% (v/v) Tween20) for 10 min at room temperature. 75 nM of the binary complex were added to 10 nM of fluorescently labeled short target DNA in a total volume of 10 μl and incubated at 37°C for 1 h. To avoid unspecific interactions of Cas12a with nucleic acids, Heparin with a final concentration of 32 μg/ml was added and the reaction incubated for additional 10 min at room temperature. Gel loading dye (final concentration: 62.5 mM Tris–HCl pH 6.8, 2.5% (w/v) Ficoll400) was added and the samples separated by a non-denaturing Tris-glycine gel electrophoresis (6% PAA, 230 V, 30 min). The gel was visualized using a ChemiDoc Imaging System (Bio-Rad).
Plasmid cleavage assay
To perform plasmid cleavage assays, the binary complex composed of Cas12a and crRNA was pre-formed by incubating 100 nM of protein and 100 nM crRNA for 10 min at room temperature in 1× Cas buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 2% (v/v) glycerol, 1 mM DTT, 0.05% (v/v) Tween20). 5 or 37.5 nM of preincubated binary complexes were added to 5 nM of target DNA (eGFP-hAgo2 plasmid (Addgene #21981) linearized with SmaI). The reaction was incubated in 1× Cas buffer in a total volume of 15 μl for 1 h at 37°C. The reaction was stopped with 0.36 U Proteinase K. After incubation for 30 min at 55°C, the reaction was mixed with loading dye, and loaded onto a 0.8% agarose 1× TAE gel (100 V, 30 min).
In order to follow cleavage kinetics, 25 nM of the binary complex were added to 5 nM of target DNA and incubated at 28°C. After 15 s, 30 s, 1 min, 2 min, 3 min, 4 min, 5 min, 10 min, 15 min, 30 min, 1 h and 2 h aliquots of the reaction were stopped with EDTA (final concentration 83.3 mM) and 0.36 U Proteinase K. After incubation for 30 min at 55°C, the reaction was mixed with loading dye, and loaded onto a 0.8% agarose 1× TAE gel (100 V, 30 min).
Short DNA target cleavage assay
The binary complex of 200 nM Cas12a protein and 200 nM crRNA was pre-formed in 1× Cas buffer for 10 min at room temperature. In a final concentration of 75 nM, the binary complex was added to 10 nM fluorescently labeled double-stranded short target DNA in a total volume of 10 μl and incubated at 37°C for 1 h. The reaction was stopped by the addition of 0.36 U Proteinase K and incubated at 55°C for 30 min. Loading dye (final concentration: 47.5% (v/v) formamide, 0.01% (w/v) SDS, 0.01% (w/v) bromophenol blue, 0.005% (w/v) xylene cyanol, 0.5 mM EDTA) was added and the samples were separated on a pre-heated 15% PAA, 6 M urea, 1× TBE gel (300 V, 30 min). Fluorescent signals were visualized using a ChemiDoc Imaging System (Bio-Rad).
In order to follow cleavage kinetics, reactions were stopped after 1 s, 30 s, 1 min, 3 min, 5 min, 10 min, 20 min, 30 min, 45 min and 60 min with EDTA (final concentration 125 mM) and 0.36 U Proteinase K. Further steps were conducted according to the cleavage assay as described above.
The cleavage reactions for the high-resolution sequencing gel (15% PAA, 6 M urea, 1× TBE) were carried out with 375 nM of the binary complex and 50 nM of fluorescently labeled target DNA. After 1 h at 37°C, the reaction was stopped by the addition of 0.36 U Proteinase K and incubated at 55°C for 30 min. Loading dye (final concentration: 47.5% (v/v) formamide, 0.01% (w/v) SDS, 0.01% (w/v) bromophenol blue, 0.005% (w/v) xylene cyanol, 0.5 mM EDTA) was added and the samples were separated on the pre-heated sequencing gel (45°C, 65 W, 1 h). Fluorescence signals were visualized using a ChemiDoc Imaging System (Bio-Rad).
Protein Thermal Shift™ melting curves
Melting curves of Cas12a variants were generated using the Protein Thermal Shift™ kit (ThermoFisher) using the Rotor-Gene Q qPCR cycler from Qiagen and the HRM (High Resolution Melt) software. The proteins were diluted in water and measurements were performed in quadruplicates. The data were analyzed using the Origin software (ADDITIVE Friedrichsdorf, Germany).
Confocal single-molecule FRET measurements
Sample chambers for confocal measurements (CELLview Slide, Greiner Bio-One) were passivated with a passivation solution (2 mg/ml BSA in PBS) for 10 min and subsequently washed with 1× Cas buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 2% (v/v) glycerin, 1 mM DTT, 0.05% (v/v) Tween20). Labeled Cas12a apo protein was diluted to picomolar concentrations in 1× Cas buffer and transferred into the sample chamber. The binary complex composed of Cas12a protein with crRNA (final concentration 0.5 nM, 1 nM, 2 nM, or 4 nM) was pre-incubated for 30 min at room temperature in a small volume of 20 μl and prior to the measurement diluted by a factor of 10 with Cas buffer to yield a final volume of 200 μl. For the ternary complexes, the binary complex composed of Cas12a with crRNA (final concentration 1 nM) was pre-incubated for 10 min prior to addition of double-stranded or single-stranded target DNA (final concentration 1, 2 or 4 nM) and incubated in a volume of 20 μl for 20 min. The sample was diluted by a factor of 10 with 1× Cas buffer to yield the final volume of 200 μl. Single-molecule FRET measurements of freely diffusing molecules were carried out for 30 min.
The single-molecule measurements were conducted on a MicroTime 200 confocal microscope (PicoQuant) with pulsed laser diodes (532 nm: LDH-P-FA-530B; 636 nm: LDH-D-C-640; PicoQuant / clean-up filter: ZET 635; Chroma). Excitation of the fluorophores was done via pulsed interleaved excitation (PIE) with a laser intensity of 20 μW. To collect the emitted fluorescence, a 1.2 NA, x60 microscope objective (UplanSApo ×60/1.2W; Olympus) and a 50 μm confocal pinhole were used. The donor and acceptor fluorescence were separated by a dichroic mirror (T635lpxr; Chroma) and bandpass filters (donor: ff01-582/64; Chroma; acceptor: H690/70; Chroma). An individual APD (SPCM-AQRH-14-TR, Excelitas Technologies) for each photon stream detected the separated photon streams, which were subsequently analyzed by a TCSPC-capable PicoQuant HydraHarp 400 module.
Data analysis
Confocal smFRET data of diffusing molecules were analyzed with the software package PAM (38). Photon bursts were extracted by an all-photon burst search (APBS, parameters: L = 100, M = 15, T = 500 μs) and a dual-channel burst search (DCBS, parameters: L = 100, MGG+GR = 15, MRR = 15, T = 500 μs) (39). The obtained burst data were corrected for donor leakage and acceptor direct excitation, determined from the APBS data (40). β and γ factors were determined by applying an internal fit on distinct FRET populations in the ES-histograms obtained by the DCBS algorithm. The corrected DCBS data were binned (bin size = 0.032) and the mean value of triplicate measurements plotted as an E-histogram. The histograms were fitted with either one, two or three Gaussian fits using the Origin software. FRET efficiency histograms with standard deviations of the triplicates are shown in Supplementary Figure S18.
RESULTS
Mutations in the bridge helix of Cas12a alter the trimming activity and cleavage rate
In order to shed light on the function and importance of the bridge helix (BH), a series of mutations were introduced in FnCas12a (Figure 1B). The I960P variant was generated with the intention to disrupt the α-helical nature of the BH. The tryptophan at position 971 is accommodated in the hydrophobic pocket of the REC2 domain (31,32) (Figure 1C). This residue was mutated to a range of amino acids with different chemical properties (W971A, W971D, W971K, W971F), e.g. amino acids that differ in size and charge, to investigate whether the anchoring of the bridge helix is important for the structural integrity and catalytic function of Cas12a. Additionally, we created a variant in which the BH is deleted (ΔBH, ΔY953-K969, deletion of 17 amino acids). All variants could be purified to homogeneity (Supplementary Figure S1) and exhibited comparable stability as assessed by a thermal shift assay (Supplementary Figure S2).
The activity of wildtype (WT) Cas12a and the BH variants was analyzed in plasmid cleavage assays utilizing linearized plasmid DNA as target (Figure 1D, Supplementary Figure S3). Addition of WT Cas12a resulted in the expected formation of two cleavage products (Figure 1D). All BH variants were catalytically active but showed clear differences in cleavage efficiency. The ΔBH variant was the least active mutant with only 15% of target DNA being cleaved as compared to 91% cleaved by the WT enzyme after 60 min (Figure 1E). EMSAs revealed that the ΔBH variant is not impaired in DNA binding (Supplementary Figure S4) suggesting that the reduced cleavage activity is solely caused by a catalytic defect of the variants. Variants W971A and W971F are not impaired in their activity. The I960P variant was only marginally less active than the WT. Variants W971D and W971K remained active and showed ∼77% of the WT activity. A detailed study of the reaction kinetics was performed at 28°C to be able to follow the reaction with a good time resolution (Supplementary Figure S3). Here, cleavage products appeared already after 2 min for the WT enzyme. Variants W971K, W971D as well as I960P cleaved the DNA considerably slower than the WT. Interestingly, variant W971F cleaved the target slightly faster than the wildtype with 80% of the substrate being cleaved (WT: 45%) after 2 h of incubation. The ΔBH was not catalytically active under these conditions.
In order to evaluate the cleavage pattern created by the BH variants, we made use of a cleavage assay established by Swarts et al. based on fluorescently labeled short DNA target strands (58 nt, Figure 2A) (21). To follow the cleavage reaction on both strands simultaneously, a Cy3 and Cy5 label was attached to the 5′-end of the non-target strand (NTS) and the target strand (TS), respectively. Cleavage products were resolved on a denaturing sequencing gel in order to analyze cleavage products with single-nucleotide resolution (Figure 2B, Supplementary Figure S5 and S6). In case of the WT enzyme, a predominant cleavage product is visible and two minor products with one and two additional nucleotides are detected. In contrast, the variants show mainly longer cleavage products (up to 5 nt longer than the main product produced by the WT). Only the W971A and W971F variants generated products as short as the WT. Notably, this is not the main product of these variants. Kinetic studies using the WT enzyme (Supplementary Figure S5C, E) showed that also the WT produces longer NTS cleavage products at the start of the reaction. Over time, the long cleavage products are shortened to yield the main product. This is the result of a trimming activity that shortens the NTS by up to 4 nt (19). After cleavage of the target DNA, the PAM distal part of the DNA is released but Cas12a remains bound to the PAM proximal part (24) (here labeled with Cy3 at the NTS). While still bound, Cas12a further shortens the NTS via the trimming activity. Trimming of the NTS was also observed for the W971A and W971F variants yielding two or three main cleavage products including the final cleavage product observed for the WT (Supplementary Figure S6). However, trimming of the NTS by the mutated enzymes is less efficient as the final product is not the dominant product even after prolonged reaction times. In contrast, all other variants never yielded the final cleavage product of the WT enzyme. This suggests that variants I960P, W971D, W971K and ΔBH lack the trimming activity altogether. The cleavage pattern for the TS is the same for all Cas12a variants and shows one main cleavage product and two additional products 1 nt longer and 1 nt shorter than the main product. Our data are in agreement with previous studies that reported the imprecise cleavage of the TS by AsCas12a and FnCas12a (19,20).

DNA cleavage assay reports on cleavage accuracy and trimming activity of Cas12a variants. Cleavage assay of WT Cas12a and Cas12a bridge helix variants using a 7.5-fold excess of protein and crRNA (375 nM) relative to the target DNA (50 nM). The reaction was incubated at 37°C for 1 h. The short target DNA (58 nt) is doubly labeled with a Cy3 label (green) at the non-target strand (NTS) and a Cy5 label (red) at the target strand (TS). (A) Cy3 (green sphere) and Cy5 (red sphere) labels are attached to the 5′-end of the DNA strands. (B) The cleaved NTS shows a different cleavage pattern for the various Cas12a variants. The TS cleavage pattern is the same for all Cas12a variants. (C) Cy3 (green sphere) and Cy5 (red sphere) labels positioned at the 3′-end of the DNA strands. (D) The cleavage patterns of target and non-target strand are the same for all Cas12a variants. Samples were analyzed on a high-resolution denaturing 15% PAA gel.
The same target DNA construct carrying the fluorescent labels at the 3′-ends was used to analyze the cleavage pattern on the PAM-proximal part (Figure 2C,D). We found a comparable cleavage pattern for all Cas12a variants (Figure 2B). The 3′-labeled TS is processed yielding three products as already observed for the 5′-labeled DNA variant. Kinetic analysis shows that no trimming takes place at the PAM-distal part of the TS (Supplementary Figure S5D, F). In agreement with data by Swarts et al., we detected two cleavage products for the NTS that simultaneously appear after a very short cleavage time (2). This suggests an initial imprecise cleavage at two different positions, before the trimming of the PAM-proximal part takes place (19). These two initial cleavage sites were detected for all Cas12a bridge helix variants (Figure 2D).
Taken together, mutations of the BH shift the final cleavage position of Cas12a on the NTS towards the 3′ end of the DNA strand and the BH is crucially involved in the trimming activity of Cas12a.
Cas12a bridge helix variants with improved sensitivity to mismatches
BH variants of SpyCas9 have been shown to be more or less sensitive to mismatches between crRNA and target DNA (41,42). We tested whether mismatches influence the cleavage specificity of WT FnCas12a and the BH variants in an additional set of cleavage assays. To this end, we used substrates that carried a single mismatch at position 2 (MM 2, mismatch in the seed region), 10 (MM 10) or 20 (MM 20) of the R-loop (Figure 3A). WT FnCas12a showed a moderate sensitivity to substrates that carried a mismatch at position 2 or 10 but the MM 20 substrate was cleaved equally well as the fully complementary substrate (MM 0). A mismatch at position 2 led to a decrease in cleavage efficiency for the wildtype and to a similar extent in all BH variants of FnCas12a (Figure 3 B,C). The exception is the W971F variant that was significantly less active when using the MM 2 substrate as compared to the wildtype whereas this mutant was equally active as the WT when using the MM 0 substrate. All BH variants were very sensitive to a mismatch at position 10. Additionally, BH variants W971D and W971K did discriminate against a substrate with a mismatch at position 20. Notably, in case of FnCas12a variants, the relative reduction in cleavage efficiencies was more pronounced when using mismatched substrates as compared to fully complementary substrates (e.g. W971D reduction to 50% of WT level in case of MM 0 as compared to 3.6% in case of MM 10). Taken together, these data show that the target selectivity of FnCas12a is modulated by the BH.
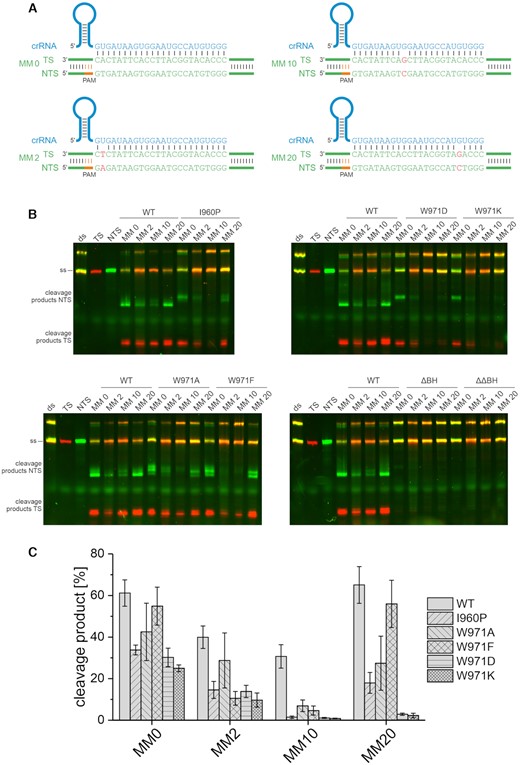
DNA cleavage specificity of Cas12a variants at mismatched substrates. (A) The short target DNA (58 nt) is doubly labeled with a Cy3 label (green) at the non-target strand (NTS) and a Cy5 label (red) at the target strand (TS). In addition to the fully complementary substrate (MM 0), substrates that carry a single mismatch at position 2 (MM 2), 10 (MM 10) or 20 (MM 20) were used. (B) Cleavage assay of WT Cas12a and Cas12a bridge helix variants using a 7.5-fold excess of protein and crRNA (75 nM) relative to the target DNA (10 nM). The reaction was incubated at 37°C for 1 h. (C) Quantification of cleavage efficiency of the NTS for the WT and Cas12a variants using fully complementary and mismatched substrates. Shown is the average, error bars show the standard deviation of three independent assays. While a substrate mismatched at position 10 is still cleaved by WT Cas12a, the BH variants do not cleave this substrate. BH variants W971D/K additionally do not cleave a substrate with a mismatch at position 20.
Cas12a adopts three conformational states during its activity cycle
A number of X-ray and cryo-EM structures are available for Cas12a that show the architecture of binary and ternary Cas12a complexes (2,17,20,21,30–32,43–48) and elucidated that a transition from the binary to ternary complex is accompanied by a structural re-arrangement from a closed to a more open state of the enzyme (Supplementary Figure S7). However, no structural information is available for the apo Cas12a enzyme and for the pre-crRNA-Cas12a complex in the absence of a DNA target. In order to follow the conformational transitions of Cas12a and to evaluate whether the BH mutations affect the conformation of Cas12a, we performed smFRET measurements on diffusing molecules. To determine relative intramolecular distances in WT Cas12a, we site-specifically incorporated the unnatural amino acid para-azido-l-phenylalanine (AzF) via the Amber-suppressor strategy (33,34). We placed the label either in the WED domain (K647AzF), the Nuc (T1222AzF) or REC lobe (D470AzF) yielding the following variants: Cas12aK647AzF/T1222AzF (termed Cas12aWED-Nuc), Cas12aD470AzF/K647AzF (referred to Cas12aWED-REC) and Cas12aD470AzF/T1222AzF (referred to Cas12aREC-Nuc) (Supplementary Figure S7). Coupling of the fluorescent dyes DyLight550 and DyLight650 to the azide group of AzF was performed via the Staudinger–Bertozzi ligation (36) (Supplementary Figure S8). We used a stochastic labeling approach that yields proteins that carry a donor and acceptor dye pair or proteins that carry only the donor or acceptor. Cas12a molecules that carry the donor-acceptor pair can be spectroscopically sorted based on the (PIE) excitation scheme (49) with separate detection channels for donor and acceptor emission. Crystal structures show the following distances between the chosen residues in the binary and ternary complex, respectively: positions K647 and T1222: 78.1 and 79.8 Å, D470 and K647: 98.0 and 104.4 Å, and D470 and T1222 30.8 and 36.0 Å (2,21) (Supplementary Figure S7). EMSAs and activity assays demonstrated that all variants were able to bind nucleic acids and to cleave the target DNA (Supplementary Figures S4 and S9).
First, we used the Cas12aWED-Nuc and Cas12aWED-REC variants to follow conformational changes of Cas12a along the WED-Nuc lobe axis and the WED-REC lobe axis, respectively. To this end, we performed smFRET measurements using (i) the apo enzyme, (ii) the binary complex composed of Cas12a plus pre-crRNA, (iii) the binary complex composed of Cas12a plus mature crRNA, (iv) the ternary complex (Cas12a + mature crRNA + dsDNA target) and (v) a ternary complex loaded with a single-stranded target. The Cas12aWED-REC and Cas12aWED-Nuc variants showed a single low FRET population for the apo enzyme, the binary and both variants of the ternary complex, respectively (Figure 4B). This indicates that the distance between WED and Nuc domain and the WED domain and REC lobe do not significantly change when Cas12a progresses through its activity cycle. This is in agreement with structural studies (2,21) and molecular dynamics simulations (50), in which only marginal changes in distances between these positions can be detected (Supplementary Figure S7). Using the Cas12aNuc-REC variant, a low FRET population with a FRET efficiency of 0.12 (± 0.04) and a high FRET population (E = 0.97 ± 0.01) can be detected for the nucleic-acid free Cas12a. This indicates that the apo form can adopt a closed and open conformation with the majority of the molecules (68 ± 7%) found in the closed state. Addition of crRNA to the protein shifts the molecules to the high FRET population (84 ± 2%). This effect is noticeable after the addition of mature crRNA, but also upon addition of pre-crRNA (Supplementary Figure S10A). Cleavage assays using samples that contained pre-crRNA or the mature crRNA (Supplementary Figure S10C,D) yielded comparable cleavage efficiencies. This demonstrates that the pre-crRNA is processed by Cas12a as only the mature crRNA allows loading and cleavage of dsDNA (16). Even though we cannot rule out that the pre-crRNA is partly processed during the incubation and single-molecule measurement time, the data nevertheless indicate that the pre-crRNA/Cas12a complex does not give rise to an additional FRET population and hence, it can be assumed that the pre-crRNA/Cas12a complex does not adopt a distinct conformation. This is in agreement with structural data (2,21). Notably, the formation of the binary complex is dependent on the crRNA concentration added to Cas12a underscoring the observation that the high FRET population reflects the binary complex (Supplementary Figure S11A). crRNA-binding therefore induces the closure of the protein reflecting the closed state of Cas12a (2) (Supplementary Figure S7). Upon loading of the DNA, a third population with a FRET efficiency of 0.82 (± 0.02) emerges. This indicates that the protein re-opens thereby providing enough space for the binding of target DNA. The relative number of molecules in the middle FRET population increased while the number of molecules that adopt the high FRET state decreased with increasing target DNA concentration indicating that the loading of DNA triggers the emergence of the middle FRET population (Supplementary Figure S11B). The measurements were also performed with the catalytically inactive variant of Cas12a (dCas12a, Supplementary Figure S12). Here, highly comparable data were obtained. This demonstrates that the medium FRET population corresponds to the ternary complex with bound target DNA and that the conformations of the ternary complex before cleavage (dCas12a) and after cleavage (WT Cas12a) of the target DNA do not significantly differ. Interestingly, loading is only moderately efficient and ∼30–50% of the proteins loaded with a crRNA remain in the closed FRET state (Supplementary Figure S11B). Additionally, we probed a ternary complex that lacks the NTS. Cas12a can process the TS even in the absence of the NTS (Figure 4, Supplementary Figure S13) (20). Omitting the NTS, the high FRET population drops to 9% (in the presence of NTS: 40%) and the middle FRET population increases to 60% indicating that loading of the sterically less demanding single-stranded TS is very efficient allowing a successful transition from the binary to the ternary complex. The FRET efficiency of the middle FRET population, however, does not significantly change suggesting that loading of the TS is sufficient to induce the opening of DNA cleft. Taken together, we show that the smFRET measurements re-capitulate the structural data of the binary complex (closed state) and the opening movement of Cas12a upon target DNA loading (semi-closed state). A single-stranded TS is sufficient and more efficient to induce the transition into the semi-closed state. Moreover, we can assess the structural state of the apo enzyme and show that apo Cas12a can adopt an open and closed conformation.
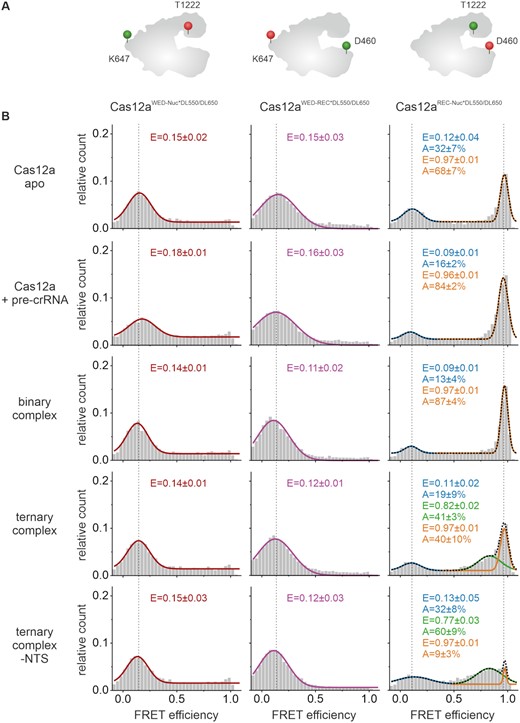
Förster resonance energy transfer (FRET) measurements reveal conformational states of Cas12a. Single-molecule FRET measurements using doubly labeled Cas12a variants were performed on freely diffusing molecules. (A) Schematic model of Cas12a showing the labeling positions at D470 (REC domain), K647 (WED domain), and T1222 (Nuc domain). (B) FRET efficiency distributions for the Cas12a apo enzyme, the Cas12a with pre-crRNA (1 nM), the binary complex (1 nM crRNA), the ternary complex (1 nM crRNA, 1 nM target DNA), and the ternary complex without NTS (1 nM crRNA, 1 nM TS DNA). Cas12aWED-Nuc*DL550/DL650 and Cas12aWED-REC*DL550/DL650 display one single FRET population for the apo, RNA- and RNA-and-DNA-bound state at a FRET efficiency of 0.15 ± 0.01 and 0.13 ± 0.02, respectively. Cas12aREC-Nuc*DL550/DL650 shows two FRET populations for the apo complex and the RNA-bound state at E1 = 0.12 ± 0.04 and 0.09 ± 0.01 and E2 = 0.97 ± 0.01 or 0.96 ± 0.01 with the high FRET population increasing with the addition of RNA. In the ternary complex, Cas12aREC-Nuc*DL550/DL650 shows three FRET populations with an additional population at 0.82 ± 0.02 FRET efficiency. Histograms show the data of three independent measurements. The histograms were fitted with a single, double or triple Gaussian function and the mean FRET efficiencies (E) and the percentage distribution of the populations (A) are given with standard errors in the histograms. Measurements were performed in triplicates (see also Supplementary Figure S18A).
An intact BH is required for efficient binary complex formation
Being able to follow the opening and closure of the protein using the Cas12aNuc-REC variant, we additionally interrogated all BH variants to understand whether the BH is involved in the stabilization of the conformational states of Cas12a (Figure 5 and Supplementary Figure S18B). For all BH variants, low, middle and high FRET populations were detected. However, differences in the relative distribution of these states were observed. In the apo state, all W971 variants showed a shift towards the low FRET population suggesting that a destabilization or modulation of the anchoring of the BH in the REC lobe increases the probability to adopt the open state. Interestingly, in case of the ΔBH variant, the FRET efficiencies of the respective populations did not change and high and low FRET states were almost equally populated. As the residue W971 is still present in this variant, we hypothesized that this might be sufficient to maintain the opened and closed state. In order to test this hypothesis, we made use of the so-called ΔΔBH in which not only residues 953–969 of the BH but in addition residues 970/971 were deleted resulting in a Cas12a variant fully devoid of the BH. Here, the relative distribution of the FRET states and the FRET efficiencies were not significantly altered (Supplementary Figure S14). For all Cas12a variants, no dynamic switching between the open and closed state was observed during the millisecond timescale that we capture with our measurements. While a shift towards the low FRET population was observed for the W971 variants, the defined structural states represented by the low and high FRET population can still be adopted by the BH deletion variants. We therefore suggest that the BH is not the main structural element that defines the open and closed state of Cas12a.
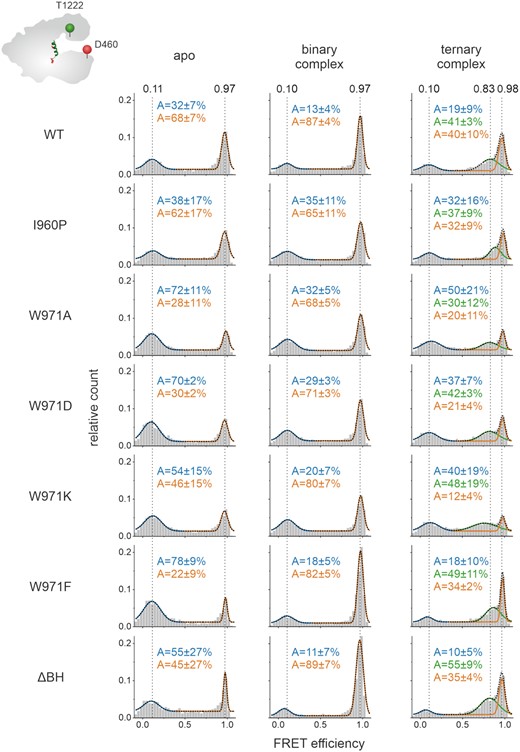
Förster resonance energy transfer (FRET) measurements reveal conformational states of Cas12a bridge helix variants. Single-molecule FRET measurements on freely diffusing molecules were performed using the doubly labeled Cas12aREC-Nuc*DL550/DL650 variant that carried additional mutations in the bridge helix. FRET efficiency distributions for the apo enzyme, the binary complex (1 nM crRNA), and the ternary complex (1 nM crRNA, 1 nM target DNA) are shown. The apo histograms exhibits two FRET populations corresponding to an open and closed state of Cas12a, respectively. The addition of crRNA leads to an increase of the high FRET population in the binary complex. Addition of crRNA and target DNA give rise to a third population (semi-closed conformation). The histograms were fitted with a double or triple Gaussian function and the averaged mean FRET efficiencies (E) and the percentage distribution of the populations (A) are given with SEs in the histograms. Measurements were performed in triplicates (see also Supplementary Figure S18B). FRET efficiencies for the single protein variants are listed in Supplementary table S2.
All variants adopt the closed conformation once the crRNA is loaded into the enzyme. crRNA titration experiments revealed, however, that the formation of the high FRET population is less efficient for variants I960P, W971A/D/K (Supplementary Figure S11A). In contrast, variant W971F and the ΔBH variant readily formed the closed state upon crRNA addition even at low crRNA concentrations (Supplementary Figure S11A). If a variant preferentially adopted the open state in the apo form, the loading of crRNA was inefficient. This suggests that for a subset of BH variants binding of the crRNA or the conformational transition from the open to the closed state is impaired resulting in reduced amounts of the binary complex (Supplementary Figure S15). In contrast, formation of the ternary complex is efficiently induced upon target DNA addition in all BH variants (Figure 5 and Supplementary Figure S11B) implying that the mutations in the BH do not affect the loading of the target DNA. Please note that in all Cas12a variants, the middle FRET population shows relatively broad distribution suggesting that this state is more flexible as compared to the high FRET state (Supplementary Figure S16).
Taken together, the smFRET data indicate that a subset of BH variants is not only impaired in their cleavage activity but that changes in the BH also disturb efficient binary complex formation.
DISCUSSION
The single effector nucleases Cas9 and Cas12a of the CRISPR–Cas type II system form a characteristic bilobal structure comprised of the REC and Nuc lobe, which are connected by the bridge helix. In Cas9, the BH reaches deep into the REC lobe and contacts directly or indirectly the target DNA, crRNA and tracrRNA (51–56). An arginine-rich patch in the BH orientates the seed nucleotides of the crRNA to render them solvent-exposed. Thus, the crRNA can readily base-pair with the target DNA strand. Consequently, mutations in the BH of Cas9 of Streptococcus pyogenes (SpyCas9) influence RNA and target DNA binding, mismatch sensing and RNA-loop stability (41,42,56,57). Several SpyCas9 BH variants showed increased specificity rendering them more suitable for genetic engineering purposes (41). The BH of FnCas12a also connects the REC and Nuc lobe but does not insert as deeply into the REC lobe as observed for SpyCas9 (2,21). The BH of FnCas12a is not in close contact with the target DNA or crRNA and only contacts the sugar-phosphate backbone of the crRNA (31). The seed region of the crRNA is arranged by the REC1 domain instead. Nonetheless, our mutational analysis shows that the BH of Cas12a contributes to the catalytic activity and specificity of the enzyme. Deletion of the entire BH severely reduced the catalytic activity. Similarly, a BH deletion variant of SpyCas9 was inactive (58). This shows that the BH is an essential segment for both enzymes. Additionally, we evaluated whether the helical nature of the BH or the anchoring of the BH in the REC domain is a pre-requisite for full catalytic activity. Introduction of a proline residue at position 960 in the BH resulted in a reduced NTS-trimming activity showing that the helical nature is required to reach the full activity spectrum of Cas12a. Disturbing the anchoring of the BH in the REC domain affected the cleavage reaction rate and/or trimming activity. Charged amino acids led to a reduction of the DNA cleavage rate. Additionally, the trimming activity of Cas12a was lost. In contrast, an alanine substitution did not affect the cleavage and slightly reduced the trimming activity. Replacement of W971 by a phenylalanine residue only marginally influenced the cleavage activity. Trimming of the NTS was only slightly affected. The differing effect of the mutations can be rationalized from a biochemical perspective: charged residues cause a repulsion of the BH from the hydrophobic pocket in the REC2 domain whereas the small and uncharged alanine substitution might still fit in the pocket. Phenylalanine is also a hydrophobic residue and can most likely functionally replace tryptophan. Consequently, the variant is only mildly affected. Similar to some variants evaluated in this work, Swarts et al. identified Cas12a mutants that exhibited reduced trimming activity (e.g. R918, D920, S1083, R1218, D1255, D1227). However, these variants are constituting the active site or are in close contact to the catalytic residues. This is not the case for the variants tested in this study. Notably, all BH variants showed a higher selectivity in DNA targeting. In contrast to the wildtype, all BH variants did not tolerate substrates with a mismatch at position 10 of the R-loop. Additionally, BH variants W971K and W971D did discriminate against substrates with a mismatch at position 20. Swarts et al. tested the cleavage efficiency of FnCas12a using linearized plasmid substrate variants with mismatches to the crRNA (2). These experiments showed that a mismatch at position 2 prevented cleavage of the DNA while a mismatch at position 10 and 20 resulted in approximately 50% and 100% cleavage, respectively. The results for mismatch position 10 and 20 are in agreement with our data. However, we still observe DNA target cleavage with the MM 2 substrate (65% relative to the MM 0 substrate). Possibly, the nature of the substrate (linearized plasmid versus short dsDNA) causes these differences. Strohkendl et al. showed that mismatches throughout the R-loop slow down binding of AsCas12a to the DNA (19). As R-loop formation is the rate-limiting step for cleavage, the reduced rate of R-loop formation impacts the substrate cleavage rate. In case of AsCas12a, cleavage rates were most dramatically decreased when a substrate with mismatches at position 2, 4 or 16 was used while mismatches at position 10, 18 and 21 only led to a slight decrease in cleavage rate. Our data therefore reveal differences in the relative tolerance of AsCas12a and FnCas12a towards mismatches throughout the R-loop (e.g. a mismatch at position 2 was well tolerated by FnCas12a but led to an almost 100-fold reduction in cleavage rate in case of AsCas12a). In case of SpyCas9, introduction of a double proline substitution in the BH led to an increase in specificity. However, these effects can be rationalized by the stabilizing role of the BH in R-loop formation in SpyCas9 (41,42). The BH of FnCas12a only contacts the phosphate backbone of the crRNA especially in the pseudoknot region (2). We therefore hypothesize that the BH stabilizes the R-loop in an indirect fashion. Moreover, destabilization of the BH anchoring seems to increase the mismatch sensitivity. Given that the W971K/D variants also showed a reduced cleavage and trimming activity it seems plausible that the anchoring of the BH is essential for the stable formation of the R-loop.
In order to rationalize the observed trimming defects and mismatch sensitivity based on structural data, we carefully inspected all Cas12a structures available. Strikingly, we found that the BH length expands upon ternary complex formation. Notably, structural changes in the BH occur in tandem with structural changes in the adjacent helix 1 of the RuvC domain (Figure 6A–D). The re-positioning of the binding pocket in the REC domain requires the positional shift of tryptophan 971 by 8.7 Å to allow docking of the residue in its binding pocket also in the ternary complex (Figure 6A, B). To promote this movement of W971, the residue has to be released from helix 1 as observed in ternary complex structures that show that helix 1 is shortened by six residues thereby releasing W971 into a flexible loop that connects the BH and helix 1. Concomitantly, the BH is extended by approximately two residues and four residues in FnCas12a and LbCas12a, respectively. Hence, these two helices act in concert as ‘suspension helices’ to compensate for the large conformational changes of the REC domains upon transition to the ternary complex. Part of the BH is not resolved in the binary complex structure indicating that this is a flexible region in the Cas12a–crRNA complex. Furthermore, a conserved lysine residue (K978 in FnCas12a) in helix 1 contacts the backbone of the target DNA in the ternary complex. Due to a shift in the position of helix 1 upon transition to the ternary complex, K978 is ideally orientated for the interaction with the DNA. While K978 is positioned in the middle of helix 1 in the binary complex, it is shifted to the N-terminal end of the re-modelled helix 1 and not released into the flexible loop in the ternary complex. This indicates that the integration of K978 into helix 1 is essential to function as DNA-contacting base. It seems likely that some of the mutations introduced into the BH of FnCas12a affect the tandem movement of the BH and helix 1 and consequently, DNA contacts and the position of the REC domains are slightly altered. This might include contacts of helix 1 residues with the phosphate backbone of the target DNA in the R-loop (e.g. K978) (2). In turn, this might have a negative impact on the R-loop formation process and thereby increases the mismatch sensitivity of FnCas12a. Altogether, these disruptions most likely cause the observed trimming defect. For example, formation of an extended BH in the ternary complex might be prevented by the I960P variant that is located in the middle of the BH. However, while it is assumed that a proline breaks a helix, we cannot rule out that the introduction of a proline can be accommodated by the flexible nature of the BH. Similarly, the absence of the entire BH completely disrupts the coupled helical restructuring events. In addition to repulsion of the W971D or W971K residues from the hydrophobic binding pocket, these substitutions might also affect the formation of the extended BH as position 971 re-positions to the very beginning of the loop (next to the end of the BH). Notably, the tandem helix arrangement is conserved in all Cas12a variants studied so far (Supplementary Figure S17).
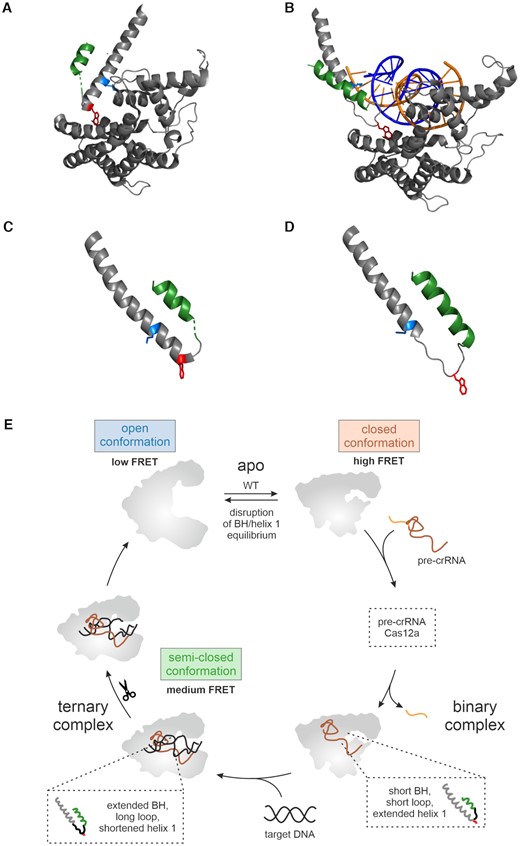
Impact of the bridge helix on the conformational flexibility and catalytic activity of Cas12a. (A) and (B) Comparison of the structural rearrangement of the BH and helix 1 relative to the REC domain upon transition from the binary to ternary complex. (A) The structural elements in the binary (PDB: 5NG6) and (B) in the ternary (PDB: 6I1K) FnCas12a complex. The structures were aligned relative to the REC domain as reference point. For clarity, only the bridge helix (BH, green), helix 1 and the REC domain (dark grey) are shown. Tryptophan 971 is highlighted in red and lysine 978 in blue. (C and D) Comparison of the length of BH, helix 1 and connecting linker in the (C) binary (PDB: 5NG6) and (D) ternary complex (PDB: 6I1K). Tryptophan 971 is highlighted in red and lysine 978 in blue. (E) In the apo state, FnCas12a can adopt two conformations: a closed (high FRET population) and an open state (low FRET population). Binding of (pre-)crRNA induces the closure of the enzyme. In order to bind a single-stranded or double-stranded target DNA, Cas12a re-opens again giving rise to a medium FRET population in single-molecule FRET measurements. The bridge helix (BH, in green) is a central structural element in Cas12a that connects the REC and Nuc lobe with the tethering residue tryptophan 971 (shown in red) that anchors the BH in a hydrophobic pocket in the REC lobe. The length of the BH (green), the adjacent helix 1 (gray) and the interconnecting linker (black) as well as the position of W971 re-adjust upon transition from the binary to the ternary complex (see insets). Mutations in the BH or replacement of W971 affect trimming activity of Cas12a and influence the equilibrium between open and closed state of Cas12a.
Crystal and cryo-EM structures (2,17,20,21,30–32,43–48) as well as smFRET studies (20,23,59) indicated that not only the protein itself but also the bound nucleic acids undergo substantial conformational changes. Hence, we employed smFRET measurements to follow the movement of the REC and Nuc lobe of Cas12a WT and the BH variants. Our data faithfully replicate the structural states of the binary and ternary complex known from crystal structures and add hitherto missing information about the conformational state of the apo enzyme. More specifically, we observe a closure of the Cas12a protein upon loading of the crRNA and a slight re-opening upon loading of the DNA. This is in agreement with structural data for FnCas12a and AsCas12a (2,17,20,21,30–32,43–48). Dong et al. observed that LbCas12a is conformationally flexible in its apo state and that addition of crRNA to LbCas12a led to a defined structural state that could be analyzed using cryo-electron microscopy (30). These data are in line with our smFRET data that led to the detection of two conformational states for the FnCas12a apo enzyme and a defined conformational state for the binary complex (reflected by the high FRET population). Our finding that the ternary complex is more flexible than the binary complex agrees with recent molecular dynamics simulations that showed the increased flexibility of the REC and Nuc lobe in the ternary complex (50). We found that loading of the DNA target is more efficient when a ssDNA target is provided suggesting that the formation of a Cas12a–crRNA–dsDNA complex is energetically demanding. In line with crystal structures, the expansion of REC and Nuc lobe upon transition to the ternary complex does not depend on the single-stranded or double-stranded nature of the DNA and loading of the TS is sufficient to induce the conformational change in Cas12a (21). Conformational heterogeneity was detected for the apo enzyme. Here, Cas12a can adopt an open or closed state. No crystal or high resolution cryo-EM structure of the apo enzyme is available so far. But the existence of an open state has been proposed based on smFRET studies by Stella et al. While we performed smFRET measurements on diffusing molecules, Stella et al. employed immobilized Cas12a proteins to follow the relative distance between the REC and Nuc lobe (20). The apo enzyme and all complexes tested in the study by Stella et al. showed a wide range of conformations that could not be discerned as discrete states. However, in the apo state, a relatively high fraction of the molecules was found in a low FRET state supporting the finding of an open state in this study. Considering the central position of the BH, it seemed feasible that a deletion of the BH might result in a collapsed state of Cas12a. However, this was not observed in our single-molecule FRET measurements. Interestingly, the structural integrity is preserved even in the ΔBH/ΔΔBH variants and these variants can load crRNA and target DNA efficiently. Considering the previous finding that the BH structurally re-arranges in concert with helix 1 of the RuvC I domain it seems plausible that an intact helix 1 (possibly in addition to structural elements in the PI and WED domains) is responsible for the correct distance between the REC and Nuc lobe. Consequently, the BH only influences the equilibrium between the open and closed state in the apo enzyme. Additional structural data that elucidate the structural state of the BH and helix 1 in the apo enzyme would be of high interest in this context.
Noteworthy, the variants that cause a reduced trimming activity produce a more precise cleavage pattern as cleavage occurs mainly at two sites. In contrast, the WT enzyme produces at least five intermediate products on the way to the two final products. Moreover, especially the W971D and W971K variants additionally discriminate better against mismatched substrates rendering these Cas12a variants more specific. While crRNA loading seems to occur with reduced efficiency in these variants, they do not affect the conformational changes required for the catalytic cycle of Cas12a. Hence, these variants might be better suited for gene editing applications than the WT as they might warrant DNA cleavage with increased specificity and selectivity.
DATA AVAILABILITY
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We gratefully thank Dr Sarah Willkomm and Dr Kevin Kramm for fruitful discussions and Elisabeth Piechatschek and Elke Papst for technical assistance.
Author contributions: D.G. conceived the study. E.W., L.J. and A.S. performed the single-molecule measurements. E.W., L.J. and A.S. analyzed the single-molecule data. E.W., L.J., A.S. and G.Z. purified the proteins and created Cas12a variants. E.W., L.J., A.S. and G.Z. performed EMSAs and cleavage assays. E.W. and D.G. wrote the paper. All authors commented on the paper.
FUNDING
Deutsche Forschungsgemeinschaft [SPP2141 and SFB960-TP7 to D.G.]. Funding for open access charge: Deutsche Forschungsgemeinschaft [SPP2141 and SFB960-TP7 to D.G.].
Conflict of interest statement. None declared.
Notes
Present address: Leonhard Jakob, Department of Pharmacology and Toxicology, Institute of Pharmacy, University of Regensburg, 93053 Regensburg, Germany

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments