





Abstract
Replicative helicases are essential proteins that unwind DNA in front of replication forks. Their loading depends on accessory proteins and in bacteria, DnaC and DnaI are well characterized loaders. However, most bacteria do not express either of these two proteins. Instead, they are proposed to rely on DciA, an ancestral protein unrelated to DnaC/I. While the DciA structure from Vibrio cholerae shares no homology with DnaC, it reveals similarities with DnaA and DnaX, two proteins involved during replication initiation. As other bacterial replicative helicases, VcDnaB adopts a toroid-shaped homo-hexameric structure, but with a slightly open dynamic conformation in the free state. We show that VcDnaB can load itself on DNA in vitro and that VcDciA stimulates this function, resulting in an increased DNA unwinding. VcDciA interacts with VcDnaB with a 3/6 stoichiometry and we show that a determinant residue, which discriminates DciA- and DnaC/I-helicases, is critical in vivo. Our work is the first step toward the understanding of the ancestral mode of loading of bacterial replicative helicases on DNA. It sheds light on the strategy employed by phage helicase loaders to hijack bacterial replicative helicases and may explain the recurrent domestication of dnaC/I through evolution in bacteria.

DciA is the antecedent but unrelated to the helicase loaders DnaC/I. Structurally, the N-terminal domain of DciA is related to the NTD of DnaA, whereas its CTD is unfolded. DciA associates with DnaB according to a 3/6 stoichiometry which loads the helicase more efficiently on DNA. We identified in DnaB proteins a determinant residue that discriminates DciA- and DnaC/I-helicases and which happens to be critical in vivo. The determinant residue is located at the center of the DciA binding site.
INTRODUCTION
Replicative helicases are essential enzymes that unwind double-stranded DNA in front of the replication fork in the three domains of life. Bidirectional replication initiated at the origin of replication of the chromosome is conditional on the recruitment and the loading of two replicative helicases, installed head to head at oriC to form a stable nucleoprotein platform on which the replication machinery will then be assembled to generate two processive replisomes moving outwards from oriC (1). The loading and the activation of replicative helicases onto DNA are critical steps that are strictly regulated to prevent the initiation of unforeseen replications or improper assembly of functional replisomes.
Replicative helicases of bacteria and eukaryotes distribute into two distinct groups of unrelated origin. Bacterial replicative helicases (DnaB) belong to the superfamily 4 (SF4) and translocate on the lagging strand in the 5′→3′ direction, whereas the MCM (Mini Chromosome Maintenance) helicases of Archaea and Eukaryotes belong to the superfamily 6 (SF6) and translocate in the 3′→5′ direction on the leading strand (2). The MCM2–7 helicases are ring-shaped hetero-hexamers (3,4), while bacterial replicative helicases are toroid-shaped homo-hexamers (5–7). Each DnaB subunit consists of an N-terminal domain (NTD) connected through an α−helical linker (LH) to a C-terminal domain (CTD) that harbors a RecA-like fold (6). Adjacent NTDs form homodimers through a 4-helix bundle arrangement and associate into a trimer of dimers, whereas CTDs form an assembly with approximately hexameric symmetry (7–9).
In Eukaryotes, loading and activation of the MCM helicases are uncoupled and strictly insulated in the G1 and S phases of the cell cycle, respectively (10). In bacteria, the delivery of replicative helicase at oriC during the replication initiation was studied in model organisms from the standpoint of the two domesticated phage helicase loaders: DnaC and DnaI (11,12). This led to the characterization of two loading strategies. In organisms such as Escherichia coli, the helicase is cracked open by DnaC in a ‘ring-breaker’ strategy. In others, like Bacillus subtilis, DnaI assists the assembly of the helicase around DNA according to a ‘ring-maker’ scenario (13–15). In these bacterial systems, loading and activation of replicative helicases are believed to be coupled, resulting from a consecutive structural reorganization of the helicase following its binding on ssDNA. The control of replication initiation in bacteria is proposed to be implemented at an earlier stage, when the initiator protein DnaA oligomerizes at the unique origin of replication of the chromosome, oriC, to form the open complex (16).
Yet, most bacterial genomes lack dnaC or dnaI. Out of the 26 phyla investigated, only five contain species in which we identified dnaC or dnaI. The other species possess instead dciA, a gene of ancestral and bacterial origin that encodes a protein shown to interact with the DnaB replicative helicase and to be essential in the early steps of replication initiation (11,12). Phylogenetic analyses showed that the emergence of the dnaC/I gene in the bacterial domain occurred by waves of domestication of phage helicase loaders and systematically at the expense of dciA (12). The fast elimination of dciA upon domestication of dnaC, suggesting incompatibility between the two proteins, remains elusive. Yet, the frequent replacement of dciA by a phage-related gene through evolution suggests that the activities of these two proteins could fulfill the same function (11,12). Although phylogenetically related, DnaC (from E. coli) and DnaI (from B. subtilis) manage the replicative helicase quite differently (14). The characterization of the function of DciA, its implication on the formation of the DNA•helicase complex and the comparative analysis with DnaC might help refine our understanding of the function of these proteins in the management of the replicative helicase during the bacterial replication initiation.
To describe the specificity of the DciA-based system, we conducted a biochemical and biophysical study of DciA and DnaB from Vibrio cholerae (Vc). We show that even though the hexameric structure of VcDnaB is closely related to that of DnaB from E. coli, a well-characterized DnaC-assisted helicase, the helicase of V. cholerae is able to load itself on DNA. Also, we show that VcDciA has a clear stimulatory effect on the loading of VcDnaB, and that the two proteins interact with each other in a 3/6 (DciA/DnaB) stoichiometry. Finally, we identified a residue of the replicative helicase (the so-called ‘determinant’ residue), which is strictly co-occurring with either DciA- or DnaC, and appears to be critical in vivo.
MATERIALS AND METHODS
Protein samples preparation
Cloning of dnaB, dciA, dciA[1–111] and dnaC coding regions was performed using genomic DNA from Vibrio cholerae ATCC 39315 strain and E. coli F11 strain as templates for PCR. Six histidine codons were added at the 3′-end of each dnaB (Vc and Ec) and of dnaC constructs, and at the 5′ end of dciA constructs, during the PCR process. The fragments were inserted into the NdeI-XhoI sites of pET21 or pET29 vectors (Novagen), respectively for dnaB and dciA/dnaC. To construct the VcDnaBS296G and the EcDnaBG299S mutants, site directed mutagenesis was performed using non-overlapping and 5′-phosphorylated oligonucleotides (Eurofins), which introduce the desired changes. The complete plasmid is amplified by PCR in a linear product and is circularized by ligation.
Over-expression of the various constructs was performed in 800 ml 2xYT Erlenmeyer flasks in the BL21(DE3)Gold strain o/n at 15°C, after induction with 0.5 mM IPTG (Sigma) and co-expression of chaperone proteins in the case of VcDnaB (17). Cells were harvested by centrifugation, resuspended in buffer A (NaCl 200 mM, Tris–HCl 20 mM (pH 7.5)) for all of the constructs and stored at –20°C. Cell lysis was completed by sonication (probe-tip sonicator Branson). After centrifugation at 20,000 g and 8°C for 30 min, the His-tagged proteins were purified on a Ni-NTA column (Qiagen Inc.), eluted with imidazole in buffer A but complemented by ATP 1 mM + MgCl2 3 mM in the case of all the helicases. VcDciA, VcDciA[1–111] or EcDnaC were then loaded onto a Heparin column in a phosphate buffer [NH2PO4 20 mM, NaCl 100 mM (pH 5.8)] and eluted with a gradient of NaCl (from 0.1 to 1 M). The elution fractions centered at 500 and 250 mM NaCl respectively for VcDciA and EcDnaC were selected. All the helicases were loaded onto a Superdex TM200 column (GE), equilibrated against NaCl 100 mM, ATP 1 mM, MgCl2 3 mM, Tris–HCl 20 mM (pH 8.8). Their purification was completed with a MonoQ column (Amersham Pharmacia Biotech) in Tris–HCl (20 mM, pH 8.8) and a gradient of NaCl (from 0.1 to 1 M). The elution fractions centered at 450 mM NaCl were selected. All the proteins were desalted up to 200 mM NaCl and concentrated using 5000 or 30 000 nominal molecular weight limit cut-off (respectively for DciA/DnaC and DnaB) centrifugal concentrators (Vivaspin), aliquoted, flash frozen in liquid nitrogen and stored at –80°C, or dialyzed in a 50% (vol/vol) glycerol buffer for a storage at –20°C.
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) was measured using a Proteon XPR36 instrument (Bio-Rad). The measurements were performed at 30°C in a PBS buffer (Bio-Rad) added with 0.01% of Tween 20 (PBST) and with or without ATP 0.5 mM. NLC sensor chips (Bio-Rad) were used to immobilize the oligonucleotides oso13 or oso15 (Supplementary Table S1) through their biotin-tag. For immobilization, DNA were diluted in PBST and attached to the chip to obtain 50 RU in different orientations (3′ free for oso13 or 5′ free for oso15). For the observation of kinetic data, two different kinds of experiments were performed. In experiments where loading and translocation were coupled, proteins were injected in PBST with 0.5 mM ATP and 10 mM MgCl2 at 50 μl/min during 240 sec, and dissociation was run during 900 sec. In experiments where loading and translocation of the helicase on DNA were uncoupled, proteins were injected in PBST with 0.5 mM ATP during same duration, and the 900 sec dissociation was followed by injection of PBST with ATP 0.5 mM plus MgCl2 10 mM during 459 sec. In all experiments, the proteins were injected at different concentrations and different molar ratios, as indicated in the Results section. After each interaction test, the chip was regenerated using 0.5% of SDS. After corrected by subtraction of the uncoated reference channel, the sensorgrams were analyzed and compared.
Crystal structure
Purified VcDnaB was preincubated for 10 min at 4°C at a concentration of 0.115 mM (monomer) with 2 mM GDP + 5 mM NaF + 0.5 mM AlCl3 + 5 mM MgCl2 for crystallization. Native protein crystals were grown in sitting drops by mixing the protein with reservoir solution in a 1:1 ratio. Crystals of VcDnaB•GDP:AlF4:Mg2+ appeared after 20 days at 18°C in 0.1M MES (pH6.0) + 10% (v/v) MPD Glycerol cryo-protected crystals (two steps at 15 and 30%) were flash frozen in liquid nitrogen.
Diffraction data and refinement statistics are given in Supplementary Table S2. Crystallographic data were collected at the PROXIMA-2 beamlines from Synchrotron SOLEIL (Saint-Aubin, France) and processed with XDS (18) through XDSME (https://github.com/legrandp/xdsme). Diffraction anisotropy was corrected using the STARANISO server (http://staraniso.globalphasing.org). The VcDnaB•GDP:AlF4:Mg2+ crystal structure was solved by molecular replacement with the MOLREP program (19) using the X-ray structures of the NTD [L24-D176] and CTD [Y211-G465] isolated domains of E. coli DnaB as search models (PDB ID 6QEL). Iterative improvements of the initial model were obtained by normal-mode refinement using iMODFIT (20) in triclinic expanded electron-density maps calculated after refinement with the BUSTER program (21). The model refinement was conducted with the BUSTER program using TLS and strong non-crystallographic symmetry restraints (22) keeping the CTD and NTD separated. The model was corrected and completed using COOT (23).
Small angle X-ray scattering
Proteins and protein complexes were prepared as described above and in the Supplementary Material and Methods section. SAXS experiments were performed on the SWING beamline at the synchrotron SOLEIL (St-Aubin, France). The sample-to-detector (Eiger 4M Dectris) distance was set to 1789 mm and the wavelength λ to 1.0 Å, allowing useful data collection over the momentum transfer range of 0.005Å−1 < q < 0.5 Å−1. SAXS data were collected directly after elution of the proteins or protein complexes through the on-line size-exclusion high-performance liquid chromatography column (Superdex Increase 200 10/300, GE Healthcare) equilibrated in Tris–HCl 20 mM (pH 8.8), NaCl 100 mM, with or without ATP 1 mM (without Mg2+). 100 μl of protein samples were injected at 15°C at an initial concentration close to 10 mg/ml. The flow rate was 0.5 ml/min, frame duration was 2.99 s and the dead time between frames was 0.01 s. Scattering of the elution buffer before void volume was recorded and subtracted from all protein scattering curves. The scattered intensities were displayed on an absolute scale using the scattering by water. For each frame, the protein concentration (below 1 mg/ml at the top of elution peak) was estimated from UV absorption at 280 nm using a spectrophotometer located immediately upstream of the SAXS measuring cell. Data were first analyzed using Foxtrot, the Swing in-house software, and then using the US-SOMO HPLC module (24). This program provided for each SAXS frame the values of the scattering intensity I(0) and of the radius of gyration Rg by applying the Guinier analysis together with a calculation of the approximate molar mass using the Rambo and Tainer approach (25). Identical frames under the main elution peak were selected using Cormap (26) and averaged for further analysis. Finally, the distance distribution functions P(r) were calculated from the averaged scattering curves using the GNOM program from the ATSAS suite (27). The determination of the all-atoms model of VcDciA from the SAXS curves was performed using the program DADIMODO available at SOLEIL (28,29). This program starts from the atomic structure obtained using crystallography or NMR and models and refines the missing parts by adjusting the calculated curve on the model to the experimental curve. All the SAXS values are given in Supplementary Table S3.
NMR experiments
Production of 15N and 13C labeled VcDciA[1–111] was obtained using the same protocol as described above except that the cellular culture was performed in a minimal medium supplemented with 15N ammonium chloride and 13C glucose (Eurisotop) (30). NMR experiments were carried out on a Bruker AvanceII-700 MHz spectrometer. For assignment experiments, purified 15N–13C VcDciA[1–111] was concentrated up to 0.9 mM, in the NMR buffer (20 mM phosphate buffer (NH2PO4) pH 5.8, NaCl 50 mM, 2 μM EDTA, 0.2 mM DSS, 0.1 mg/ml NaN3, 10% D2O). Data for assignment of the backbone resonances were collected at 293 K using standard 1H-15N HSQC, 15N-edited NOESY-HSQC, TOCSY-HSQC, HBHA(CO)NH, HN(CA)N, HNCO, HNCA, HN(CO)CA, HN(CA)CO and CBCA(CO)NH experiments. Proton chemical shifts (in ppm) were referenced relative to internal DSS and 15N and 13C references were set indirectly relative to DSS using frequency ratios (31). NMR data were processed using Topspin (Bruker) and analyzed using SPARKY 3 (T.D. Goddard and D.G. Kneller, UCSF).
Chemical shift indices and structural model of VcDciA[1-111] from NMR data
Chemical shift indices were calculated for VcDciA[1–111] based on comparison of the chemical shift values obtained from NMR experiments with random coil chemical shifts calculated with sequence correction factors, temperature and pH corrections and GGXGG-based neighbor correction for glycines (32). Structural models of VcDciA[1–111] were computed with CS-ROSETTA version 1.01 (33). First, the MFR program from NMRpipe (34) was used to search a structural database for best matched fragments based on the protein backbone 15N, 13C, 13CA, 13CB, 1HN and 1HA chemical shifts of VcDciA[1–111]. Then the ROSETTA 3.8 software was used to generate 5844 models by fragment assembly and full-atom relaxation. The 200 models with best ROSETTA full-atom score were rescored by comparing the experimental chemical shifts with the chemical shifts predicted by SPARTA (35) for each model. The best model after rescoring was chosen as a representative NMR model and used for structural representation, for SAXS data interpretation and for X-ray structure molecular replacement.
DNA unwinding assays
Forked DNA substrates were built by the annealing of the two oligonucleotides oso3 and oso16 (see sequences in Supplementary Table S1). In case of streptavidin (SA) blocking, SA from Sigma was added to biotinylated fork with an excess of four. DNA substrates (40 fmol) were then incubated in 10 μl at 37°C with indicated amounts of proteins in buffer B (Hepes 50 mM (pH7), NaCl 150 mM, ATP 1 mM, MgCl2 10 mM, DTT 1 mM, glycerol 10%) supplemented with oligonucleotide oso7 (Supplementary Table S1) at 2 mM. This oligonucleotide is partially complementary to oso16 and prevents new annealing after unwinding activity. Reactions were stopped by addition of 150 mM EDTA and 0.5% SDS (final concentration) and incubated for 30 min with 1 μg of proteinase K (Thermofisher). Reaction products were then resolved by electrophoresis on a 8% native polyacrylamide gel in Tris-borate-EDTA (TBE 1X) buffer. Biotinylated DNA are very strongly attached to streptavidin proteins, which are organized in very stable tetramers (36). At the end of the experiment, streptavidin proteolysis is carried out in a mixture SDS/proteinase K. Yet, the complete hydrolysis of the complex was never achieved in our hands; some protomers of streptavidin perdured, associated with DNA molecules. The results are revealed by the fusion FX camera (Vilbert Loumat). Fork opening was quantified by ImageJ (37).
Negative staining electron microscopy
VcDnaB solution was diluted in salt buffer (NaCl 200 mM, Tris–HCl 10 mM (pH 8)) at 1.3 μM in the absence or presence of ATP 250 μM (without Mg2+). One μl of VcDnaB solutions was mixed with 130 nM of VcDciA or/and 250 μM of ATP in a final volume of 10 μl during 2 min at 4°C in final buffer (NaCl 70 mM, Tris-HCl 10 mM (pH8)). Five μl of the different mixtures were adsorbed onto 300 mesh copper grids coated with a collodion film covered by a thin carbon film, activated by glow-discharge. After 1 min, grids were washed with aqueous 2% w/vol uranyl acetate (Merck, France) and then dried with ashless filter paper (VWR, France). Transmission electron microscopy (TEM) observations were carried out with a Zeiss 912AB transmission electron microscope in filtered zero loss mode. Electron micrographs were obtained using a Tengra digital camera (Olympus) and Soft Imaging Software system (iTEM). The helicase surface density was estimated by counting the number of particles per surface unit (SU) of 770 nm × 770 nm. The results presented here correspond to the average of 50 independent such analyses.
Protein interaction analysis by thermal shift assay and intrinsic fluorescence variation
Intrinsic fluorescence changes of tryptophan (and tyrosine at a lower level) is recorded at 330 and 350 nm while heating the protein sample from 35 to 95°C at a rate of 3°C/min. The emission profile of a tryptophan is shifted to the red emissions when it is released to the solvent during the thermal denaturing of the protein. We used Tycho analysis (Tycho NT.6, NanoTemper Technologies) to follow interaction between VcDnaB and VcDciA, and between VcDnaB and VcDciA[1–111]. Interactions were performed in Hepes 50 mM at pH7,5, NaCl 150 mM, ATP 1 mM, with 20 μM of each protein, in capillary tubes of 10 μl. Four to five replicates were obtained to increase results confidence. To detect binding, we compared the 350/330 nm ratio of fluorescence of the complex with the predicted ratio we should obtain in absence of interaction (S brightness at 350 nm/S brightness at 330 nm).
Phylogenetic analyses
An alignment of proteins (a crude alignment generated using the program ClustalW and refined by hand) was fueled into PhyLM (v. 3.0) and 100 bootstrap replicates were generated for each analysis (38). A consensus tree was eventually obtained by running the program CONSENSE and fed as an input tree into PhyML. Significant bootstrap scores (arbitrarily above 70%) of key nodes are indicated (39).
V. cholerae cells, plasmids, and media
Three different media were used: LB (Bactotryptone 10 g/l, Yeast extract 5 g/l, NaCl 5 g/l), natural transformation medium (KH2PO4 3 g/l, Na2HPO4 6 g/l, NaCl 0.5 g/l, NH4Cl 2 g/l, MgCl2 32 mM, CaCl2 5 mM), and minimal medium (KH2PO4 3 g/l, Na2HPO4 6 g/l, NaCl 0.5 g/l, Thiamine 5 μg/ml, D-fructose 0.4%, MgSO4 1 mM, CaCl2 0.1 mM). The V. cholerae strains used in this study derive from E7946 (wild type V. cholerae O1 El Tor). The plasmids containing the different alleles of dnaB were introduced in E7946 by conjugation (40). These plasmids were constructed as follow: the oriT region of RP4 was introduced at the XbaI – HindIII sites of pBAD18 (41) to create pREP725. An EcoRI–SacI PCR fragment containing the WT VcdnaB (pREP728) and the mutated VcdnaBS296G copy of the gene (pREP729) were introduced in pRP725 to place the expression of the helicase under the control of the Para promoter. Three V. cholerae strains were thus obtained: YAV366 (E7946/pREP725), YAV369 (E7946/pREP728) and YAV372 (E7946/pREP729).
Natural transformation
Vibrio cholerae cells were cultivated in LB medium at 37°C until an OD550nm of 0.1. Then, they were harvested and resuspended in an equivalent volume of natural transformation medium (KH2PO4 3 g/l, Na2HPO4 6 g/l, NaCl 0.5 g/l, NH4Cl 2 g/l, MgCl2 32 mM, CaCl2 5 mM) and incubated for 24 h at 30°C with chitin chips to induce cell competency. Between 200 and 500 ng of DNA (PCR amplification) was added to 1 ml of this solution and further incubated for 24 h at 30°C. The cells were then plated on minimal medium (KH2PO4 3 g/l, Na2HPO4 6 g/l, NaCl 0.5 g/l, Thiamine 5 μg/ml, d-fructose 0.4%, MgSO4 1 mM, CaCl2 0.1 mM) complemented with the appropriate antibiotic and incubated at 30°C. The plates were daily inspected for the appearance of new colonies during seven days.
RESULTS
VcDnaB can self-load onto DNA
We investigated by SPR the loading of VcDnaB on ssDNA substrates. We designed 3′ and 5′ protruding end DNA substrates to record both loading and threading of the helicase on DNA (Supplementary Table S1 and Materials and Methods). Once around ssDNA, the replicative helicase translocates in the 5′ to 3′ direction. Hence, the enzyme is either released from the DNA when a 3′ free end substrate is used (Figure 1A, left) or trapped at the bottom of the DNA strand bound to the matrix when the 5′ free end substrate is used (Figure 1A right). We performed the experiment on both 3′ and 5′ extremities, in ‘coupled’ (both loading and translocation can occur) and in ‘uncoupled’ conditions (the Mg2+, which triggers the translocation, is added after the loading of DnaB around the DNA; Materials and Methods).
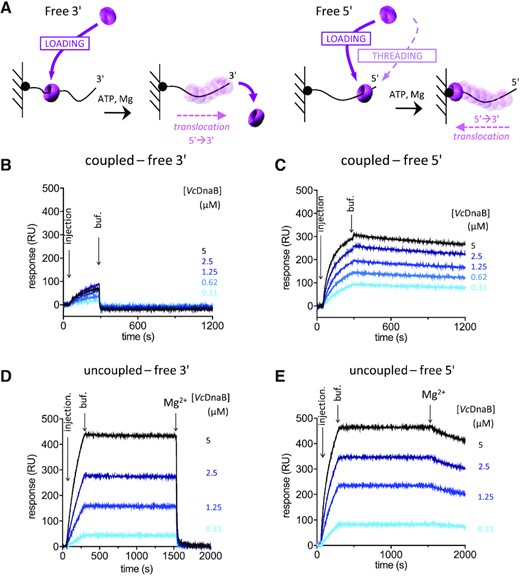
Loading of VcDnaB on DNA. (A) Experimental design of the SPR experiments. The single stranded DNA molecules were bound to the SPR matrix through their 5′ (left) or their 3′ (right) biotinylated extremity, leaving a 3′ or 5′-end accessible. After addition of the helicase to the reactive mixture, the loading of the helicase on DNA is expected to give rise to a translocation motion in the 5′ → 3′ direction, dropping from the DNA when the 3′ is free (left) or trapping the helicase at the interface between the matrix and the DNA substrate when the 5′ is free (right). In coupled conditions, proteins were injected in the presence of 1 mM ATP and 10 mM MgCl2, and dissociation was carried out in the same buffer, allowing helicase translocation as soon as the helicase was loaded. In uncoupled conditions, proteins were injected in the presence of 1 mM ATP, a first dissociation was observed in the same buffer (buff.), before injection of a buffer containing 1 mM ATP and 10 mM MgCl2 (Mg2+). Then, in uncoupled conditions, the loading can be observed before translocation that occurs only when MgCl2 is added. (B to E) VcDnaB at the indicated concentrations (from 0.31 μM to 5 μM) was injected, from light blue to black.
In coupled conditions, the binding of VcDnaB on a 3′ protruding end DNA substrate is much lower than with a 5′ protruding probe (less than 100 versus 300 RU) and is immediately washed away with buffer (Figure 1B). This suggests that VcDnaB translocates once loaded on DNA, to eventually drop off the DNA substrate. On the free 5′ end substrate, the amount of VcDnaB trapped on DNA increases as a function of the quantity of helicase injected (Figure 1C). To confirm that the low binding observed on the free 3′ end substrate is due to the VcDnaB translocation activity, we realized the same SPR experiments in conditions where the loading and the translocation activities were uncoupled. Under these conditions, the loading was performed in the presence of ATP alone, pursued by a first wash in the same buffer before addition of a solution containing ATP and MgCl2, which enables translocation. Under these new experimental conditions, the helicase efficiently loaded itself on DNA and remained strongly associated to it until Mg2+ was added (Figure 1D and E). After addition of Mg2+, the helicase was either quickly released from the 3′ end substrate (Figure 1D) or trapped at the bottom of the 5′ end substrate (Figure 1E), indicating that the helicase was efficiently loaded onto DNA and strongly attached to it before addition of Mg2+. Moreover, the amount of bound helicase is roughly similar on both DNA whether the free DNA end is accessible (5′) or not (3′) for threading, suggesting that the helicase threading is negligible. Altogether, these results indicate that the helicase can load itself on DNA without any assistance.
Free VcDnaB hexamers adopt a lock washer conformation
The ability of VcDnaB to self-load on DNA contrasts with the inability of the helicase of E. coli (EcDnaB) to do so under the same experimental and physiological conditions (Supplementary Figure S1). Granted that EcDnaC crack-opens the EcDnaB ring in order to allow its loading on DNA, we wondered whether VcDnaB could organize itself as an extended-spiral and therefore open structure, bypassing the requirement for a loader. To investigate this hypothesis, we attempted to crystallize VcDnaB. We obtained crystals of the helicase in presence of GDP:AlF4 and Mg2+, that diffracted to 3.9 Å resolution (PDB ID 6T66, Figure 2A and Supplementary Table S2). As expected at this resolution, most of the flexible loops and side chains are outside of density. However the density of the overall tertiary fold and secondary structural elements is clearly visible and enables a reliable fitting, with the help of a good homology model used in molecular replacement (PDB 6QEM, RMSD = 1.6 Å for 441 aligned residues (42)). Consistent with other bacterial replicative helicase ring-like structures, VcDnaB is organized in two layers, corresponding respectively to the NTD [I21-N173] and CTD [F208-G462] domains of the protein, connected through a linker α-helix (LH, segment [D180-K195], in yellow in Figure 2A and B). The NTD ring is composed of a trimer of homodimers, whereas the RecA-fold CTD are assembled with a 6-fold symmetry. In each VcDnaB subunit, the CTD is located 60° away from the NTD, placing the LH above the CTD of the next VcDnaB subunit. The LH of a subunit makes antiparallel interactions with a α−helix (segment [D287-K305], in green in Figure 2A and B, so-called ‘determinant helix DH’ below) of the CTD of the neighboring subunit, forming the so-called ‘DH-LH module’.

VcDnaB is a slightly spiral open and dynamic hexamer (A) Crystal structure of the VcDnaB•GDP:AlF4:Mg2+ complex (PDB ID 6T66). The six GDP:AlF4-bound-VcDnaB subunits are represented in pink (B), light blue (C), magenta (D), cyan (E) or grey (A and F). The red arrow indicates where the hexameric helicase ring opens, forming a spiral shape. The LH and so-called DH helices, forming what we have named the ‘DH-LH module’ of the helicase, are in yellow and green, respectively. The positions of the six GDP (light orange dots) in the crystal structure are also indicated. (B) Superimposition of the B+C and D+E CTD homodimers. B and D monomers (pink and magenta) were aligned to highlight the shift (indicated by a red double-headed arrow) between the C and E monomers (light blue and cyan), in response to the opening of the hexamer ring between the D and the E subunits. (C) Evolution of the gyration radius Rg (black line) during the SEC-SAXS elution of VcDnaB•ATP:Mg2+, together with the molar mass (red line) and the forward scattered intensity I(0) (dashed black line). The dark grey rectangle corresponds to the zone of elution where the successive SAXS curves are rigorously identical and their average was analyzed using the US-SOMO HPLC module. (D) Superimposition of the SAXS curve calculated on the all-atoms model of VcDnaB•ATP:Mg2+ (red curve, the missing fragments of our VcDnaB•GDP:AlF4:Mg2+ crystal structure were added using the program DADIMODO (28,29)) against experimental data (black dots). The corresponding distribution of reduced residuals, reflecting the variation between the two profiles, is presented at the bottom of the panel figure.
Interestingly, the ring-shaped hexamer of VcDnaB (internal chamber = 26 Å), which contains six GDP:AlF4 molecules and six Mg2+ ions, is slightly spiral open at the interface of the CTD domains between the D and E monomers (red arrow, Figure 2A). Practically, when the B and D subunits of the B+C and the D+E dimers (Figure 2B) are perfectly superimposed, the subunit E is shifted 17 Å away from subunit D than is subunit C from subunit B. This is caused by the reorientation of the entire linker domain, including the LH and the unstructured flexible region, with respect to the DH helix of an adjacent DnaB subunit. In complete support with the crystal structure of VcDnaB•GDP:AlF4:Mg2+, in a small range at the beginning of the SEC-SAXS elution peak of VcDnaB•ATP (Figure 2C), the radius of gyration decreased slightly whereas the mass—corresponding to that of the hexamer—remained constant throughout the elution peak. This suggests that the hexamer ring oscillates around this slightly opened form in solution. This dynamic structure may also explain the slight difference (χ2 = 3.83, Figure 2D) between the calculated (red curve) and the experimental SAXS curves (VcDnaB•ATP, black dots, Supplementary Table S3).
Yet, the wideness of the aperture of the hexameric ring-like conformation of the free VcDnaB is not sufficient for the helicase to accommodate single strand DNA into the internal chamber. This would require a larger modification of the conformation of the enzyme, as those observed in the EcDnaB•EcDnaC complex.
DciA shares structural similarities with DnaA but not with DnaC
The identification of dciA in dnaC-lacking bacteria and the implication of DciA and DnaC at an early step of replication initiation led to the hypothesis that DciA and DnaC carry the same function (12). Yet, while DnaC/I contain an AAA+ domain (43), HHpred (44,45) does not detect a AAA+ domain in VcDciA (residues 1–111), but rather structural similarities with the NTD of DnaA (PDB ID 4TPS, 2E0G, (46)), the CTD of DnaX (τ5, PDB ID 2AYA, (47)) and the flagellar hook-length control protein FliK (PDB ID 2RRL, (48)) (Materials and Methods). The C-terminal region of VcDciA (residues 112–157) is predicted to be disordered (49).
To determine the 3D structure of the NTD domain of VcDciA encompassing residues M1 to A111, we purified 13C–15N labeled protein (Materials and Methods and Supplementary Figure S2A) and collected nuclear magnetic resonance (NMR) chemical shift data for the backbone. We then computed chemical shift indices to identify secondary structure elements (Supplementary Figure S2B and C) and to generate a 3D structural model (Figure 3A). The NTD domain is characterized by a mostly globular fold containing a long NTD helix spanning residues L12 to I38, a mixed three-strands β-sheet comprising residues V47 to R53, N56 to A61 and S91 to V96, one α−helix from residues A64 to N84, kinked around positions 73–74, and another one between residues E99 to Y101 (Figure 3A). Comparison of VcDciA with the NTD of DnaA (the closest structural homolog according to PDBeFold server (50)) or with the CTD of DnaX shows that their globular parts superpose well with an organization typical of the KH type II domain (51) (Figure 3B).

The NTD of VcDciA shares a common type-II KH-domain fold with the DnaB-binding domain of DnaA and with the DnaX domain V. (A) Representative structural model for VcDciA NTD (residues 1 to 111), built from NMR backbone chemical shifts. Corresponding secondary structure elements are displayed in red for α-helices and yellow for β-strands. The regions without defined secondary structure are in green. VcDciA NTD exhibits a type-II KH-domain fold topology (51). (B) Structural superimposition between the representative NMR-based structural model of VcDciA (residues 1–111, cyan) and the homologous NTD of BsDnaA (left, PDB ID 4TPS, chain B, pink) and EcDnaX CTD (right, PDB ID 2AYA, model 1, residues 1–96, purple). (C) Distance distribution function P(r) of VcDciA resulting from SEC-SAXS analysis of the full-length protein. (Inset) Four superimposed models of the VcDciA full-length protein generated from the SAXS curve starting from the NMR model (NTD) and modeling the missing CTD residues using the program DADIMODO (see Materials and Methods and Supplementary Table S3) (28,60). The four models display random coiled VcDciA CTD.
Structural information about the CTD domain of VcDciA (corresponding to the residues 112–157) was deduced from SAXS data (49). The distance distribution function P(r) of full-length VcDciA calculated from the SAXS curve I(q) is highly asymmetric, with an extension towards larger r-values (Figure 3C, Supplementary Table S3). The maximal extension Dmax in solution of VcDciA is of the order of 110 Å, as compared with Dmax ≈ 50 Å for the structured part of the protein (NTD, residues 8–111) as determined using NMR. Four typical models are represented (inset in Figure 3C). Our results strongly suggest that the CTD region of VcDciA is highly unfolded in solution, but local and transient helical structures were highlighted by molecular dynamics and circular dichroism (49).
VcDciA stimulates the unwinding activity of VcDnaB
The ability of VcDnaB to self-load on DNA in vitro raises the question of the need and function of VcDciA during the replication initiation. We tested the requirement for VcDciA in the DNA-unwinding activity of the replicative helicase VcDnaB. We followed the unwinding of a Cy5-labeled branched DNA substrate, whose 5′-end was accessible for the helicase either for loading or threading (Figure 4A, left and Materials and Methods). When VcDnaB was added to the reaction mixture, a faster migrating product corresponding to the Cy5-labeled oligonucleotide appeared at the bottom of the gel (Figure 4B and F). Unwinding activity was not detected in these experimental conditions without protein (Figure 4F). By covering the biotin extremity of one of the branches of the DNA substrate with streptavidin, the access to the 5′ protruding end was blocked, thereby preventing the threading of the helicase through the 5′ end of the DNA substrate and solely enabling the loading of the helicase (Figure 4A right and Materials and Methods). Under these conditions, DNA unwinding was still observed as about 15% of the free Cy5-labeled oligonucleotide was still produced after addition of VcDnaB (Figure 4C and G). Hence, these results confirm that VcDnaB can self-load on DNA as shown previously (Figure 1), even if the efficiency of the helicase to load itself on DNA in our SPR experiments contrasts a bit with that recorded with the streptavidin labeled substrate. We hypothesize that the difference could result from the steric hinderance created by the bulky streptavidin.

VcDciA stimulates the unwinding activity of VcDnaB. (A) Experimental design of the DNA unwinding assays. The branched DNA substrate used in these series of experiments consisted in a 25 bp-long double-stranded DNA molecule prolonged by two non-complementary single stranded DNA (25 and 50 nucleotides long at 3′ and 5′ extremities, respectively). A Cy5 fluorophore was added at the 5′ end of the 50 nucleotides long oligonucleotide (blunt end of the branched molecule) to enable the detection of DNA molecules. In presence of ATP and Mg2+, the loading/threading of the helicase on this substrate and its translocation in the 5′ → 3′ direction lead to DNA unwinding. Biotin was added at the 5′ protruding end of the 75 nucleotides long oligonucleotide (fo3-16bio, left) to enable its blocking by streptavidin thereafter and to prevent threading of the helicase (fo3-16SA, right). (B, C) The unwinding assays were performed with VcDnaB (125 nM) with fo3-16bio (B) or fo3-16SA (C) oligonucleotides (4nM). At regular time intervals, samples were taken from the reaction mixture, mixed with a STOP solution and run on a 8% acrylamide gel. (D, E) The unwinding assays were performed with VcDnaB (125 nM) with fo3-16bio (D) or fo3-16SA (E), in presence of VcDciA (42 nM), for a final ratio of six DnaB monomers for three DciA. At regular time intervals, samples were taken from the reaction mixture, mixed with a STOP solution and run on a 8% acrylamide gel. (F, G) DNA unwinding kinetics. A graphic representation of the accumulation of Cy5-unwound products over the time is presented for VcDnaB (light blue), with different ratio of VcDciA (purple, pink and red) and without helicase (negative control in grey) (three independent experiments, mean +/- SEM).
The ability of VcDnaB to self-load and unwind DNA in vitro raises the question of the need and function of VcDciA during the replication initiation. We therefore analyzed the implication of VcDciA in the DNA unwinding activity of VcDnaB. Whether the 5′-end of the branched DNA substrate was accessible or not, the addition of VcDciA improved the production of free Cy5-labeled oligonucleotide (Figure 4D-G). Kinetics of unwound product accumulation, performed with various VcDnaB/VcDciA ratios, showed that most of the accumulation was already achieved at a 6/1 VcDnaB/VcDciA ratio. A maximum unwinding yield was obtained for a VcDnaB/VcDciA ratio of 6/3 (Figure 4F and G).
VcDciA stimulates the loading of VcDnaB on DNA
We then investigated by SPR the loading of VcDnaB on ssDNA substrates in the presence of VcDciA. Under these experimental conditions, the binding of VcDciA on DNA was negligible (Supplementary Figure S3). As observed for the helicase alone, when VcDciA was injected together with VcDnaB in the presence of ATP and Mg2+ on a free 3′ end substrate (coupled conditions), the translocation of the helicase resulted in its fast release from the DNA (Figure 5A). In contrast, on the free 5′ end ssDNA, we observed that VcDciA stimulated the binding of the helicase on DNA by a factor of 3 to 4. The maximum of stimulation was obtained at a VcDciA/VcDnaB ratio of 3/6 (Figure 5B). This stimulation was clearly similar to that observed in the unwinding assay (Figure 4D–G), suggesting that VcDciA stimulates the unwinding activity of VcDnaB by increasing its loading on DNA.
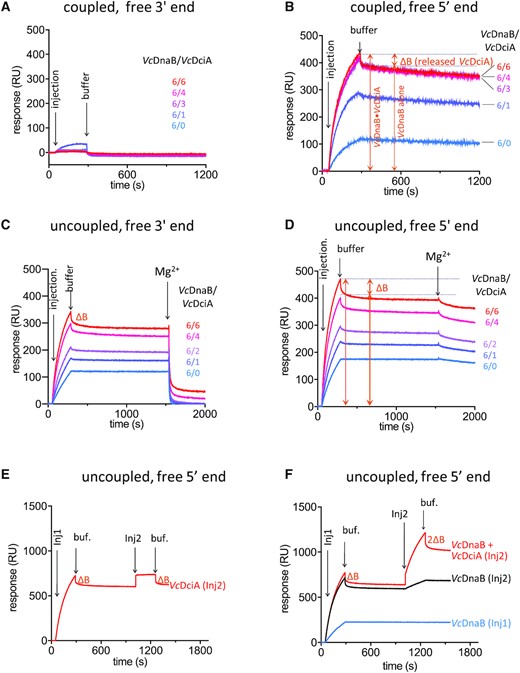
VcDciA stimulates the loading of VcDnaB on DNA. (A, B) SPR analysis of the loading of VcDnaB on ssDNA with free 3′ (A) and free 5′ (B) ends, in the presence of VcDciA. VcDnaB was injected at a concentration of 625 nM during 240s (injection) in a buffer solution containing ATP and MgCl2 (coupled conditions, Materials and Methods). Dissociation was assessed in the same buffer (buffer). The amount of DNA-bound protein is proportional to the Response Units (RU on the ordinate axis). Different VcDnaB/VcDciA ratios (from 6/0 to 6/6 in subunits) were analyzed. ΔB represents the mass of VcDciA proteins that dissociated immediately after the loading of the VcDciA•VcDnaB complex on DNA. (C, D) Same as in A and B, except that the experiments are monitored in the ‘uncoupled’ conditions, where the Mg2+ was omitted during the loading step and added after stabilization of the helicase onto the DNA. The translocation is triggered by the Mg2+. (E, F) SPR analysis of the dissociation of VcDciA from the VcDciA•VcDnaB complex on free 5′-ssDNA substrate. In absence of Mg2+, after the first interaction with DNA of VcDnaB + VcDciA (Inj1, 1.25 μM in subunits) during 240 s, dissociation of VcDciA (ΔB) was observed for 750 s. A second injection of various protein composition (Inj2) was then carried out during 240 s and produced a secondary response. (E) A second injection of an equimolar amount of VcDciA was performed (Inj2) giving rise to an equivalent transient binding of VcDciA (same ΔB) on the VcDnaB•DNA complex. (F) A second injection containing VcDnaB alone (equimolar amount, Inj2, black) produced a secondary response similar to the blue curve which corresponds to the profile of a single injection of VcDnaB alone. A second injection of the VcDciA•VcDnaB complex (equimolar amount, Inj2, red) resulted in twice as much transient binding of VcDciA (2ΔB) on the VcDnaB•DNA complex.
The SPR profiles observed in the presence of VcDciA systematically revealed a biphasic dissociation curve (Figure 5B). An abrupt reduction of the amount of DNA-bound protein was observed immediately after the injection, followed by a slight and continuous slope diagnostic of the slow dissociation of VcDnaB observed earlier (Figure 1C). Similar observation was made during the study of the binding of the GsDnaB•BsDnaI hybrid complex on DNA (Gs for Geobacillus stearothermophilus and Bs for Bacillus subtilis). The first and rapid dissociation was reported to correspond to the release of BsDnaI (52). To determine whether VcDciA dissociates upon binding of the VcDnaB•VcDciA complex on DNA, we uncoupled loading and translocation by injecting VcDciA and VcDnaB in a Mg2+-less buffer containing ATP in order to prevent VcDnaB translocation on DNA. Interestingly, the SPR profiles showed that some protein mass was also released immediately upon injection, like in the experiment performed in the presence of Mg2+ (compare Figure 5C and D with Figure 5B). This suggests that the same DNA•protein complex was built in the absence and in the presence of Mg2+ (compare Injection in Figure 5C and D with Injection in Figure 5B). Mg2+ was added after stabilization of the nucleoprotein complex (Figure 5C and D, uncoupled conditions). The second fast release of protein mass upon addition of Mg2+ strongly suggests that the nucleoprotein complex was indeed the DNA-bound helicase. To determine which protein was first released upon binding of the VcDnaB•VcDciA complex on DNA, we performed a second injection of either VcDciA, VcDnaB or both proteins under uncoupled conditions (Inj2, Figure 5E and F). The injection of VcDciA alone led to an evanescent rise of the binding response of an amplitude identical to that recorded during the first injection. This binding lasts solely during the time of the injection 2, indicating that VcDciA interacted transiently with the DNA•protein complex (Figure 5E). In contrast, when VcDnaB was reinjected, more helicase was strongly bound to DNA, confirming that once bound on DNA in absence of Mg2+, the helicase remained strongly associated with it (compare Inj2 of black curve with blue curve, Figure 5F). When the VcDnaB•VcDciA complex was reinjected, the binding profile was similar to that obtained during the injection 1 of the VcDnaB•VcDciA complex (red curve, Figure 5F). These results show unambiguously that VcDciA, and not VcDnaB, is the sole protein to be released immediately upon binding of the protein complex on DNA. It further suggests that the conformation of the VcDnaB•VcDciA complex is modified upon binding on DNA, causing the release of VcDciA in an ATP hydrolysis-independent manner.
Assuming that the fast protein release observed after binding of the VcDnaB•VcDciA complex on DNA corresponds to that of VcDciA, the variation of the binding response (ΔB) becomes a fair indicator of the protein mass released and hence of the protein stoichiometry in the VcDnaB•VcDciA complex. Our assessment of ΔB is consistent with a VcDnaB/VcDciA stoichiometry of 6/3 (6xVcDnaB MW = 317 kDa, 3xVcDciA MW = 52.2 kDa) (Figure 5B).
VcDciA interacts with VcDnaB in a 3/6 stoichiometry
We examined the influence of the binding of VcDciA on the organization of the helicase by negative staining electron microscopy. In the absence of ATP and Mg2+, the same amount of VcDnaB rings per unit area (603 nm2) was detected by EM with or without VcDciA (33 ± 8/su without VcDciA versus 35 ± 7/su with VcDciA, Figure 6A). When VcDnaB was preincubated with ATP, however, the addition of VcDciA doubles the amount of ring-shaped structures (67 ± 8/su without VcDciA versus 150 ± 12/su with VcDciA, Figure 6A). These results suggest that VcDciA recognizes and stabilizes pre-arranged hexamers or that VcDciA helps assemble hexamers and that ATP stabilizes the assembled state. We analyzed by SEC-SAXS the complex formed between VcDnaB and VcDciA at a 2-fold molar excess of VcDciA, in the presence of ATP and Mg2+ (Supplementary Table S3 and Materials and Methods). We detected a VcDnaB•VcDciA complex at the beginning of the elution peak of the gel filtration chromatography whose mass stabilized at a MW of 381 ± 20 kDa (Figure 6B left and Supplementary Table S3). Assuming observed MW for the VcDnaB hexamer and for a monomer of VcDciA of 328 ± 20 kDa and 18 ± 2 kDa, respectively (Figure 2C and Supplementary Table S3), the stoichiometry of the complex is therefore estimated to be of 3 VcDciA molecules per helicase ring. These results have to be compared with the theoretical masses of 317 and 18.4 kDa of the His-tagged proteins. These calculations are in good agreement with the measured values obtained from SEC-MALS experiments (Supplementary Information and Supplementary Figure S4).

VcDciA binds and stabilizes VcDnaB hexamers. (A) Negative staining electron microscopy was performed on apo-VcDnaB (upper left) or after preincubation with ATP without Mg2+ (upper middle). The VcDnaB•VcDciA complex, without (lower left) or with ATP (but without Mg2+, lower middle), was also visualized. A great diversity of oligomeric structures was visible without nucleotide, but hexameric ring structures increased in presence of ATP. The white scale bar on these four representative electron micrographs represents 100 nm. The number of hexameric particles per surface unit of VcDnaB alone, in presence of VcDciA, without or with ATP, is plotted on the graph (right). (B) Evolution of the gyration radius Rg (black line) during the SEC-SAXS elution of VcDnaB•VcDciA•ATP:Mg2+ with the molar mass (green line) and the forward scattered intensity I(0) (dashed black line) (left). The dark grey rectangle corresponds to the zone of elution where the successive SAXS curves are rigorously identical and their average was analyzed using the US-SOMO HPLC module (24). A VcDnaB•VcDciA•ATP complex compatible with a 6/3 stoichiometry (M = 381 ± 20 kDa, Supplementary Table S3) was first eluted (the corresponding frames are marked out by the dark grey rectangle), but dissociated rapidly as indicated by the decrease of M (green line) (left). Superimposition of the experimental SAXS curves of VcDnaB•ATP:Mg2+ (red curve) and VcDnaB•VcDciA•ATP:Mg2+ (green curve) (right). (C) Localization of the VcDnaB W291 used to highlight the interaction between VcDnaB and VcDciA by fluorescence spectroscopy (see Tycho analysis in D). W291 (in orange stick) is located on the so-called DH helix (in green), named after the determinant residue (S296 in red stick). The LH (in yellow) of the adjacent subunit makes antiparallel interactions with the DH, forming together the DH-LH module of the helicase. (D) Characterization of the interaction between VcDnaB and VcDciA by Tycho NT.6 analysis. In red is reported the 350/330 nm ratio obtained for the complex and in black is reported the predicted ratio for an absence of solvent protection. The curves correspond of the mean ± SEM of four to five analyses. Mann-Whitney U test statistics reveal a significant difference between predicted and measured ratios (P< 0.0001), indicating an interaction between the two proteins.
As expected, the experimental SAXS curve of the VcDnaB•VcDciA complex does not overlap with that of VcDnaB (Figure 6B right), confirming that they form different assemblies in solution. To get insight into the mode of interaction between VcDnaB and VcDciA, we followed the variation of the intrinsic protein fluorescence (i.e. of tryptophan residues) as a thermal ramp is applied (Tycho NT.6, NanoTemper Technologies). VcDnaB contains only two tryptophans: W45 is buried inside the NTD domain, while W291 in the CTD (indicated in orange in Figure 6C) is exposed to the solvent and located on the DH α−helix (see below for description and in green in Figure 6C). On the other hand, VcDciA does not contain any tryptophan. The interaction between the two proteins is revealed by the curve profile observed at the 350 nm/330 nm ratio. The initial 350 nm/330 nm ratio of the experimental curve (in red) is low compared to that of the theoretical curve (in black) representing the absence of solvent protection (Figure 6D). Our data shows unambiguously that W291 is strongly protected in the VcDnaB•VcDciA complex, indicating that VcDciA interacts with the helicase around the DH α−helix. Hence, VcDciA might likely interact with the DH α−helix of VcDnaB, like EcDnaC with EcDnaB (53).
Serine 296 of VcDnaB is essential in vivo
In a phylogenetic study of DciA- and DnaC-dependent helicases aimed at identifying critical residues, we noticed serine 296 of VcDnaB as a residue that could have been modified during the transition process from dciA to dnaC as an adaptive response to DnaC (Materials and Methods).
The initial analysis was restricted to the replicative helicase protein sequences of the Enterobacterales. This order of bacteria regroups closely related species that possess either DciA or DnaC. An alignment of replicative helicase protein sequences revealed that a unique residue strictly distinguishes helicases from dnaC- and dciA-species (Figure 7A and Supplementary Figure S5A). This residue (the determinant residue, indicated in red in Figure 6C), at position 296 in VcDnaB, is located in the above described α−helix, parallel to the LH (Figures 2A and 6C), which we named the determinant helix (DH). In dciA-species, this residue is always a serine whereas it is mostly a glycine in dnaC-species (Supplementary Figure S5A). Two additional observations substantiate the relevance of this co-occurrence. Firstly, we identified two monophyletic branches of DnaC proteins in Enterobacterales; each branch derives from an independent dnaC domestication event (Supplementary Figure S5B), indicating that coincidentally with the domestication of dnaC, the determinant residue was replaced by a glycine or an alanine in the replicative helicase (Supplementary Figure S5C). Secondly, the same type of analysis performed with DnaB sequences of Alteromonadales, which is the second identified bacterial order that regroups dnaC- and dciA-species (12), revealed that the determinant residue discriminates dnaC- and dciA-containing species as well. The determinant residue is systematically a serine in dciA-containing species and another residue – mostly an asparagine – in the dnaC-containing species (Supplementary Figure S5D).

In vivo analyses of the determinant residue. (A) Primary structure of VcDnaB displaying the NTD and the CTD of the helicase. Relevant motifs and residues are displayed. The determinant residue is indicated at position 296. In dciA-species, this residue is always a serine, while this residue is mostly a glycine in dnaC-species (Supplementary Figures S5A-D). (B) Design of the in vivo mutagenesis of VcdnaB. WT cells were transformed with a plasmid carrying either nothing, the WT VcdnaB or the mutated VcdnaBS296G gene under the control of the arabinose promoter before the deletion of the chromosomal copy of VcdnaB. Deletion was induced by natural transformation of these cells with a PCR fragment carrying a kanamycin resistance gene instead of VcdnaB (see Materials and Methods). The resulting cells were plated on selective medium (kanamycin) containing or not arabinose. The frequency of natural transformation is given for each condition (average of 3 independent experiments).
To evaluate the relevance of this determinant residue on the function of the helicase in vivo, we undertook to construct a VcdnaBS296G mutant. Our attempt to replace the WT copy of VcdnaB by VcdnaBS296G by natural transformation failed (Materials and Methods). To verify that VcDnaBS296G could not replace VcDnaB, we set up a two-step strategy (Figure 7B left). We introduced in WT cells a plasmid carrying either VcDnaB (B/B), VcDnaBS296G (B/BSG) or nothing (B/-) (Materials and Methods and Figure 7B right). Then, we introduced by natural transformation a piece of DNA carrying the kanamycin resistance gene bordered by sequences homologous to those found upstream and downstream of dnaB on the chromosome to induce the recombination-dependent replacement of the WT copy of dnaB by the kanamycin resistance gene (Materials and Methods). The transformation products were plated on selective medium. After 24h of incubation, the only transformants that gave rise to colonies were those that contained a WT copy of VcDnaB on a plasmid, indicating that a VcdnaBS296G mutant is unviable (Figure 7B, right). The expression of the mutated helicase VcdnaBS296G was ascertained by Mass Spectrometry analysis performed on NanoLC-MS/MS Tim TOF PRO (data not shown).
This in vivo study demonstrates that the residue at position 296 is critical for the function of the helicase, so important that its modification is lethal. We wonder whether this phenotype may result from the involvement of this residue in the loading or the translocation of the helicase.
The determinant residue of DnaB is important for the interaction with DnaC
We first characterized the interaction of EcDnaB on DNA by SPR and observed that under uncoupled, and therefore non-physiological conditions, the replicative helicase of E. coli can bind on DNA (compare Supplementary Figure S6A and B to Supplementary Figure S1A and B). Under these conditions, EcDnaC stimulates the loading of EcDnaB (Supplementary Figure S6C and D) but alone does not bind on DNA (Supplementary Figure S6E and F).
To assess the implication of the determinant residue during the loading of the helicase and the DNA unwinding in vitro, we then produced and purified mutant proteins of the replicative helicase of E. coli and V. cholerae, carrying a serine instead of a glycine at position 299 (EcDnaBG299S) or a glycine instead of a serine at position 296 (VcDnaBS296G), respectively.
We established through our DNA unwinding assay that the substitution of the determinant residue did not affect the capacity of both mutant proteins either to be loaded on DNA or to translocate on DNA. Furthermore, we observed that these mutations did not affect the ability of VcDciA or EcDnaC to stimulate or enable, respectively, the loading of their cognate mutant helicases on DNA (Supplementary Figure S6G–J).
We investigated the impact of the mutation of the determinant residue on the kinetics of dissociation of VcDciA and EcDnaC from their respective helicase complex once bound on DNA. The mutation G299S on EcDnaB resulted in a faster (twice as fast) dissociation of EcDnaC from the DNA-bound helicase (compare first injection response in Figure 8A and B, Supplementary Table S4). This indicates that the DNA-bound mutant helicase has a lower affinity for EcDnaC and that the determinant residue modulates the release of EcDnaC during the loading of the EcDnaB•EcDnaC complex on DNA. The cryo-EM structure of the EcDnaB•EcDnaC complex, showing that G299 is in the immediate vicinity of the NTD of EcDnaC, gives weight to this hypothesis (53). When a second injection was carried out with EcDnaC only, EcDnaC reassociated with the DNA-bound helicase. Following the second injection, the dissociation of EcDnaC was again faster from EcDnaBG299S than from EcDnaB bound on DNA (compare Figure 8A red and B red), in agreement with the lower affinity recorded of EcDnaC for the DNA-bound mutated helicase. When the second injection was carried out with VcDciA instead of EcDnaC, we observed a negligible interaction of VcDciA with EcDnaBG299S and EcDnaB (compare Figure 8A blue and B blue).
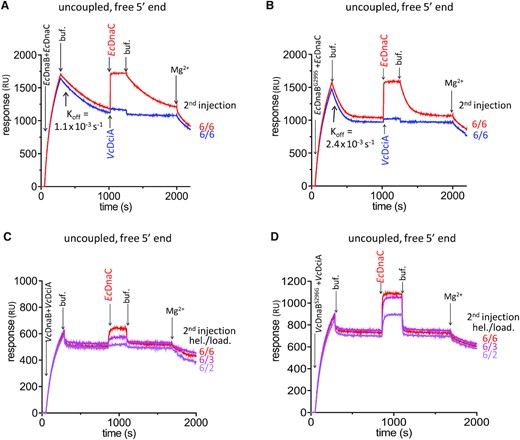
The determinant residue modulates the binding of EcDnaC loader on replicative helicases. SPR analyses on 5′ free ssDNA substrate were conducted as uncoupled experiments (see Materials and Methods) to assess the role of the determinant residue in the specificity of the replicative helicases for DnaC or DciA. In absence of MgCl2 (uncoupled conditions), after the first interaction with DNA of VcDnaB + VcDciA or of EcDnaB + EcDnaC during 240 s, dissociation was observed for 570 s to 750 s. A second injection of EcDnaC or VcDciA at the indicated ratio was then carried out during 240 s and produced a second response. (A) The first injection and dissociation of EcDnaB + EcDnaC (625 nM each) were followed by a second injection of EcDnaC at 625 nM (red) or VcDciA at 625 nM (blue). (B) Same as in (A) except that the first injection was carried out with EcDnaBG299S instead of EcDnaB. The dissociation constants (koff) are indicated for EcDnaC from EcDnaB (A) and from EcDnaBG299S (B). (C) The first injection and the dissociation of VcDnaB + VcDciA (1.25 μM each) were followed by a second injection of EcDnaC (0.4, 0.6 and 1.25 μM from pink to red, respectively). (D) Same as in (C) except that the first injection was carried out with VcDnaBS296G instead of VcDnaB (see Supplementary Table S4).
Conversely, we analyzed the mutation S296G on VcDnaB. The mutation seemed not to affect the speed at which VcDciA dissociated from the DNA-bound helicase (compare first injection response in Figure 8C and D, Supplementary Table S4). Yet, we found a difference between VcDnaBS296G and VcDnaB, when EcDnaC was injected secondarily on the DNA-bound helicase (Figure 8C and D). Indeed, at each EcDnaC concentration tested, more EcDnaC protein associated with VcDnaBS296G than with VcDnaB, when the helicase is bound on DNA (compare second injection signals in Figure 8C and D).
These data show that EcDnaC interacts more strongly with VcDnaBS296G than with VcDnaB. Also, EcDnaC dissociates faster from EcDnaBG299S than from EcDnaB. Altogether, these results suggest that the determinant residue of DnaB is strongly involved in modulating the interaction with DnaC, and that its modification from Ser to Gly was an important step during the adaptation of the cell to DnaC.
DISCUSSION
Alternative loading of the replicative helicase under DciA’s rule?
Converging pieces of evidence support the hypothesis that the determinant residue located within the DH-LH module of DnaB plays a critical role in the interaction with DciA and DnaC and during replication initiation. First, the protection of the W291 enclosed in the DH α−helix by VcDciA, as measured by Tycho, indicates that VcDciA binds VcDnaB in the immediate vicinity of the determinant residue (Serine at position 296). The second evidence comes from the recent cryo-EM structure of the EcDnaB•EcDnaC complex, showing that the determinant residue G299 is located close to where the extended NTD of EcDnaC interacts with the helicase (53). The characterization of a thermosensitive mutant of the E. coli replicative helicase, EcdnaB252, is the third direct evidence (54). The mutation was identified in the codon of the determinant residue, replacing the glycine at position 299 by an aspartate in EcDnaB. This mutation confers a thermosensitivity phenotype to the helicase for replication initiation specifically (54) since the EcDnaB252 protein is fully competent for the recruitment of the primase and for the binding on ssDNA. In addition, EcDnaB252 retains WT ATPase and helicase activities (55). The fourth evidence is indirect. It comes from our failure to construct mutant cells in which the determinant residue of VcDnaB was replaced by a glycine. Similarly, we could not construct a VcDnaBS296D mutant protein in which the serine was replaced by an aspartate (data not shown), despite the lack of effect associated with the modification of this residue on the loading and the translocation of the helicase in vitro. The last evidence comes from the impact of the determinant residue on the release of EcDnaC and VcDciA after the helicase complex is loaded on DNA. An instant release of VcDciA from the DNA-bound helicase complex is observed whether the assay was carried out with the WT or the VcDnaBS296G mutant protein. In contrast, the release of EcDnaC upon binding of the helicase complex on DNA is much slower. It is noteworthy that this release is quickened when the assay is carried out with EcDnaBG299S, suggesting that the slow release of EcDnaC once the helicase is bound on DNA is an adaptive feature that was acquired by DnaC-using helicases in the course of evolution. Yet, the purpose of this feature remains elusive. We hypothesize that the different speed at which DnaC and DciA are released upon loading of their respective helicase on DNA reflects differences in the loading mechanisms of DciA- and DnaC-helicases that remain to be elucidated.
The recent resolution at a molecular level of the DnaB•DnaC and DnaB•λP complexes also revealed that the helicase is opened by breakage of a DH-LH module, i.e. by a clear disjunction of these two α−helices belonging to two neighboring subunits of the helicase. In the same vein, we showed that a crack in a DH-LH module is the reason why the free VcDnaB structure is dynamic in solution. This crack also makes plausible the hypothesis according to which the loading of DciA-helicases requires the disjunction between LH and DH. Yet, the opening of the free VcDnaB ring-like structure is not wide enough to accommodate ssDNA into the internal chamber. Hence, we propose that the ability of VcDnaB to self-load on DNA (like other DciA-helicases: Pseudomonas aeruginosa, Azotobacter vinelandii, Azotobacter chroococcum and Deinococcus radiodurans, data not shown and (56)) relies on DNA:protein interactions. The assistance of DciA during the loading of the helicase, together with the predicted interaction between DciA and the DH-LH module of DnaB, remain the open questions that need to be deciphered.
The hijacking of DnaB by DnaC and λP
DnaC and DnaI derive from mobile elements genes that were domesticated at least seven times through evolution at the expense of the ancestral protein DciA (11,12). Phages such as T4 come with a complete set of replication proteins and mostly rely on their own proteins to propagate. Other phages depend on host factors to replicate their genome. For example, the lambda phage genome does not encode a replicative helicase but instead an accessory protein, λP, whose function is to hijack the host helicase: λP replaces DnaC in the DnaB•DnaC complex creating a new helicase complex that loads onto the λ initiation complex (57). Other lambdoid phages may encode other accessory proteins to fulfill the function of λP, such as the phage helicase loader DnaC protein (12). The recent resolution of the structures of the DnaB•DnaC and DnaB•λP complexes revealed that in E. coli, the wide opening of the helicase is promoted by the intercalation of the small N- or C-terminal α−helix of DnaC or λP, respectively, between the LH and the DH α−helices of two neighboring subunits, forming the DH-LH module of the helicase (53,58,59).
Despite the total lack of structural homology between DciA and DnaC/λP, these proteins seem to target the same helicase protein motif; the DH-LH module might be the central point of the regulation of loading/activation of the bacterial replicative helicases. Therefore, it seems likely that the binding of phage loaders such as DnaC or λP will finalize the hijacking of the host helicase by forcing DciA out while inducing the transition of the helicase into an extended-spiral open conformation, competent for a direct loading on DNA. Following our hypothesis proposing that DciA-helicases can load themselves through DNA-protein contacts, phage loaders would need not only to chase out DciA but also to hide the DNA contact zone of the helicase to prevent the helicase to be recruited at oriC.
The function of helicase loaders specified by domesticated phage proteins like DnaC derives from the activity of its phage ancestor. We can only speculate that after domestication of a phage loader gene like dnaC, the weak ability to self-load on DNA of the replicative helicase continuously regressed, because the domesticated phage loader rendered this activity superfluous. It would be ironic that such a strategy, aimed at diverting a host protein from its cellular function, could have permitted the functional complementation of an accidental and otherwise lethal loss of DciA and could explain that the domestication of various phages has happened several times (11,12).
DATA AVAILABILITY
The entry code in the Biological Magnetic Resonance Data Bank (BMRB) for the NMR chemical shifts of VcDciA[1–111] is 27689. The PDB accession number for the crystal structure reported in this paper is 6T66 (VcDnaB•GDP:AlF4:Mg2+). The molecular models and experimental SAXS data have been deposited on SASBDB (Small Angle Scattering Biological Data Bank) under the SAS codes: SASDGP5 (VcDciA), SASDGQ5 (VcDnaB•ATP:Mg2+) and SASDGR5 (VcDnaB•VcDciA•ATP:Mg2+).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
X-ray diffraction and SAXS data were collected at Synchrotron SOLEIL (Saint-Aubin, France; beamlines PROXIMA-2 and SWING). We thank the beamline staff for assistance and advice during data collection. This work has benefited from the expertise of the Macromolecular interactions measurements Platform (PIM) of I2BC (Gif-sur-Yvette, France) for the SPR and SEC-MALS experiments; we thank Michel Desmadril for help and discussions. We thank the Proteomic-Gif SICaPS platform (I2BC) supported by IBiSA, Ile de France Region, Plan Cancer, CNRS and Paris-Saclay University, and in particular Virginie Redeker, David Cornu and Laïla Sago for their expertise. We are thankful to Herman van Tilbeurgh, Sylvie Nessler, Julien Bischerour, Isabelle Vallet-Gely and Frédéric Boccard for critical reading of the manuscript and helpful comments. We thank Marie-Thérèse Paternostre, Agnès Delahodde, Jean-Luc Pernodet and Yves Gaudin for their help and support during the writing process.
Author contributions: J.-L.F. and S.Q.-C. conceived this study; A.H., Y.A. and S.Q.-C. performed the cloning and mutagenesis; C.C. and S.Q.-C. purified and crystallized the proteins; S.M. and M.A.-N. performed the SPR and biochemical assays; H.W., I.G.-L.S. and P.L. performed the X-ray structure determinations and refinements; J.A. and F.O. performed the NMR structural study; E.L.C. and S.B. performed the electron microscopy experiments; D.D. performed the SEC-SAXS experiments; C.V. performed the SEC-MALS experiments; J.-L.F. performed the phylogenetic analyses; Y.A., C.P. and J.-L.F .performed the in vivo experiments; J.-L.F., S.Q.-C. and S.M. wrote the paper with input from all authors.
FUNDING
French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INBS-05]; Centre National de la Recherche Scientifique (CNRS); Region Ile de France; C.C. was supported by a Ph.D. fellowship from the French Ministry of Education; Y.A. by the DIM Malinf program from the Conseil Régional d’Ile de France. Funding for open access charge: Centre National de la Recherche Scientifique (CNRS).
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments