





Abstract
The detailed analysis of the impact of deletions on proteins or nucleic acids can reveal important functional regions and lead to variants with improved macromolecular properties. We present a method to generate large libraries of mutants with deletions of varying length that are randomly distributed throughout a given gene. This technique facilitates the identification of crucial sequence regions in nucleic acids or proteins. The approach utilizes in vitro transposition to generate 5΄ and 3΄ fragment sub-libraries of a given gene, which are then randomly recombined to yield a final library comprising both terminal and internal deletions. The method is easy to implement and can generate libraries in three to four days. We used this approach to produce a library of >9000 random deletion mutants of an artificial RNA ligase enzyme representing 32% of all possible deletions. The quality of the library was assessed by next-generation sequencing and detailed bioinformatics analysis. Finally, we subjected this library to in vitro selection and obtained fully functional variants with deletions of up to 18 amino acids of the parental enzyme that had been 95 amino acids in length.
INTRODUCTION
Numerous studies have highlighted the importance of deletions in protein evolution. For example, analysis of natural proteins or proteins derived from in vitro evolution showed that deletions of up to 40 amino acids within loop regions can be structurally tolerated (1). The Indel PDB database of structural insertions and deletions (2) provides abundant examples for deletions that are not only found in loops and unstructured regions, but also in α-helices and β-sheets. Proteins with indels are also highly represented among essential proteins, and are very common in protein networks, where they show a high level of connectivity, suggesting important regulatory roles (3). Deletions can also result in improved biophysical properties and increased enzymatic activity of a protein (4,5), or in a change in substrate specificity for enzymes. For example, single amino-acid deletions at several positions in a secondary structural element of β-lactamase, resulted in increased activity toward ceftazidime, a poor substrate of the wild-type protein (6). Deletions have also been useful to reveal the importance of protein dynamics to both folding and activity. A well folded protein like EGFP adapted to single amino acid deletions in β-strands by conformational rearrangements influencing a network of amino acids close and far away from the deletion site (7). In another case, the deletion of a loop region in tRNA nucleotidyltransferases resulted in the reduction of protein flexibility and caused the addition of the dinucleotide CC rather than the triplet CCA (8). Furthermore, deletions can also have important evolutionary roles by favoring the emergence of new functions through the change of substrate specificity (8,9).
Besides the clear importance to protein biochemistry, deletion studies have also been invaluable to understand and improve the function of nucleic acid aptamers, ribozymes and deoxyribozymes generated by in vitro selection (10–14). Commonly, the starting point for the in vitro selection are libraries of random nucleic acids with a length between dozens to a few hundred nucleotides. For the identification of crucial regions and to enable practical applications of these functional nucleic acids, a subsequent shortening is often desirable.
The importance of deletions and their evident impact on properties of proteins and nucleic acid demands a detailed investigation in order to better comprehend evolutionary mechanisms, and, ultimately, accelerate the discovery of improved variants by molecular engineering. The starting point for directed evolution experiments most commonly is a large library of variants created by introducing point mutations to a given gene (15) or by recombination of different gene variants (16). In contrast, there are only few examples for the generation of deletion variants, which are usually rationally based on structural information (5,17,18). While this deletion approach has resulted in variants, e.g. with increased stability (5) or catalytic activity (12), a more thorough and efficient investigation of the effects of deletions would require the generation of large libraries of deletion mutants in a combinatorial fashion. Although a few methodologies to generate such libraries have been described, these methods have several undesirable drawbacks. Some of these methods are sequence specific and require the laborious design of primer pairs for each deletion mutant to be generated (19). Other methods employ nucleases such as DNaseI or exonuclease III in a first step to partially degrade the target gene into fragments with a range of lengths (4,20,21). The activity of these nuclease enzymes is difficult to control, and reaction conditions such as time or enzyme concentration usually require extensive optimization to avoid over-digestion (4,20,21). Other methods, despite their simplicity, introduce extraneous sequences at the deletion site (22), are restricted to very short deletions (23–25) or require custom chemical oligonucleotide synthesis capability (12,26).
In order to simplify the procedures required to build large libraries of deletion variants, we developed a method that overcomes the drawbacks mentioned above and generates a library of deletions of varying lengths randomly distributed throughout the parental gene. This method is based on in vitro transposition mediated by the MuA transposase (27) and is therefore sequence-independent without relying on the difficult use of nucleases. The well-characterized MuA transposition system is well-suited for the development of a generally applicable method to create random deletions for several reasons. First, the target site preference of MuA is very low, with insertions occurring essentially at random along the target sequence (27–29). Second, robust standard reaction conditions for MuA have been defined which can be employed for any target DNA, hence no optimization of the transposition reaction is needed. Furthermore, single insertions are obtained when transposon and enzyme are mixed in the presence of an excess of target for a short time (27). The MuA transposition system has been used for several applications such as DNA sequencing (30) and protein engineering (23–25), highlighting its efficiency and wide applicability.
We validated our method by creating a deletion library of the artificial RNA ligase enzyme 10C. This enzyme had previously been generated de novo by in vitro selection from a randomized non-catalytic protein library (31,32). Building on these previous findings, our goal now was to identify truncated active enzyme variants that might be more amenable to crystallization, and to improve ligase properties for potential biomedical applications (33). Previous structure determination of ligase 10C by nuclear magnetic resonance spectroscopy revealed a well-structured protein core framed by highly dynamic termini. In addition, the structured core also contained a flexible internal loop region (31). The flexibility of those three regions likely prevented previous attempts of crystallizing ligase 10C. However, it is conceivable that some of those conformationally dynamic regions are less important for catalytic function and might therefore be dispensable. Hence, deletions in these regions could potentially promote crystallization and even increase activity by reducing alternative conformations that may be inactive. Yet, rational prediction of tolerable deletions has been difficult in general, and for ligase 10C in particular as the active site of this enzyme is still unknown. Therefore, we decided to employ the combinatorial deletion approach described here in conjunction with directed evolution. We used the deletion library of ligase 10C as input for an in vitro selection by mRNA display to isolate shorter active enzyme variants.
In order to build the deletion library, we first generated two separate sub-libraries of 5΄ and 3΄ terminal gene fragments by transposition and PCR. These sub-libraries were randomly recombined by restriction digestion and ligation to obtain the final library comprising both terminal and internal deletions. We analyzed the resulting library by next generation DNA sequencing and confirmed an even coverage and distribution of deletions throughout the parent gene. This library of deletion variants was subjected to six rounds of in vitro selection for active ligase enzymes. We identified ligase variants that were up to one-fifth shorter while exhibiting similar or even slightly increased enzymatic activity.
MATERIALS AND METHODS
The Template Generation System II Kit from Finnzymes was used to perform the transposition reactions. TOPO TA cloning Kit and pUC19 vector (2.68 kb) were purchased from Invitrogen. Phusion High Fidelity DNA Polymerase from New England Biolabs (NEB) was used in all PCR reactions in Phusion buffer (20 mM Tris-HCl pH 8.8, 20 mM KCl, 20 mM, (NH4)2SO4, 2 mM MgCl2, 100 μg/ml bovine serum albumin, 1.25 M betaine, 0.1% Triton X-100). Deoxynucleoside triphosphate (dNTP) solutions and restriction enzymes were purchased from NEB; primers were purchased from Integrated DNA Technologies, and ethylenediaminetetraacetic acid (EDTA) (0.5 M, pH 8.0) was purchased from AccuGENE. Gel Extraction Kit and PCR Purification Kit (QIAGEN) were used according to manufacturer instructions. DNA concentrations were determined by measuring absorbance at 260 nm on a Nano Drop spectrophotometer 2000c (Thermo Scientific). A total of 10X Tris-Borate EDTA (TBE) buffer from National Diagnostics contained 0.89 M Tris Borate pH 8.3 and 20 mM EDTA disodium salt. Gel electrophoresis was conducted in 0.5X Tris-Borate EDTA buffer (TBE). DNA ladders were purchased from NEB.
Preparation of DNA starting materials needed for subsequent library construction
All PCR amplifications were performed in presence of 200 μM of each dNTP, 1X PCR buffer, 0.5 μM of each primer, 0.01 U/ μl of Phusion high fidelity DNA polymerase. Reaction conditions were as follows: 3 min at 94°C followed by the reported number of cycles with 30 s at 94°C, 45 s at 55°C and 1–3 min at 72°C. The DNA encoding the original ligase 10C (31) was amplified using primers AM003 Fwd and AM016C Rev from a pCRII-TOPO, with an extension time of 1 min for 20 cycles. The resulting amplicon constituted what we will from here on refer to as ligase 10C parent. The transposon was amplified from the CamR-3 transposon provided with the Template Generation System II Kit, with 2 min extension time for 24 cycles. The linker DNA pUC19 fragment (GenBank: M77789.2, position 1704–394, named pUC19*) was amplified from a solution of 6 fM pUC19 vector for 26 cycles in a 1 ml PCR reaction, with an extension time of 3 min, with primers AM014 Fwd and AM013 Rev. The DNA obtained was gel purified, desalted and used as template for 8 additional cycles of PCR in the same conditions as above, but with an increased template concentration of 1 nM. The β-lactamase gene was amplified from a pBAD plasmid for 20 cycles with 1 min extension time. The glycerophosphodiester phosphodiesterase (GDPD) gene was amplified from the pET28/GDPDwt plasmid (34) for 30 cycles, with 1 min extension time. Primers are listed in Supplementary Table S1, along with the length of the PCR products. Each PCR product was gel purified and subsequently desalted using gel extraction and PCR clean-up kits (QIAGEN) according to manufacturer's instruction. The transposon was subsequently digested with BglII as described previously (23) and desalted prior to the transposition reaction. The pUC19* fragment was cut with BsaI, and desalted as above.
Agarose gel electrophoresis
Samples were mixed with Ficoll to a final concentration of 2.5% (w/v) prior to loading. Electrophoresis was conducted at 120 V, for 30–50 min. DNA was visualized by UV light at 254 nm and either including 0.5 ng/μl ethidium bromide in the gel prior to pouring, or post-electrophoresis by soaking the gel in 0.5 ng/μl ethidium bromide for 20 min, and subsequently destaining in water for 20 min.
Transposition reactions
Transposition reactions were performed using the Template Generation System II Kit from Finnzymes. Reactions (20 μl) were assembled using 12 ng of ligase 10C parent DNA (64 fmol), 20 ng of transposon (24 fmol), reaction buffer to a final concentration of 1X (25 mM Tris-HCl pH 8.0 at 20°C; 10 mM MgCl2; 110 mM NaCl; 0.05% Triton X-100; 10% glycerol) and 220 ng of MuA transposase. The reaction was incubated at 30°C for 1 h, stopped by deactivating the enzyme at 75°C for 10 min and either stored at −20°C or directly used as template for PCR. Negative control reaction was inhibited by addition of EDTA (final concentration 10 mM), and by heating at 75°C for 10 min prior to addition of the target sequence, and then incubated at 30°C for 1 h.
Transposition for β-lactamase and GDPD was carried out under the same conditions as for ligase 10C, with the amount of target gene adjusted accordingly (32 ng for GDPD and 35 ng for β-lactamase).
Deletion library construction
Two separate PCR reactions were carried out to generate 5΄ and 3΄ fragment sub-libraries of the ligase 10C gene. Amplification was performed for 30 cycles with 200 μM dNTP, 1X PCR buffer, 0.02 U/μl Phusion polymerase, 0.5 μl of transposition reaction/50 μl of PCR reaction and 0.5 μM of the following primers: For the 5΄ library, the AM015 Fwd primer binds to the N terminus of the parent ligase 10C gene and the AM009P Rev primer allows amplification of a 908 bp fragment of the transposon (Supplementary Table S1). For the 3΄ library, the AM010S Fwd primer amplifies 1198 bp of transposon and the AM016 Rev binds to the C terminus of ligase 10C (Supplementary Table S1).
The two PCR products were gel purified and desalted as above. The purified products were both digested using BsaI to generate sticky ends compatible with the ends of the pUC19 fragment. Each reaction contained 100 ng/μl for the 3΄ fragment sub-library, and 83 ng/μl for the 5΄ fragment sub-library, 0.375 U/μl of restriction enzyme, 1X cut smart buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/ml bovine serum albumin) and was incubated at 37°C for 12 h, stopped by heat inactivating the enzyme at 65°C for 20 min. The product was purified and desalted as above. For the subsequent ligation, both digested sub-libraries were mixed at a molar ratio of at least 1:1 with pUC19* that had also been cut with BsaI (≥4.6 pmol of each library containing DNA of varying length and 4.6 pmol of pUC19*). Specifically, the ligation contained 5΄ fragment sub-library at a final concentration of 67 ng/μl, the 3΄ fragment sub-library at a final concentration of 84 ng/μl, pUC19* at a final concentration of 74 μg/μl, 40 U/μl T4 DNA ligase in 1X ligation buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM adenosine triphosphate (ATP), 10 mM dithiothreitol). The reaction was incubated at 23°C for 15 min and the product was gel purified and desalted as above. To remove the transposon sequences, the product was digested with MlyI for 11 h at 37°C in a reaction containing 15 ng/μl of ligated product, 0.3 U/μl of MlyI and 1X cut smart buffer. The desired product was gel purified and desalted. The DNA was circularized by intramolecular ligation overnight at 16°C in a 50 μl reaction containing: 100 ng DNA (2 ng/μl), 40 U/μl of T4 DNA ligase (from a 2000 U/μl ligase stock) and 1X cut smart buffer supplemented with 50 μM ATP. The crude ligation reaction served as template to amplify the final deletion library with the AM015E Fwd and AM016C Rev primers for ligase 10C (Supplementary Table S1) using Phusion Polymerase. The following reaction conditions were applied: 3 min at 94°C followed by 30 cycles with 30 s at 94°C, 15 s at 55°C and 1 min at 72°C. The final deletion library in the range of interest (53–288 bp) was gel purified, desalted by ethanol precipitation and subjected to Illumina next generation sequencing for 150 bp paired ends at the University of Minnesota Genomics Center (UMGC) according to standard procedures.
PCRs of the sub-libraries for β-lactamase and GDPD were performed as described for ligase 10C, with appropriate primers for the respective gene termini as listed in Supplementary Table S1.
Analysis of next generation sequencing data
The software tool Trimmomatic (35) was used to remove the Illumina specific adapter contamination from paired 150 bp long reads produced by a MiSeq. Reads that were longer than 36 bp and maintained their paired status (both R1 and R2 were still viable) were retained for downstream analysis. Paired reads were merged using usearch –fastq_mergepairs (36). Reads to be merged were truncated at the first base with a quality score below 3 and merged reads were allowed to have up to 5 mismatches. Once merged, the sequences were then filtered to remove sequences that contained too many low quality base calls using usearch –fastq_filter. A strict filter was used (–fastq_maxee 0.5) to only include the highest quality sequences with very low error rates.
The resulting high quality sequences (queries) were aligned to the parental full-length sequence of ligase 10C using SSEARCH36 from the FASTA36 (37,38) package using a +1/−3 match/mismatch scoring matrix and −1000/−1000 as gap open and gap extend penalties. SSEARCH36 using the options above created high identity local alignments between the queries and the parental full-length sequence. For each query/parent pair, the high gap penalties resulted in two statistically significant alignments on either side of the deletion. By combining the two alignments, the position and length of the deletion in each query sequence was determined. Combined alignments that did not include the last 5 positions of the 5΄ constant region and the first 5 positions of the 3΄ constant regions, combined alignments that had insertions in the query, combined alignments that did not include the entire query sequence, combined alignments where part of the constant regions was included in the deletions and query/parent pairs that did not result in exactly 2 alignments to be combined were removed. The unique deletions (deletions occurring at a specific location and of a specific length) that resulted from the alignments that met all of the above criteria were then identified and used to track deleted positions and deletions lengths.
N = Number of nucleotides in gene region targeted for deletions.
L = deletion length, N = number of nucleotides in gene region targeted for deletions.
The first part of Equation (4) represents the total number of deletions up to a maximum length (s) that can occur at a position (p). The middle section removes deletions that cannot occur because the deletion length would reach beyond 5΄ end of the targeted gene region. The last section removes those deletions that cannot occur because deletion length would reach beyond the 3΄ end of the targeted gene region.
In vitro selection for ligase variants by mRNA display
mRNA display was performed as described previously (39,40), except that wherever Triton X-100 was present, its concentration was increased from 0.01% to 0.25%, primer AM030 (5΄-pTTTTTTTTTTTTTTTTTTCCCAGATCC) was used instead of BS50 to synthesize the reverse transcription primer and the RNA was only purified by lithium chloride precipitation before photo-crosslinking to the puromycin oligonucleotide (39,41).
The deletion library described above was used as starting material for the mRNA display selection together with two additional libraries: a double deletion library obtained by subjecting the deletion library to the deletion generation procedure for a second time; and a deletion library produced from the original ligase 10C after it had been randomly mutated by two consecutive error prone PCRs using the GeneMorph II kit from Agilent and was appended with an N-terminal Flag-epitope tag using primers AM003C and AM016C (Supplementary Table S1). Prior to the first round of selection, each of the three libraries were amplified in two successive PCRs using primers BS3longb″ Fwd and AM016C Rev and then primers BS3 long Fwd and AM016C Rev (Supplementary Table S1) in order to add the terminal constant regions that are necessary for mRNA display. The three libraries (deletion library, double deletion library, deletion and random mutation library) were transcribed and cross-linked to the puromycin oligonucleotide. Equimolar amounts of the cross-linked RNA from each library were combined and used as input for the first round of the mRNA display.
For each round of selection, a 0.5 ml translation was performed and the resulting mRNA–protein fusions were purified using oligo-dT cellulose and Flag affinity chromatography. Fusions were then reverse transcribed in presence of 32P-dATP for more sensitive radioactive detection and dialyzed 3 times against 1 l of Flag buffer (50 mM HEPES pH 7.4, 150 mM KCl, 5 mM 2-mercaptoethanol, 0.25% Triton X-100) and purified again by Flag affinity chromatography. The resulting eluate was incubated in two separate reactions for 5 min and 60 min, respectively, in presence of the photocleavable biotinylated 3΄-OH-substrate and a complementary splint for the ligation step (39,40). The ligation was quenched by adding EDTA to a final concentration of 10 mM and mixed with solid urea to reach a final concentration of 8 M. The sample was heated at 90°C for 3 min, immediately transferred on ice and purified by streptavidin–biotin affinity chromatography (streptavidin agarose beads, Thermo Scientific). After intensive washing with SA-binding buffer, SA urea washing buffer and SA basic wash solution, water and phosphate buffered saline as described in (40), beads were recovered with 300 μl phosphate buffered saline and irradiated for 15 min at 365 nm while shaking to release the bound cDNA. The released cDNA from the 5 min and 60 min selection was separately ethanol-precipitated after addition of 1 μl glycogen (20 mg/ml), washed with 70% ethanol and dissolved in 100 μl doubly distilled water and amplified in a 1 ml PCR reaction using primers BS3longb Fwd and AM016C Rev (Supplementary Table S1). For each round of the selection, aliquots were taken at the end of each selection step and analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis (4–12% Bis-Tris acrylamide) for quality control. The procedure was repeated for six rounds.
Analysis of enriched ligase sequences from round 6 of the ‘5 min’ and ‘60 min’ selections
DNA obtained after round 6 from both selections was cloned into a pCR-blunt-II TOPO vector, transformed into Escherichia coli and plated according to manufacturer instructions (Invitrogen). Plates were sent to Beckman Coulter for colony picking and sequencing. The resulting sequences were analyzed with the software CLC genomics. Sequences were aligned to the ligase 10C parent gene while some obviously aberrant sequence artefacts were discarded.
Cloning, expression and purification of individual ligase variants
Selected ligase genes were amplified by PCR from the pCR-blunt-II TOPO vector with primers listed in Supplementary Table S1 to introduce NdeI and EcoRI restriction sites and remove the N-terminal Flag tag. The genes were then cloned into pET24a expression vector and transformed into E. coli BL21-DE3 Rosetta strain cells (Novagen). For protein expression, cells containing the respective plasmid were grown in LB medium with 36 μg/ml kanamycin overnight at 37°C. This culture was then used to inoculate 400 ml of LB medium containing 36 μg/ml kanamycin. The cultures were grown to an OD600 nm of 0.6–0.9 at 37°C, induced with 1 mM isopropyl-β-D-thiogalactopyranoside and shaken at 37°C for an additional 6 h. Cultures were centrifuged and the cell pellet was stored at −20°C.
Cell pellets were resuspended in lysis buffer (20 mM HEPES, 400 mM NaCl, 100 μM ZnCl2, 100 mg/l Triton X-100, 5 mM 2-mercaptoethanol, pH 7.4) and lysed by sonication. The lysates were centrifuged, the supernatant recovered and subjected to affinity chromatography using HisPur Ni-NTA resin (Thermo Fisher Scientific) as all ligases contained the sequence HHQHHH, which functioned similarly to a His6-tag. The proteins were eluted with acidic elution buffer (20 mM NaOAc, 400 mM NaCl, 0.1 mM ZnCl2, 100 mg/l Triton X-100, 5 mM 2-mercaptoethanol, pH 4.5) directly into a proper amount of 1 M HEPES pH 7.5 to result in a final concentration of 100 mM HEPES by immediate mixing to re-adjust the pH back to 7.5. All fractions were stored at 4°C. Protein purification was evaluated by SDS-PAGE on NuPAGE Bis-Tris precast gels (Thermo Fisher Scientific). Elution fractions containing pure ligase protein were combined, concentrated and buffer exchanged into ligation buffer (20 mM HEPES, 150 mM NaCl, 0.12 mM ZnCl2 and 0.1 mM 2-mercaptoethanol, pH 7.5) with Amicon Ultra-15 Centrifugal Filter device 3000-MWCO (Merck-Millipore). Protein concentrations were determined by measuring absorbance at 280 nm using a NanoDrop spectrophotometer 2000c (Thermo Fisher Scientific).
Activity measurements of ligase enzymes
Ligase 10C or its selected deletion variants (5 μM) were incubated with 10 μM 32P-labeled 5΄-triphosphorylated PPP-substrate (5΄-pppGGAGACUCUUU), 15 μM complementary splint (5΄-GAGTCTCCGCGAACGT) and 20 μM 3΄-HO-substrate (5΄-CUAACGUUCGC) in ligase buffer (20 mM HEPES (pH 7.5), 150 mM NaCl, 100 μM 2-mercaptoethanol and 120 μM ZnCl2). Reactions were incubated at 24°C and aliquots of the ligation reactions were quenched after 0, 10, 20, 30 and 40 min by mixing with the same volume of 20 mM EDTA/8 M urea and heating to 95°C for 5 min. The ligation progress was analyzed by 20% denaturing PAGE and using a GE Healthcare (Amersham Bioscience) Phosphorimager and ImageQuant software (Amersham Bioscience).
To determine the rate constant (kobs), the slope of the linear fit of percentage of ligation over time was multiplied by the ratio of PPP-substrate/enzyme (10 μM/5 μM) to adjust for the enzyme concentration. To enable a linear fit, the product formation was kept below 15% for all time points. Furthermore, it was assumed that all protein was in its active conformation. The reported values are an average of 3 or 4 independent biological replicates (proteins were separately expressed and purified). For each biological replicate, 2–4 technical replicates were performed. Standard error was calculated on averages of all replicates for a given variant.
RESULTS
Library construction
The goal of this work was to create a library of random internal and terminal deletions of variable length within a given gene (Figure 1). For that purpose, a transposition reaction mediated by the MuA transposase was performed first to insert an artificial transposon (EntranceposonTM, CamR-3) into the target gene (ligase 10C parent) at random positions (Figure 1). Subsequent amplification of the transposition products in two separate PCR reactions using distinct sets of primers generated two ensembles of gene fragments – one for each gene terminus. These two 5΄ and 3΄ fragment sub-libraries had the expected range of lengths as can be seen as diffuse DNA bands in the agarose gel (Figure 2, lane TR) (957–1217 bp for the 5΄ fragment sub-library; 1248–1514 bp for the 3΄ fragment sub-library). No DNA was detected in that size range for any control PCR reactions that included the inhibited transposition reaction (IT) (MuA transposase was inhibited by addition of 10 mM EDTA, and an incubation at 75°C for 10 min before adding the target gene), the transposon DNA alone or the target gene ligase 10C alone. These results demonstrated that the diffuse DNA bands were the product of the specific amplification of the transposition product.
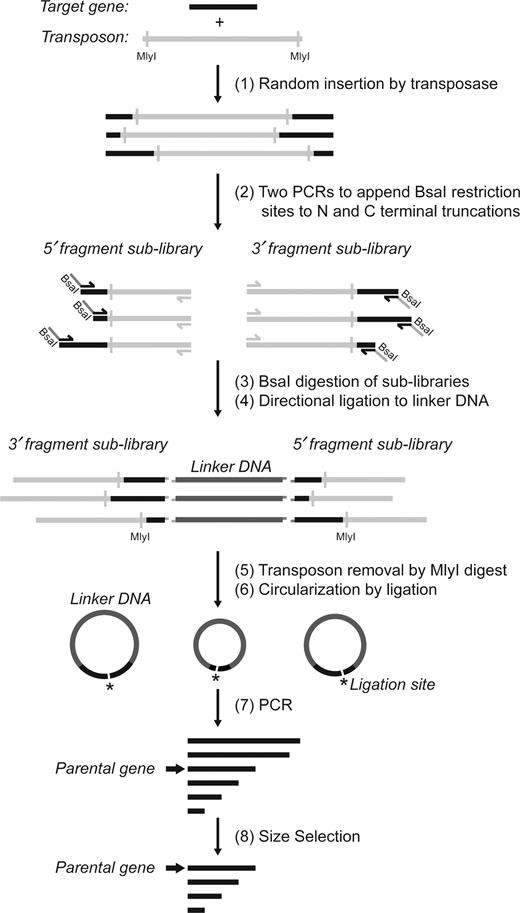
Overview of method to generate random deletions in a target gene. (1) A transposition reaction was performed to obtain random insertions of the transposon into the target gene. (2) 5΄ and 3΄ fragment sub-libraries of the gene were amplified in two separate PCR reactions. In each reaction, one primer was complementary to the 5΄ or 3΄ constant regions of the target gene, and the other to a region on the transposon. (3) The digestion with BsaI created unique overhangs in each library complementary to unique overhangs in the linker DNA (pUC19*-fragment) to favor directional ligation. (4) Libraries were ligated to the linker DNA, which was free of MlyI sites. (5) The product of ligation was treated with MlyI to remove the transposon sequence. (6) Intramolecular blunt-end ligation joined the 5΄ and 3΄ terminal fragments of the gene. (7) This circular library was linearized by PCR with primers complementary to the termini of the parental gene. (8) The final library of deletion variants of the desired size range was isolated by gel electrophoresis.
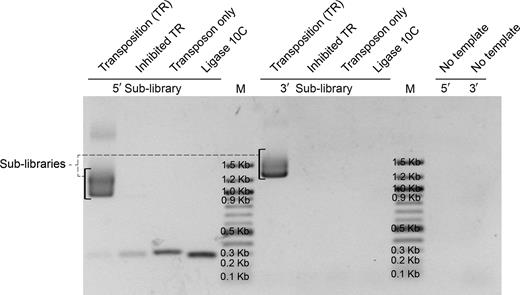
5΄ and 3΄ fragment sub-libraries generated from the transposition reaction (TR) by PCR amplification and analyzed on 1% agarose gel. The transposition reaction was performed according to manufacturer instruction and directly used as PCR template. The ‘Inhibited TR’ sample was heated at 75°C for 10 min in the presence of 10 mM ethylenediaminetetraacetic acid prior to the addition of the target gene, incubated at 30°C, and then used for PCR. The transposon alone was used as template for the PCR reaction for the ‘Transposon only’ sample, and ligase 10C parent alone as template for the PCR reaction for the ‘ligase 10C’ sample. M = Marker (100 bp DNA ladder).
During the amplification of the 5΄ fragment sub-library or the transposon alone, by-products at about 300 bp and 350 bp were observed, respectively, likely resulting from mispriming as indicated by annealing temperature optimization experiments (data not shown).
Following the PCR amplification, the two sub-libraries were digested with BsaI, and ligated to a linear linker DNA produced by PCR amplification of part of the plasmid pUC19 (position 1704–394) as seen in Figure 1. The BsaI type IIS restriction enzyme cuts outside its recognition sequence and allowed the design of a unique cut site for each of the two sub-libraries. The resulting different four-nucleotide overhangs enabled directional ligation to the linker DNA that contained suitable complementary overhangs. This ligation therefore connected one fragment from each of the 5΄ and the 3΄ sub-libraries, which can be seen as a band at ∼4000 bp in Supplementary Figure S1. This is in agreement with the expected size range for this construct of 3491 to 4017 bp. In the following step, the ligation product was digested by the restriction endonuclease MlyI to cleave off the transposon sequences from both ends of this construct.
Supplementary Figure S2 shows the successful removal of the transposon yielding DNA with the expected size range between 1385 bp and 1911 bp, albeit with a slight overrepresentation of shorter sequences. The two additional bands observed at ∼900 bp and ∼ 1200 bp (calculated 908 bp and 1198 bp) confirmed the successful removal of the transposon sequences. The digestion product DNA of ∼1400–2000 bp was excised from the gel, purified, circularized by ligation and used as template to amplify the final deletion library. From this PCR reaction, DNA of all deletion variants in the range from about 53 to 288 bp was gel purified, corresponding to the deletion of the whole gene except for the primer binding regions (32 bp + 21 bp), and the full length gene, respectively. The final deletion library after size-selection can be seen in Figure 3 as a smear from about 300 bp to less than 100 bp. This library of deletion variants of ligase 10C was subjected to next generation sequencing in order to ascertain the quality of the library.
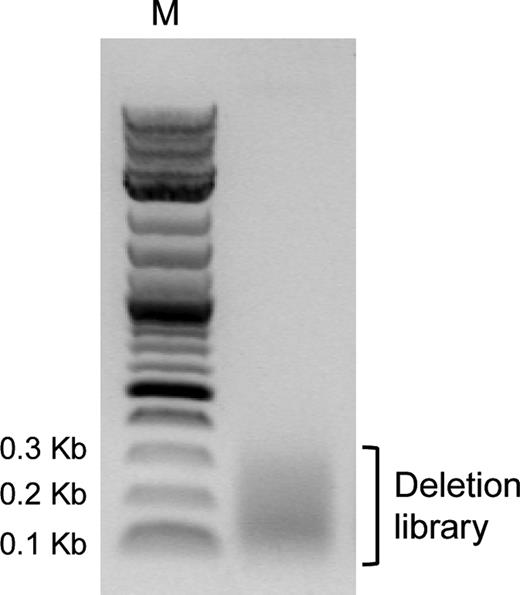
DNA of final deletion library after size-selection by gel extraction shown on a 1% agarose gel. M = Marker (2-Log DNA ladder).
In order to verify the general applicability of our strategy to generate deletion libraries, transposition reactions and PCR amplifications were also performed to produce sub-libraries for two additional unrelated genes: β-lactamase and GDPD. In both cases, the same conditions were used for transposition and PCR as for ligase 10C and successfully amplified 3΄ and 5΄ fragment sub-libraries by using primers specific for the termini of the two target genes (Supplementary Figure S3). These results demonstrated the broad applicability of this deletion library building approach.
Next generation sequencing analysis
Next generation sequencing analysis of our final deletion library of the ligase 10C was conducted to assess the efficiency of our method to generate a large number of deletion mutants randomly distributed throughout the parental sequence. High throughput DNA sequencing of 2 241 199 PCR amplicons (reads) yielded 605 673 high quality merged sequences (queries). These merged sequences contained 9006 unique deletions corresponding to 32% of the 27 730 possible unique deletions (calculated according to Equation (1) with N = 235). In order to analyze the distribution of deleted positions and deletion lengths, the 9006 sequences were aligned to the parental sequence. Figure 4 compares the number of observed deletions at each position in the parental 10C sequence (closed circle) to the number of possible deletions that could occur at that position (open circle). For example, almost 7000 deletions were observed that included position 140. These data demonstrated that the deletions were well distributed throughout the gene with a slight bias for deletion towards the 3΄ end of the parental sequence.
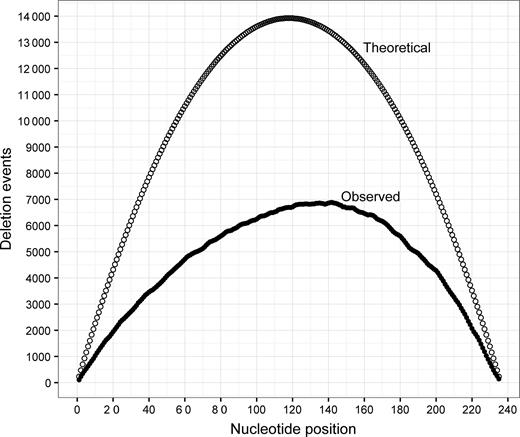
Theoretically possible and observed counts of deletions at each nucleotide position of the ligase 10C parent gene are shown as empty or open circles, respectively. The number of possible deletions were calculated using Equation (2). Observed counts were obtained from alignments between sequences found in the library and the parental sequence of ligase 10C.
We then analyzed the distribution of deletion lengths and found that the library contained variants with deletions between 6 and 235 nucleotides in length (Figure 5). Our observed deletions were enriched for longer lengths with deletions longer than 110 bp observed at 50% or greater of the number of all possible deletions. This percentage decreased with decreasing deletion length.
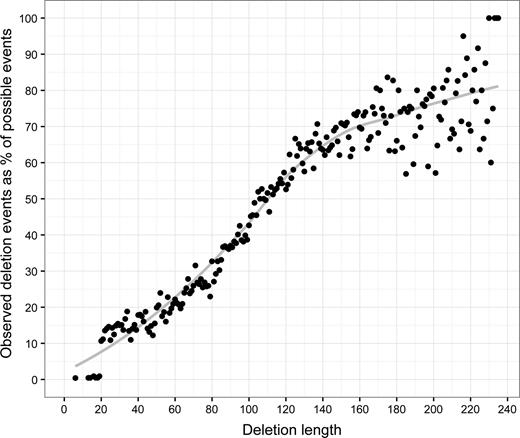
Deletion lengths observed in the library are shown as percent of theoretically possible deletions of that length (closed circle, trend line shown in grey). The number of possible deletion events for each length was calculated using Equation (3).
Each position in the parental 10C gene was observed as a member of many deletion events. Each position in the 10C gene was deleted at a rate of 42–61%, with position towards the 5΄ end of the gene showing a higher rate (Supplementary Figure S4). When sequences are divided into groups by deletion length, most positions in the parental 10C gene were found to be deleted in the context of long deletions while shorter deletions are under-represented and therefore have a lower rate of deletions at each position (Supplementary Figure S5).
In vitro selection for ligase variants
We used the final deletion library as starting point for an in vitro selection by mRNA display in order to isolate shorter variants of the ligase 10C, ideally catalyzing the reaction faster than the parental enzyme and potentially facilitating crystallization of the protein. The deletion of residues at the termini of some other proteins had been successfully employed to favor their crystallization (17). However, considering the highly dynamic overall structure of ligase 10C it was difficult to rationally identify specific disordered regions that could be deleted. We hence decided to take a directed evolution approach by mRNA display using the deletion library of ligase 10C. mRNA display is an in vitro selection technique that enables the isolation of functional variants from libraries of up to 1012 mutants (40,42–44). To further increase the diversity at the beginning of this in vitro selection, two additional libraries were also prepared: a double deletion library by reapplying the deletion generation procedure to the deletion library generated previously; and a deletion library of ligase 10C that had first been randomly mutated by error-prone PCR. The mutational load after error prone PCR was found to be 0–6 mutations per gene corresponding to an error rate of up to 2.3 mutations for 100 nucleotides (on average 1.5 mutations per gene), as assessed by sequencing 20 clones. The three libraries were separately transcribed into RNA, modified with puromycin (to enable mRNA display) and then mixed in equimolar amounts prior to in vitro translation during round 1 of the in vitro selection. We performed two in vitro selections in parallel with different levels of selection pressure by stopping the ligation reaction after 60 min or 5 min of reaction time, respectively. The longer, more permissive incubation time was expected to yield enzyme variants (including deletion variants) with activities similar to the ligase 10C, and the shorter incubation to favor potentially faster enzymes. The mRNA display procedure was performed for 6 rounds of selection and amplification similar to previously published procedures (39,40). The selection progress was monitored by calculating the percentage of cDNA bound to the streptavidin resin at the end of each round as described previously (39,40). For the ‘60 min selection’, we observed an increase in the percentage of cDNA bound from 0.18% to 5% in round 3. In the subsequent rounds 4–6 the percentage varied between 2.3% and 4% (Figure 6, grey bars). For the ‘5 min selection’, the percentage of bound cDNA increased from 0.03% after round 1 to 3.5% in round 5 and then decreased to 1.4% in round 6 (Figure 6, black bars).

Progress of in vitro selections for ligases using an incubation time during the selection step of 5 min (black bars) or 60 min (grey bars).
The PCR amplification of the cDNA after round 3 of the ‘60 min selection’ yielded a clearly discernible dominant DNA band in the agarose gel of a length similar to the full-length ligase 10C parent gene with the T7 promoter and AMV enhancer appended (332 bp), while the smear representing DNAs encoding a range of shorter deletion variants had substantially decreased (data not shown). In order to specifically favor and therefore enrich deletion variants over the full-length gene, after rounds 3–5 of the ‘60 min selection’, DNA shorter than the parental gene (∼300–100 bp) was isolated by gel electrophoresis of the PCR product and used it as input for the following round. This removal of the dominant band at 332 bp explained the drop in percentage of cDNA immobilized observed in rounds 3 to 5 (Figure 6). Despite the repeated removal by gel extraction, the band at 332 bp reappeared after every round as it was enriched during the selection step. Finally, the increase in percentage of cDNA immobilized from round 5–6 of the ‘60 min selection’ indicated substantial enrichment of active deletion variants. An SDS-PAGE analysis of aliquots taken at each step of round 6 of the ‘60 min selection’ (Figure 7A) showed the enrichment of shorter active variants not observed in previous rounds. These shorter variants can be seen as the lower of the two bands in Figure 7A, highlighted in brackets. Comparison of the ligation reaction (rightmost lane) to material from the preceding step (lane ‘Flag II’), showed the appearance of two bands at a higher molecular weight, which represented ligation product caused by the covalent linkage of the 3΄-OH-substrate to the 5΄-PPP-substrate, indicating that both bands represent catalytically active clones. In contrast, the ‘5 min selection’ was mostly enriching variants of a length similar to ligase 10C (Figure 7B) as indicated by the presence of a single band that also shifted up in the ligation reaction (Figure 7B, rightmost lane).
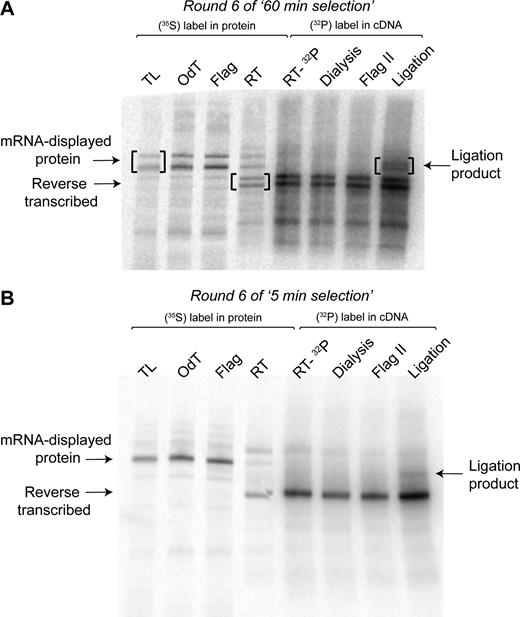
mRNA-displayed proteins after individual steps of selection round 6 analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis. (A) Gel for ‘60 min selection’. (B) Gel for ‘5 min selection’. TL: translation; OdT: eluate after purification by oligo-(dT) cellulose chromatography; Flag: eluate from Flag affinity purification; RT: reverse transcription of 35S-Met-labeled protein-RNA fusions to verify efficiency of reverse transcription; RT-32P: reverse transcription in presence of 32P-α-dATP to include 32P-labeling in the cDNA; Dialysis: sample recovered after dialysis into Flag buffer; Flag II: eluate from second Flag affinity purification. The ligation reaction was performed with biotinylated 3΄-OH-substrate in presence of complementary splint oligonucleotide.
The cDNA obtained after round 6 of both selections was amplified by PCR (Figure 8). Consistent with the results observed by SDS-PAGE gel of the mRNA-displayed proteins from round 6 of the ‘60 min selection’, two major DNA bands were observed in the agarose gel at about 350 bp and at about 300 bp, respectively. The ‘5 min selection’ yielded only a single band at about 350 bp. Figure 8 also shows a comparison of the input DNA to the DNA from round 6 of both selections, demonstrating that the selection pressure changed the composition of the smear by enriching the full-length and the deletion variant band.
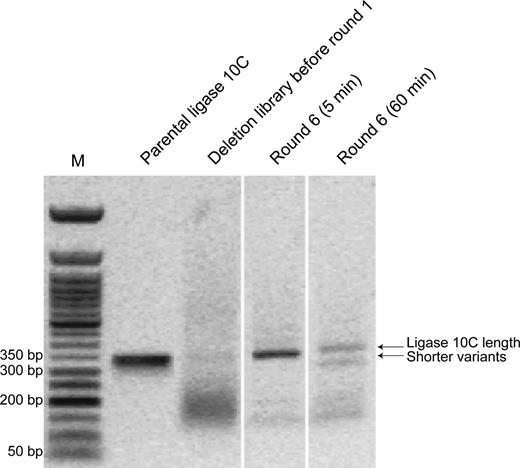
Comparison of DNA from deletion library before round 1 to DNA obtained after 6 rounds of the selections with 5 or 60 min reaction time analyzed by agarose gel electrophoresis (1%). M = Marker (50 bp ladder). It can be clearly seen that the selection process enriched variants similar in length as ligase 10C parent in the ‘5 min selection’, and in addition shorter variants were also enriched in the ‘60 min selection’. This figure is a composite gel picture combining different lanes from the same original agarose gel.
Sequence analysis of ligase clones enriched after six rounds of selection
For both selections, cDNA after round 6 was cloned and sequenced. A total of 68 sequences were obtained for the ‘5 min selection’ and 40 sequences for the ‘60 min selection’, which were aligned to the parental sequence of ligase 10C. After manual inspection of the alignments, we first discarded all short peptide sequences of 20–47 amino acids in length, which consisted of the 13 N-terminal (containing the Flag tag) and 8 C-terminal amino acids of the library, and of duplications of these sequences. These peptides were likely selection artefacts and improbable to possess activity as they were missing crucial regions of ligase 10C such as the putative zinc and substrate binding sites (31). The remaining 55 sequences from the ‘5 min selection’ were either identical to the parent ligase 10C (64%) or point-mutants thereof (38%), but no deletions variants were detected. Of the 18 sequences remaining from the ‘60 min selection’, 7 sequences were identical to the parent ligase 10C, 8 sequences were point-mutants thereof and the other 3 sequences were deletion variants missing either 13 or 18 amino acids near the N terminus (Figure 9). These deletions corresponded to a DNA length of 293 bp and 278 bp, consistent with the lower molecular weight band marked as shorter variants in Figure 8. The analysis of specific point mutations originating from the error prone PCR library that were also enriched for during this selection is beyond the focus of this manuscript and will be published elsewhere.

Sequence alignment of ligase 10C with selected deletion variants from round 6. Ligase variants containing deletions of 13 and 18 amino acids (shown as periods highlighted in gray) were isolated after six rounds of the ‘60 min selection’. The topmost sequence is the original ligase 10C (31). The second sequence shows ligase 10C parent which comprises an additional N-terminal Flag tag for purification purposes during the selection. Dashes symbolize amino acids that are identical to the original ligase 10C sequence. Numbering of residues corresponds to numbering used in the first publication of ligase 10C (31).
Analysis of ligase activity of selected deletion variants
The three ligase deletion variants identified after round 6 of the ‘60 min selection’, namely Del 13, Del 18 and Del 18 I22T (Figure 9), were amplified from their respective TOPO plasmids used for sequencing. We removed the Flag tag at this step, as the original ligase 10C did not have this feature either (32) and the epitope tag was used here during mRNA display selection only for purification purposes. Thus, we obtained the ligase variants Del 13 ΔFlag, Del 18 ΔFlag and Del 18 I22T ΔFlag. After cloning into an expression vector, the variants displayed sufficiently soluble expression in E. coli and were purified by Ni-NTA affinity chromatography, which yielded solutions of >98% pure protein (Supplementary Figure S6). The three enzymes were assayed alongside with the original ligase 10C at 24°C for up to 24 h. All deletion variants reached a maximum ligation percentage similar to ligase 10C, (66–72% of maximum ligation versus 72% for ligase 10C). To compare their catalytic efficiencies, kobs was measured for each ligase variant in the linear range of ligation percentage over time. All deletion variants had ligation rates similar to ligase 10C with variant Del 18 I22T ΔFlag even displaying a slightly increased activity (Figure 10).
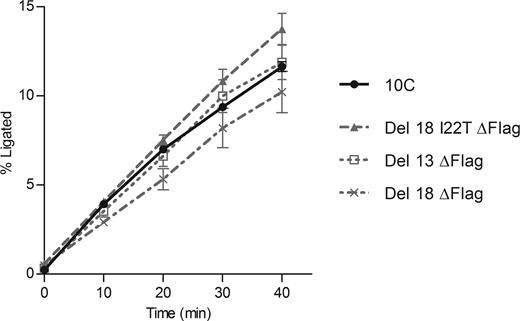
Ligation progress over time catalyzed by the three deletion variants (dashed or dotted gray lines) compared to original ligase 10C activity (solid black line). Rates of ligation were 0.387 ± 0.094 h−1, 0.304 ± 0.120 h−1 and 0.412 ± 0.059 h−1 for Del 13 ΔFlag, Del 18 ΔFlag and Del 18 I22T ΔFlag, respectively, while kobs of ligase 10C was 0.340 ± 0.020 h−1. For each variant, the error bars represent the standard error from at least 3 biological replicates for which 2–4 technical replicates were performed.
DISCUSSION
Previous deletion analyses of sequences and structures of natural proteins and proteins evolved in vitro revealed that the removal of dozens of amino acids can be accepted in some cases without significantly altering the tertiary structure of the protein (1). Furthermore, laboratory experiments showed that deletions can result in increased thermostability of proteins (5), higher transduction efficiency of viral particles utilized for gene therapy (4) and higher catalytic activity for ribozyme variants obtained by in vitro selection (12,13). These reports demonstrated that deletions are not only tolerated, but potentially improve a number of properties of proteins and nucleic acids, highlighting the need to consider deletions more frequently as a valuable source of genetic variability to be incorporated in molecular engineering projects. We devised a protocol capable of generating a large number of deletion mutants in 3–4 days. This method is ideal to routinely generate deletion libraries for directed evolution experiments because little optimization is required for implementation. All procedures used in our protocol can be performed in any laboratory equipped for basic molecular biology techniques.
In addition to improving macromolecular properties by subjecting deletion libraries of a given gene to directed evolution, deletion analysis has also been invaluable for the biochemical characterization of proteins and ribozymes. For example deletion variants were generated to identify the core sequence required for function of artificial proteins (45,46) or ribozymes (12,13), or to remove flexible regions when crystallization had proven difficult (17,18). Usually these approaches were based on detailed structural knowledge in order to determine which segments could potentially be removed. However, when structures are not available, or in molecules with a high degree of disorder like ligase 10C (31), it is difficult to predict which regions can be deleted. It is therefore more practical to generate a library of random deletions and identify functional constructs using one of the many established high throughput screening and selection technologies (47,48).
We performed a detailed analysis of the produced deletion library by next generation sequencing, which confirmed the generation of deletions that varied widely in position and length as desired, resulting in a thorough coverage of the target gene. Therefore this method is suitable to explore the effect of deletions throughout a given parental gene, with no particular targeting of any specific region. Our deletion library contained >9000 different deletion variants that corresponds to 32% of all theoretically possible deletion variants for this parental gene. Nonetheless, the analysis also revealed that while the deletions were generally well-distributed throughout the gene, the library had two slight biases: Deletions toward the 3΄ terminus of the target gene, and deletions of >20 amino acids in length were generated more efficiently. The first bias might be due to the possibility of preferential insertion sites toward the 3΄ terminus of this specific target gene sequence. This is in agreement with previous findings that, although the process of transposition by MuA transposase is mostly random, some sites can be preferred (27–29). Going forward, our method will benefit from more universal transposase enzymes currently under development with further reduced sequence preferences. However, our analysis showed that we have an overall deletion coverage at each nucleotide position of about 42% of the theoretical maximum at the 5΄ terminus of the gene, increasing to about 61% at the 3΄ terminus (Supplementary Figure S4). Therefore, this bias is only small and probably of little significance for most deletion studies due to the high overall coverage. The observed more efficient generation of gene variants with longer deletions might have been caused by the known preferential amplification of shorter sequences during PCR (49,50) because longer deletions resulted from combining of two shorter fragments of the 3΄ and 5΄ sub-libraries (Figure 1, step 2). Previous work by others also described that PCR amplification altered the length distribution of a DNA library depending on the PCR system used (50). In the future, this bias could be mitigated either by using emulsion PCR (51,52), or different polymerase-buffer systems (50). Furthermore, this bias may in part be a result of the library analysis by high-throughput sequencing as this method also includes PCR steps that would similarly deplete longer sequences containing shorter deletions as they are harder to PCR amplify.
While a gene library containing almost half of all possible deletions variants as described here will likely be sufficient for most downstream applications, it might be possible to increase this coverage even further. Transposition reaction conditions similar to ours, which were used in a previous directed evolution experiment targeting a 2.6 Kb plasmid, yielded 2000 insertion events (23). Furthermore, the rate of transposon insertion is known to decrease with decreasing length of the target gene (personal communication from transposase manufacturer). Therefore, it is conceivable that our transposition reaction did not create all possible insertions in our relatively short 288 bp target resulting in the less than complete coverage observed for our library. Using a more efficient transposase such as Hyper MuA transposase or conducting transposition reactions at a larger scale could enable further increased coverage of the theoretical possible library complexity.
We applied our deletion generation procedure to a gene coding for an artificial RNA ligase enzyme (31,32). The resulting library of deletion variants was then subjected to in vitro selections by mRNA display for active ligase variants. While the selected libraries were mostly dominated by full length variants, the extra selection step of gel-purifying and therefore favoring shorter variants during rounds 3–6 of the ‘60 min selection’ led to enzyme variants with single N-terminal deletions of 13 or 18 amino acids (Figure 9). Despite the fact that a single deletion library and a double deletion library were used as input for the in vitro selection, only these single deletions were found. The absence of variants with deletions outside of the N-terminal region suggested that these other regions of the ligase enzyme are more important for proper enzyme activity. However, the possibility of functional variants with deletion in those regions cannot entirely be ruled out as only about one-third of all possible deletion variants were represented in the deletion library.
Biochemical ligase activity assays showed that the deletions in our selected protein variants did not interfere with enzymatic activity. The largest observed N-terminal deletion of 18 amino acids removed the entire epitope tag (E-tag) that had been used for purification during the selection of the original ligase 10C and three additional amino acids (Gly15, Gly16, Lys17; numbering as in original publication) (32). Interestingly, this deletion ended just before residue His18, which we have previously shown to be the start of the highly structured core of the enzyme and involved in binding to a structurally crucial Zn2+ ion (31). In contrast, this same study of the three-dimensional structure of ligase 10C by nuclear magnetic resonance spectroscopy showed that residues N-terminal of Lys17 were essentially unstructured. This deletion study here now independently identified this same region and confirmed that these unstructured residues were not necessary for catalytic function.
Deletion libraries can also facilitate the mapping of protein–protein interactions. One protein would be subjected to the deletion procedure to generate a large library of variants, and the other protein would be used as a bait to recover shorter variants that retain binding affinity. Such an approach, in conjunction with other techniques, was employed to identify interaction surfaces and to determine the structure of the yeast nuclear pore complex Nup84 at a resolution of 1.5 nm by combining domain deletion mapping data with electron microscopy and computer modeling (53). In that study, deletion constructs were individually generated one by one, based on knowledge about the domain boundaries of the complex components. The generation of a random deletion library could be a more efficient approach if sufficient structural data were not available.
Another potential application of this protocol could be the creation of non-homologous recombinant sequences. For that purpose, separate or one-pot transposition reactions would be performed with different target DNA sequences encoding for the parental genes while using the same transposon sequence. The subsequent random recombination of the resulting gene fragments would then be facilitated by using PCR primers that introduce common restriction sites. Therefore, the same directional ligation, transposon removal and circularization ligation steps as described in the current work could be applied to generate non-homologous recombinant sequences.
CONCLUSIONS
An efficient tool was developed for generating libraries of >9000 random deletion mutants of a given gene. Next Generation Sequencing analysis confirmed that deletions ranging widely in length and position throughout the target gene can be obtained. Such a library is suitable for directed evolution experiments as the isolation of fully active deletion variants of the artificial enzyme ligase 10C demonstrated. Furthermore, this strategy of combining in vitro transposition and PCR can be applied to different targets, demonstrating broad applicability of our method. Given the ease of execution, this method will be a very useful tool for both the biochemical characterization and the engineering of proteins and functional nucleic acids, further advancing biochemical knowledge and creating biomolecules with improved properties.
GENBANK ACCESSION NUMBERS
Ligase 10C original: KY286531; ligase 10Cp-Del13 ΔFlag: KY286532; ligase 10Cp-Del18 I22T ΔFlag: KY286533; ligase 10Cp-Del18 ΔFlag: KY286534.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work is dedicated to Prof. Romas Kazlauskas on the occasion of his 60th birthday.
The authors thank J. A. Baller for advice on the sequencing data analysis, J. C. Haugner and M. Held for comments on the manuscript.
FUNDING
US National Aeronautics and Space Administration (NASA) Agreement [NNX09AH70A] through the NASA Astrobiology Institute–Ames Research Center; US National Institutes of Health [GM108703]. Funding for open access charge: U.S. Department of Health and Human Services; National Institutes of Health; National Institute of General Medical Sciences [GM108703].
Conflict of interest statement. None declared.
Present address: Aleardo Morelli, Adaptimmune Ltd., Abingdon, Oxfordshire, OX 144RY, UK.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments