





Abstract
AR9 is a giant Bacillus subtilis phage whose uracil-containing double-stranded DNA genome encodes distant homologs of β and β’ subunits of bacterial RNA polymerase (RNAP). The products of these genes are thought to assemble into two non-canonical multisubunit RNAPs - a virion RNAP (vRNAP) that is injected into the host along with phage DNA to transcribe early phage genes, and a non-virion RNAP (nvRNAP), which is synthesized during the infection and transcribes late phage genes. We purified the AR9 nvRNAP from infected B. subtilis cells and characterized its transcription activity in vitro. The AR9 nvRNAP requires uracils rather than thymines at specific conserved positions of late viral promoters. Uniquely, the nvRNAP recognizes the template strand of its promoters and is capable of specific initiation of transcription from both double- and single-stranded DNA. While the AR9 nvRNAP does not contain homologs of bacterial RNAP α subunits, it contains, in addition to the β and β’-like subunits, a phage protein gp226. The AR9 nvRNAP lacking gp226 is catalytically active but unable to bind to promoter DNA. Thus, gp226 is required for promoter recognition by the AR9 nvRNAP and may represent a new group of transcription initiation factors.
INTRODUCTION
Transcription, the synthesis of RNA from DNA template, is the first step of gene expression in all living organisms. It is catalyzed by DNA-dependent RNA polymerases (RNAPs). These enzymes can be divided into two evolutionarily unrelated classes—the single-subunit RNAPs and the multisubunit RNAPs. Members of the former class belong to a super-family of ‘right-handed’ DNA and RNA polymerases, are composed of a single catalytically active polypeptide, and transcribe genes of mitochondria, chloroplasts and some bacteriophages (1). Multisubunit enzymes, together with some RNA-dependent RNAPs, form a family of ‘two-barrel’ RNAPs (2). Multisubunit RNAPs are responsible for transcription of eubacterial, archaeal and nuclear eukaryal genes. All multisubunit RNAPs are related to each other through common ancestry. The simplest multisubunit RNAPs are encoded by eubacterial genomes. The bacterial RNAP core has a subunit composition of α2ββ’ω and a molecular weight of about 350 kDa. The basic RNAP subunit composition is preserved in archaea and eukarya, although the core complex contains several additional subunits and has a molecular weight in excess of 500 kDa (3). Three-dimensional structures of the multisubunit RNAP core enzymes are similar and resemble a ‘crab claw’ (3), with the two largest subunits, β and β’ in bacteria, forming the jaws of the claw. The catalytic center with a tightly bound Mg2+ ion is located deep in the cleft between the jaws. The dimer of bacterial α subunits or their archaeal and eukaryal homologs keeps the largest subunits together (4).
To initiate specific transcription, RNAP must recognize and bind promoters and locally melt the double-stranded DNA at and around the transcription initiation start point. To perform these steps, all multisubunit RNAPs require accessory transcription factors. Bacteria employ one of the several σ factors each of which binds the RNAP core forming a holoenzyme able to recognize promoters with different consensus elements (5,6). When bound to RNAP core, σ subunits also initiate localized promoter melting. Archaeal and eukaryal RNAPs use a set of unrelated proteins called general transcription factors that bind promoter DNA and then recruit the RNAP core (7).
In the dominant class of bacterial promoters recognized by holoenzymes containing σ subunits of the σ70 class, promoter melting occurs at the promoter-proximal consensus element (referred to as the ‘–10 element’) (8–10). The σ subunits recognize the non-template strand of this element and initiate melting at its upstream end (11). Melting is then propagated downstream, directing the template strand into the RNAP catalytic center (10,11).
Besides the well-studied ‘canonical’ cellular multisubunit RNAPs, atypical enzymes may exist (reviewed in (12)), as judged by the presence of evolutionarily conserved motifs in the products of several genes of unknown function. These include the product of the early gene 64 of Thermus thermophilus phage P23-45 (a protein that contains the metal-binding motif of the multisubunit RNAP catalytic center (13)), the large Cgl1702 protein of Corynebacterium glutamicum (a single protein which is distantly related to a fusion of the β and β' subunits of bacterial RNAP), a hypothetical RNAP encoded by cytoplasmic killer plasmids of yeast Klyveromyces lactis (composed of polypeptides homologous to parts of the β and β’ subunits subunits of bacterial RNAPs), and RNAPs of giant phiKZ-related phages (also composed of distant homologs of the largest bacterial RNAP subunits (14,15)).
The sequence of the uracil-containing double-stranded DNA genome of the giant Bacilus subtillis AR9 phage has recently been determined (16). The AR9 phage shares a core set of orthologous genes (including the RNAP genes) with phiKZ-related phages (16). Development of phiKZ and AR9 was shown to be independent of a bacterial host cell RNAP, confirming that both phages rely on their own transcription machinery for expression of their genes (15,17). Interestingly, no genes coding for the homologs of α subunit or promoter-specificity σ factors have been identified in phiKZ-related phage genomes. Whenever it has been investigated, one set of β/β’ homologs is found in the virions of phiKZ-related phages (16,18–20), likely forming a virion RNAP (vRNAP) that is injected into a bacterial cell along with phage DNA and transcribes early phage genes. Another set of β/β’ homologs forms a non-virion RNAP (nvRNAP) synthesized during subsequent stages of infection that transcribes late phage genes, including the vRNAP genes.
We have recently reported the isolation and initial biochemical characterization of the phiKZ nvRNAP (21). In this work, we purified the nvRNAP of AR9. The enzyme consists of four polypeptides together comprising the equivalents of complete bacterial β and β’ subunits and an additional fifth polypeptide. We show that the AR9 nvRNAP transcribes from late AR9 promoters in vitro and that the fifth nvRNAP subunit is required for promoter recognition. In vitro analysis of transcription initiation by the AR9 nvRNAP shows that promoter recognition depends on the presence of conserved uracils in the template strand of late AR9 promoters. Furthermore, the AR9 nvRNAP is capable of promoter-specific transcription from single-stranded DNA molecules. This ability, unprecedented for any multisubunit RNAP studied to date, provides a new vantage point for detailed functional analysis of the mechanism of transcription initiation and its evolution.
MATERIALS AND METHODS
Bacteriophage, bacterial strain and growth conditions
Information about the AR9 phage, B. subtilis strains, bacterial growth and phage infection conditions and preparation of AR9 phage lysates can be found in (16,17).
For purification of AR9 nvRNAP, 20 l of B. subtilis cells were grown up to OD595 = 1 and infected with AR9 phage at a MOI of 10. The infection was stopped after 22 min by chilling the culture on an ice water bath followed by centrifugation at 3500 g for 30 min at 4 °C. The resulting pellets were stored at –20°C.
Purification of AR9 nvRNAP
All steps of the following procedure were done on ice or at 4°C. Twenty grams of infected B. subtilis cells were disrupted by sonication in 100 ml of buffer A (40 mM Tris–HCl pH 8, 5 mM EDTA, 5 mM β-mercaptoethanol, 0.1 mM PMSF) containing 50 mM NaCl followed by centrifugation at 15 000 g for 30 min. An 8% polyethyleneimine (polymin P) solution (pH 8.0) was added with stirring to the cleared lysate to the final concentration of 0.8%. The resulting suspension was incubated on ice for 30 min and centrifuged at 10 000 g for 15 min. The supernatant was removed and the pellet was resuspended in buffer A containing 0.3 M NaCl. After 10 min incubation, the PEI pellet was formed by centrifugation as previously. Supernatant containing 0.3 M NaCl extract from the PEI pellet was saved for further analysis. Then, extraction was repeated twice with buffer A containing 0.5 M NaCl and 1 M NaCl. Eluted proteins were precipitated by addition of ammonium sulfate to 67% saturation and dissolved in buffer A without NaCl. The same procedure also was done for uninfected cells. All samples were loaded onto a 5 ml HiTrap heparin-sepharose HP column (GE Healthcare) equilibrated with buffer A with 0.1 M NaCl. The column was washed with buffer A with 0.1 M NaCl. Then, step elution with buffer A containing 0.3 M NaCl, 0.6 M NaCl and 1 M NaCl was carried out. Heparin-sepharose chromatography was done for three PEI extracts from infected and uninfected cells. All fractions were analyzed by denaturing SDS polyacrylamide gel electrophoresis (SDS-PAGE). The bands missing in samples obtained from uninfected cells were analyzed by mass-spectrometry. Following this way, fractions containing gp089 and gp154 were found. They corresponded to fractions eluted in 0.6 and 1 M NaCl, respectively, from the Heparin-sepharose column during chromatography of 1 M NaCl PEI-extract. The bacterial RNAP was separated from the nvRNAP during heparin-sepharose chromatography, where it was eluted at 0.6 M NaCl in fractions ahead of the nvRNAP. These fractions were pooled and concentrated by ultrafiltration (Amicon Ultra-4 Centrifugal Filter Unit with Ultracel-30 membrane, EMD Millipore) and loaded onto a Superdex 200 Increase 10/300 (GE Healthcare) gel filtration column equilibrated with buffer A containing 200 mM NaCl. As a final purification step, the combined nvRNAP fractions eluted from the Superdex 200 column were diluted 4-fold with buffer A and applied to a MonoQ HR 5/5 column (GE Healthcare). Bound proteins were eluted with a linear 0.25–0.45 M NaCl gradient in buffer A. The nvRNAP was eluted from the column at 0.34–0.38 M NaCl. The fractions containing nvRNAP subunits were concentrated to a final concentration 0.5 mg/ml, then glycerol was added up to 50% to the sample for storage at −20 °C.
Native gel electrophoresis
One microgram of AR9 nvRNAP was resolved by a native 5% PAGE. A single band was revealed by Coomassie blue staining. To determine the protein composition of this band, it was excised from the native gel and the gel piece was placed into a well of an SDS 8%–polyacrylamide gel, supplemented with 5–8 μl of Laemmli loading buffer and subjected to electrophoresis. The SDS gel was silver stained.
Mass-spectrometric identification of proteins after SDS-PAGE
Information about the mass-spectrometric procedure can be found in (16).
DNA templates for transcription assay
Genomic DNA of AR9, phiR1-37 and phiKZ bacteriophages for transcription assay were purified using the QIAGEN Lambda Midi Kit according to the manufacturer's instructions.
DNA templates containing late AR9 promoters and their derivatives were prepared by polymerase chain reaction (PCR). PCRs were done with Encyclo DNA polymerase (Evrogen, Moscow) and the AR9 genomic DNA as a template, with a standard concentration of dNTPs to obtain DNA fragments with thymine or in the presence of dUTP in place of dTTP to obtain DNA fragments with uracil. Oligonucleotide primers used for PCR are listed in Supplementary Table S1.
To synthesize promoter templates for analysis of the consensus sequence, PCR with oligonucleotide primers bearing single substitution at desired positions of the promoter was performed. Since thymine-containing oligonucleotide primers were used, the final templates were hybrids with respect to their thymine/uracil content (full sequences of primers and resulting templates are shown in Supplementary Table S2). Since we found that the AR9 nvRNAP efficiently and specifically transcribes from such templates containing the wild-type P007 and P077 promoters with thymines in functionally important positions of the non-template but not template strands (Figure 2B and Supplementary Figure S1B, lanes 1), we concluded that such cost-effective ‘hybrid’ strategy is appropriate for mutational analysis.
Double-stranded and partially single-stranded DNA templates containing the P007 and P077 promoters with uracils and thymines at certain positions were prepared by annealing of oligonucleotides ordered from Integrated DNA Technologies (IDT) and listed in Supplementary Table S4. To prepare specific DNA templates, two corresponding oligonucleotides were annealed together by mixing in buffer containing 20 mM Tris–HCl and 40 mM KCl, incubating at 75 °C for 1 min and cooling down to 4°C by a decrement of 1°C/min.
Single-stranded DNA templates containing the P007 promoter were ordered from IDT and listed in Supplementary Table S5.
To prepare RNA–DNA scaffold, the template DNA oligonucleotide (5΄-GGTCCTGTCTGAAATTGTTATCCGCTAC-3΄), the non-template DNA oligonucleotide (5΄-ACAATTTCAGACAGGACC-3΄) and the 32P-end-labeled RNA oligonucleotide (5΄-GUAGCGGA-3΄) were mixed in concentrations 1, 1 and 0.5 μM, respectively in a buffer containing 20 mM Tris–HCl, 40 mM KCl, 1 mM MgCl2 and 0.5 mM DTT, incubated at 65 °C for 1 min and cooled down with an increment of 1°C/min.
Primer extension and sequencing reactions
For in vitro primer extension reaction RNA was synthesized by AR9 RNAP for 15 min at 37 °C from PCR fragments containing late AR9 promoters in 50 μl of transcription buffer (20 mM Tris–HCl, 40 mM KCl, 1 mM MgCl2, 0.5 mM DTT and 100 μg/ml bovine serum albumin) in the presence of 100 μM each of ATP, CTP, GTP, UTP. RNA was purified with TRIzol reagent (Invitrogen) according to manufacturer's protocol and used for primer extension reaction. The primers indicated by asterisk in Supplementary Table S1 were labeled with [γ-32P]-ATP by phage T4 polynucleotide kinase (New England Biolabs), as recommended by the manufacturer. The purified RNA was reverse-transcribed from a 32P-end-labeled primer with Maxima enzyme (Thermo Fisher Scientific) according to the manufacturer's protocol. The reactions were stopped by addition of a loading buffer and heating at 85°C. Sequencing reactions were carried with USB Thermo Sequenase Cycle Sequencing Kit (Thermo Fisher Scientific) on the PCR products containing corresponding start sites, with the primers used for primer extension reactions. The reaction products of sequencing and reverse transcription reactions were resolved on 6–8% (w/v) denaturing polyacrylamide gels and visualized using a PhosphorImager (Molecular Dynamics).
In vitro transcription
Multiple-round run-off transcription reactions were performed in 10 μl of transcription buffer (20 mM Tris–HCl, 40 mM KCl, 1 mM MgCl2, 0.5 mM DTT and 100 μg/ml bovine serum albumin) and contained 30–50 nM AR9 nvRNAP and either 0.06 nM phage genomic DNA or 30–50 nM of indicated DNA template. The reactions were incubated for 10 min at 37 °C, followed by the addition of 100 μM each of ATP, CTP, and GTP; 10 μM UTP and 3 μCi [α-32P] UTP (3000 Ci/mmol). Where indicated, rifampicin was added to the final concentration of 10 μg/ml. Reactions proceeded for 30 min at 37°C and were terminated by the addition of an equal volume of denaturing loading buffer. The reaction products were resolved by electrophoresis on 6–20 % (w/v) denaturing 7 M urea polyacrylamide gel and visualized by PhosphorImager (Molecular Dynamics).
Abortive transcription initiation reactions were set at the same general conditions as run-off transcription reactions but supplemented with 175 μM of initiating RNA dinucleotides specified by the –1/+1 positions of promoters studied (UpG for P007 and UpA for P077 promoters were used). Reactions were incubated for 10 min at 37°C, followed by the addition of 3 μCi [α-32P] UTP (3000 Ci/mmol). The reactions were allowed to proceed for 15 min at 37 °C and terminated by the addition of an equal volume of denaturing loading buffer. Abortive initiation reaction products were resolved by electrophoresis on 20% (w/v) denaturing 7 M urea polyacrylamide gels and visualized by PhosphorImager (Molecular Dynamics).
Transcription reactions from RNA–DNA scaffold were set at the same buffer as run-off transcription reactions and contained 15 nM RNA–DNA scaffold and 15 nM AR9 nvRNAP. Reactions were incubated for 10 min at 30 °C, followed by the addition of 1 mM each of ATP, CTP, GTP and UTP. Reactions proceeded for 15 min at 37 °C and were terminated by the addition of an equal volume of denaturing loading buffer. The reaction products were resolved on 18% (w/v) denaturing 7 M urea polyacrylamide gel and visualized as described above.
Footprinting reactions
DNA templates for footprinting reactions were prepared by PCR (as templates for transcription reactions) with a 32P-end-labeled reverse primer to obtain template strand labeled or with a 32P-end-labeled forward primer to obtain non-template strand labeled (Supplementary Table S3). Promoter complexes were formed in 20-μl reactions containing 50 nM AR9 nvRNAP and 30 nM 32P-end-labeled DNA fragment in a buffer with 20 mM Tris–HCl, 40 mM KCl, 1 mM MgCl2 and 100 μg/ml bovine serum albumin. Reactions were preincubated for 10 min at 37°C. DNase footprinting reaction was initiated by addition of 1 unit of DNase I (Ambion). The reaction proceeded for 30 s at 37 °C and was terminated by addition of EDTA to 15 mM followed by phenol extraction and ethanol precipitation. For KMnO4 probing, promoter complexes were treated with KMnO4 (2 mM) for 20 s at 37°C. Reactions were terminated by addition of β-mercaptoethanol to 450 mM, followed by ethanol precipitation, and 15 min treatment with 10% piperidine at 95°C. Products of footprinting reactions were resolved by electrophoresis on 8% (w/v) denaturing 7M urea sequencing polyacrylamide gels and visualized by PhosphorImager (Molecular Dynamics).
RESULTS
Purification of multisubunit phage RNAP from AR9 infected cells
To purify phage-encoded RNAP(s), cell lysate of B. subtilis culture infected with AR9 at high multiplicity of infection (MOI) and collected midway through the infection cycle was subjected to fractionation following the standard bacterial RNAP purification scheme involving polyethyleneimine (Polymin P) fractionation, heparin-sepharose affinity chromatography, gel-filtration, and anion exchange chromatography (Figure 1A, left panel). Extraction of Polymin P pellet with a buffer containing 1.0 M NaCl yielded, after heparin-sepharose chromatography, fractions that contained two prominent protein bands with apparent molecular weights of ∼80 and ∼75 kDa (indicated by asterisks in Figure 1A, right panel, lane 3). Mass-spectrometric analysis of these bands identified them as AR9 gp089 and gp154, the presumed subunits of nvRNAP homologous to C-terminal parts of bacterial RNAP β and β’ subunits, respectively (16). By following the gp089 and gp154 bands during subsequent chromatographic steps, a fraction from a MonoQ column that contained five protein bands as judged by SDS-PAGE (Figure 1A, right panel, lane 5) was obtained. In addition to gp089 and gp154, this fraction also contained two AR9 polypeptides homologous to the N-terminal parts of bacterial RNAP β and β’ subunits, gp105 and gp270, respectively (16). The fifth polypeptide was gp226, a distant homolog of phiKZ gp68, a subunit of the recently purified phiKZ nvRNAP with unknown function (21). All five polypeptides migrated in a single band during non-denaturing gel electrophoresis (Figure 1B), indicating that they form a complex, which we will refer to as AR9 nvRNAP. The subunit composition of AR9 nvRNAP corresponds to that reported long ago for an RNAP isolated from B. subtilis culture infected with a closely related PBS2 phage, with gp089, gp154, gp105, gp226 and gp270 of AR9 likely matching PBS2 P80, P76, P58, P53 and P48, respectively (22,23).
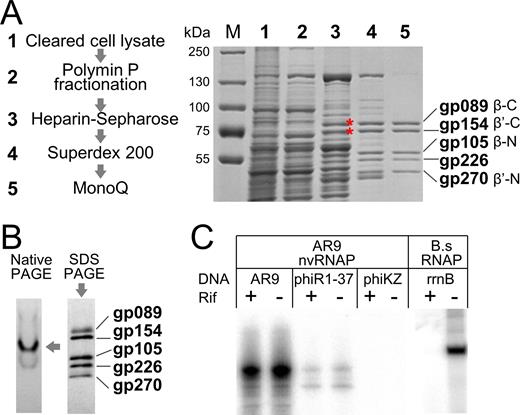
Purification of nvRNAP from AR9 infected Bacillus subtilis cells and analysis of its transcriptional activity. (A) Left: main steps of nvRNAP purification. Right: SDS-PAGE analysis of fractions containing gp089 and gp154 (marked by red asterisks) during the course of AR9 nvRNAP purification. A Coomassie-stained gel is shown; lane numbers correspond to the steps of purification shown on the left. (B) Left: a Coomassie-stained gel after native PAGE analysis of the five-subunit form of AR9 nvRNAP after the final MonoQ purification (step 5 in panel А). Right: a silver-stained gel after SDS-PAGE analysis showing polypeptides present in the native gel band marked by an arrow. (C) In vitro transcription by AR9 nvRNAP of genomic DNA of AR9, phiR1-37 and phiKZ phages in the presence and in the absence of rifampicin. Transcription by B. subtilis RNAP of a PCR-fragment containing the rrnB promoter was used as a control.
In vitro transcription by AR9 nvRNAP
The PBS2 RNAP was reported to transcribe genomic DNA of the phage in vitro (22). Transcription of several other phage genomes was much less efficient (22). We tested the AR9 nvRNAP for transcription from genomic DNA of the AR9, phiR1-37 and phiKZ phages. For each template, transcription reactions were conducted in the presence or in the absence of rifampicin, a host RNAP inhibitor. The result, shown in Figure 1C, revealed that the AR9 nvRNAP was highly active on the AR9 template, was partially active on the phiR1-37 template, and was inactive on the phiKZ template. Whenever transcription was observed, it was rifampicin-resistant. Control transcription by host RNAP was sensitive to rifampicin.
As mentioned above, the nvRNAP likely transcribes late viral genes. Late AR9 promoters were recently identified in the course of global transcript profiling of AR9-infected cells (17). When PCR fragments containing several predicted late promoters were tested as templates in in vitro transcription reactions with the nvRNAP, no transcription products were detected (Figure 2A, top panel). Since AR9 nvRNAP transcribed the AR9 and phiR1-37 genomic DNA both of which contain uracil instead of thymine (16,24), we considered whether the presence of uracil is required for promoter recognition. Accordingly, DNA templates with late promoters containing uracil instead of thymine were tested for in vitro transcription. Robust transcription by AR9 nvRNAP was observed from every template tested (Figure 2A, bottom panel).
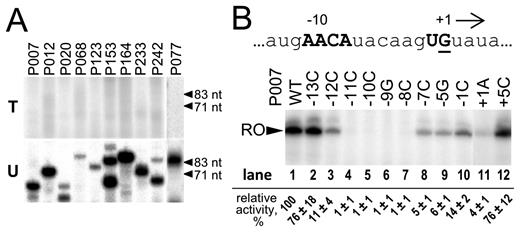
In vitro transcription by AR9 nvRNAP from late AR9 promoters and their mutant variants. (A) Multiple-round run-off transcription by AR9 nvRNAP was performed using templates containing indicated late AR9 promoters. The templates for transcription were prepared by PCR either with dTTP (top panel) or dUTP (bottom panel). The primers used to prepare the DNA templates are listed in Supplementary Table S1. (B) Mutational analysis of the AR9 P007 late promoter. The nucleotide sequence of the non-template strand at and around the TSS of the P007 promoter DNA is shown at the top. The position of the +1 start site is underlined and direction of transcription is shown by an arrow. The AR9 late promoter consensus nucleotides are shown in capital bold letters. Below, in vitro run-off transcription by AR9 nvRNAP of the DNA templates containing P007 and its derivatives (see Supplementary Table S2) is shown. ‘RO’—run-off transcripts (62 nt). Numbers indicate transcription activities relative to transcription from the wild-type P007 promoter (taken as 100%). Average values and standard deviations from three independent experiments are presented.
The 5΄ ends of transcripts generated by the AR9 nvRNAP in vitro were mapped by primer extension analysis and matched late promoter transcription start sites (TSSs) revealed in vivo (Supplementary Figure S1A). We therefore conclude that the five-subunit AR9 nvRNAP recognizes late AR9 promoters. We further conclude that AR9 nvRNAP specifically transcribes late promoter-containing templates with uracil in place of thymine.
Functional analysis of AR9 late promoter consensus element
To determine the role of the late promoter 5΄-A−11ACA-(6N)-UA/G+1-3΄ consensus motif (17) in transcription by the AR9 nvRNAP, DNA templates bearing single-substitutions at conserved and non-conserved positions of the motif were tested in an in vitro multiple-round run-off transcription assay (Figure 2B and Supplementary Figure S1B). Mutations were introduced into the P007 and P077 late phage promoters. For both promoters, substitutions at the positions –11, –10, –9 and –8 with respect to the TSS fully abolished transcription, indicating that the conserved 5΄-A−11ACA−8-3΄ motif plays a crucial role in promoter specific transcription. Substitutions at the +1 position also strongly decreased transcription. Substitutions at non-conserved promoter positions and at conserved position –1 had a smaller effect. We therefore conclude that the late promoter 5΄-A−11ACA-(6N)-UA/G+1-3΄ consensus motif is necessary for in vitro transcription by the AR9 nvRNAP.
Characterization of AR9 nvRNAP-promoter complex
To characterize AR9 nvRNAP promoter complexes we performed DNase I footprinting and KMnO4 probing. DNase I footprinting of the P077 promoter complex on uracil-containing DNA revealed that AR9 nvRNAP protected the template strand positions between ca. –20 to +20 and the non-template strand positions from ca. –20 to +13 (Figure 3, lanes 2 on the left and right panels, respectively). Some upstream positions (–25, –36, –44) became hypersensitive to DNase I attack in the presence of AR9 nvRNAP. When AR9 nvRNAP was added to thymine-containing promoter template no significant protection from DNase I digestion was observed (Figure 3, lanes 4 on the left and right panels). Thus, the absence of transcription from thymine-containing late promoters is caused by the inability of AR9 nvRNAP to bind to such templates.

Promoter binding and promoter opening by AR9 nvRNAP. DNase I footprinting and KMnO4 probing of nvRNAP complexes with the P077 promoter DNA was performed with DNA templates containing uracil (U) or thymine (T). Positions relative to the TSS (+1) are indicated. Lanes indicated as ‘AG’ show markers. Areas protected from DNase I attack are indicated in blue. A fragment of the P077 promoter sequence is shown below, with uracils that undergo oxidation by KMnO4 in the presence of AR9 nvRNAP indicated by blue triangles.
KMnO4-sensitive bands between positions –8 to +3 of the uracil-containing template were observed (Figure 3, lanes 6 on the left and right panels), delineating a transcription bubble. No KMnO4 sensitivity was observed in reactions with the thymine-containing template (Figure 3, lanes 8 on the left and right panel). Introduction of non-consensus G at the position –9 abrogates transcription (Supplementary Figure S1B) and also abolished promoter melting on uracil-containing template (Supplementary Figure S2, lane 4 in comparison to the lane 2).
The nature of uracil requirement by AR9 nvRNAP
To further investigate the uracil requirement for AR9 nvRNAP transcription we designed a set of double-stranded DNA templates based on the P007 late promoter with uracils and thymines at different positions, and tested them in a multiple-round run-off transcription assay. As expected, the nvRNAP did not transcribe the thymine-only template but efficiently transcribed from uracil-only template (Figure 4A, lanes 1 and 2, respectively). Introduction of single thymines at the –11 and –10 positions in the template strand of the consensus element 5΄-A−11ACA−8-3΄ led to dramatic decrease in transcription (Figure 4A, lanes 4 and 5, respectively) while thymines at the –14, –8 and –6 positions had little or no effect (Figure 4A, lanes 3, 6 and 7, respectively). Transcription of the thymine-containing template with uracils at positions –14, –11, –10, –8 and –6 of the template strand was even more efficient than transcription of uracil-only template (Figure 4A, lane 8). The nvRNAP also transcribed from a thymine-containing template with uracils at the –11 and –10 positions (Figure 4A, lane 9). Therefore, we conclude that the presence of uracils instead of thymines at the –11 and –10 positions of the template strand is both necessary and sufficient for promoter specific transcription by AR9 nvRNAP; the presence of neighboring uracils increases transcriptional activity.

Analysis of promoter template DNA requirements by AR9 nvRNAP. (A) In vitro run-off transcription by AR9 nvRNAP of double-stranded P007 promoter templates carrying uracils and thymines at different positions. ‘RO’—run-off transcripts (18 nt). The numbers under the gel indicate transcription activities relative to uracil-only control template. Below, DNA sequences around the TSS of the DNA templates used in the experiment are shown. Uracils and thymines are highlighted in blue and red, respectively. The position of the +1 start site is underlined. Conserved nucleotides of the late promoter are shown in capital bold letters. Average values and standard deviations from three independent experiments are presented. (B) The results of in vitro abortive initiation reactions by AR9 nvRNAP from the double-stranded and fork-junction P007-based DNA templates shown below. RNA dinucleotide monophosphate UpG was used as a primer to initiate transcription. The full DNA sequences of the templates can be found in Supplementary Table S4.
Template strand recognition by AR9 nvRNAP
We designed fork-junction templates based on the P007 and P077 promoters where parts of either template strand or non-template strand were absent, while the transcribed part was double-stranded (Figure 4B and Supplementary Figure S3). The AR9 nvRNAP transcribed from templates without the non-template strand with same efficiency as from the fully double-stranded templates (Figure 4B and Supplementary Figure S3, lanes 3 and 1, respectively). No transcription from templates with missing template strand of promoters was detected (Figure 4B and Supplementary Figure S3, lanes 2). Transcription from the partially double-stranded templates was abolished when thymines were introduced instead of uracils in the consensus positions (Figure 4B and Supplementary Figure S3, lanes 4). Thus, AR9 nvRNAP specifically recognizes single-stranded late promoter consensus in the template strand (3΄-U−11UGU−8-5΄).
Promoter specific transcription by AR9 nvRNAP from single-stranded DNA
The fact that nvRNAP recognizes the promoter consensus element in single-stranded form and in the template strand suggested that the enzyme may be capable of specific transcription of single-stranded DNA. Indeed, we observed robust multiple-round transcription by AR9 nvRNAP from single-stranded P007 promoter template containing uracil and no transcription from thymine-only template (Figure 5A, lanes 2 and 1, respectively). Introduction of thymines at the –11 and -10 positions strongly inhibited transcription from single-stranded templates containing uracils in other positions (Figure 5A, lanes 4 and 5, respectively). Introduction of thymines in several randomly chosen non-consensus positions or consensus position –8 had small or no inhibitory effect. As was the case with the double-stranded templates, introduction of uracils at the –11 and –10 positions was sufficient to allow transcription from a single-stranded template containing thymines in all other positions (Figure 5A, lane 9). Thus, the AR9 nvRNAP requirement for uracils in single-stranded and double-stranded promoters are the same.
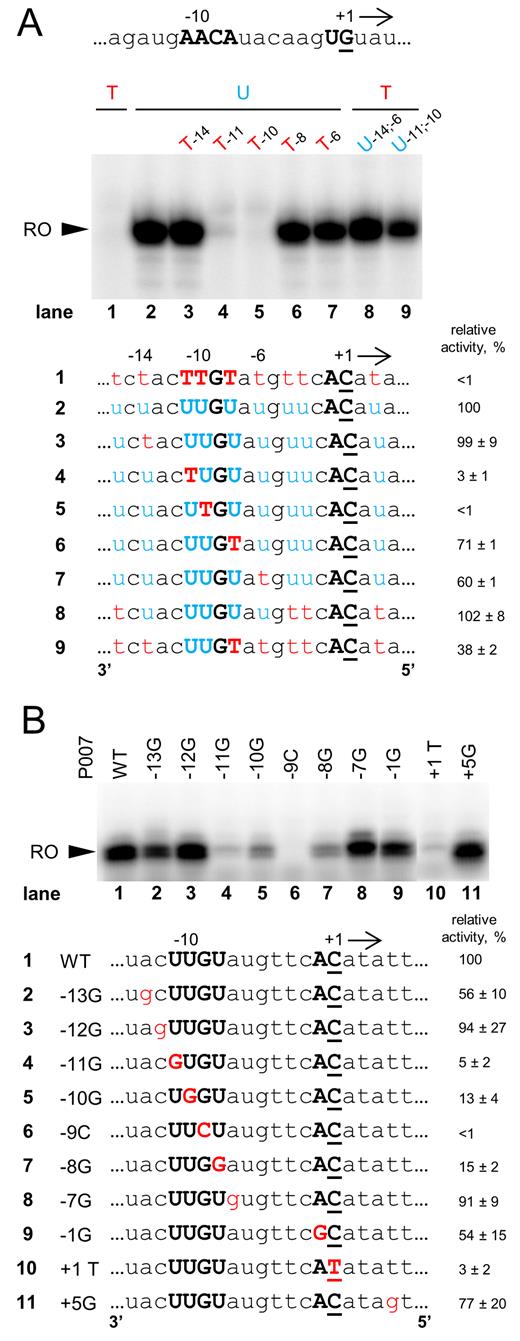
Specific transcription initiation by AR9 nvRNAP using single-stranded promoter DNA templates. (A) In vitro run-off transcription assay of AR9 nvRNAP using the single-stranded DNA templates matching the template strand of the P007 promoter carrying uracils and thymines at different positions. ‘RO’—run-off transcripts (18 nt). The nucleotide sequence of the non-template strand at and around the TSS of the P007 promoter DNA is shown at the top. Below the gel, DNA sequences around the TSS of the DNA templates used in the experiment are shown. (B) In vitro run-off transcription assay of AR9 nvRNAP using single-stranded templates based on the P007 promoter DNA and its derivatives. Average values and standard deviations from three independent experiments are presented. The full sequences of the templates can be found in Supplementary Table S5.
Mutational analysis of the promoter consensus element in the context of single-stranded DNA was also performed (Figure 5B). While the consensus requirement appeared less strong for single-stranded DNA transcription than for double-stranded DNA transcription, nevertheless, a common pattern of important positions was observed in both cases.
AR9 nvRNAP lacking gp226 subunit is catalytically active but unable to initiate transcription from promoters
At the last step of AR9 nvRNAP purification we obtained a minor fraction that contained trace amounts of gp226, the AR9 nvRNAP subunit with unknown function (Figure 6A, bottom). This finding is in agreement with the earlier observation that P53, the likely counterpart of gp226, is dissociable from the PBS2 RNAP (23). We compared AR9 nvRNAP lacking gp226 with the five-subunit form in in vitro transcription reactions from an RNA–DNA scaffold and promoter-containing templates (Figure 6B). AR9 nvRNAP lacking gp226 extended the RNA primer from the RNA–DNA scaffold with the same efficiency as the five-subunit enzyme but was unable to transcribe promoter-containing templates. No binding or melting of the promoter-containing template by AR9 nvRNAP lacking gp226 was observed when it was tested in footprinting experiments (Figure 6C). Thus, we conclude that the four-subunit AR9 nvRNAP form composed of the β/β’ bacterial homologs but lacking gp226 is catalytically active but unable to bind to promoters.
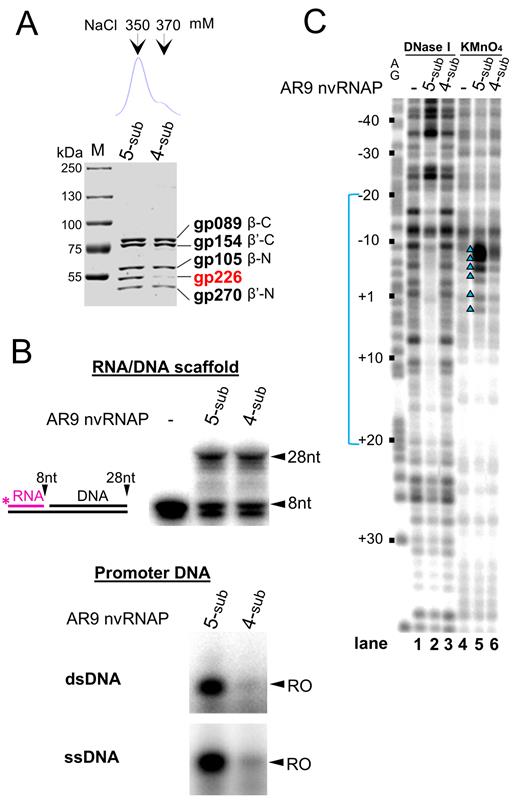
Functional analysis of the two forms of AR9 nvRNAP. (A) Atop: chromatographic profile of AR9 nvRNAP eluted from a MonoQ column with a NaCl concentration gradient. Below: a Coomassie-stained SDS gel of the MonoQ fractions containing five-subunit (5-sub) and four-subunit (4-sub) forms of AR9 nvRNAP. (B) Comparison of transcriptional activities of the 5s and 4s nvRNAP. Top panel: RNA extension assay using the RNA-DNA scaffold schematically shown on the left. Below: in vitro run-off transcription from the uracil-containing promoter P007 in double- and single-stranded DNA (dsDNA and ssDNA). ‘RO’—a run-off transcript (62 nt for dsDNA and 18 nt for ssDNA). (C) DNase I footprinting and KMnO4 probing of nvRNAP–promoter P077 complexes formed by 5s and 4s nvRNAP. The experiment was performed using the uracil-containing template (with template strand radiolabeled). See Figure 3 legend for details.
DISCUSSION
Here, we describe the purification of one of the two RNAPs encoded by a giant bacteriophage AR9 and characterize its interaction with phage promoters. Since AR9 nvRNAP subunits are the products of early phage genes the enzyme was expected to transcribe late phage genes from promoters characterized by the 5΄-A−11ACA-(6N)-UA/G+1-3΄ consensus sequence revealed by the dRNA-seq analysis of infected cells (17). This expectation was fulfilled, however, the enzyme was also found to strictly require the presence of uracils instead of thymines in the template strand positions –11 and –10 of promoter DNA (Figure 4A). The presence of thymines in other positions of promoter or transcribed DNA has little or no effect on AR9 nvRNAP transcription.
The AR9 phage possesses a double-stranded DNA genome with uracil in place of thymine (16). While unusual nucleotides are generally thought of as a strategy to overcome host defenses by restriction-modification systems (25), they can also help specific transcription of viral genes. Bacillus SP01 phage genome contains hydroxymethyl uracil instead of thymine (26). SP01 utilizes host RNAP core bound to the phage-encoded sigma factor, gp28, for transcription of its middle genes that was shown to be dependent on the presence of the modified nucleotides in the phage middle promoters (26). In the case of T4 bacteriophage, whose genome contains hydroxymethyl cytosine instead of cytosine, the phage-encoded transcription terminator factor Alc terminates transcription elongation from cytosine-containing host DNA, while transcription of viral DNA is unaffected (27). The requirement for uracils in promoter consensus element is an elegant strategy that should allow AR9 to avoid unnecessary transcription from host DNA, which contains multiple matches to the simple consensus of phage late promoter.
The requirement for uracils in the template strand of the late promoter consensus element suggested that nvRNAP recognizes the template strand. Indeed, AR9 nvRNAP specifically transcribes from the single-stranded template containing a reverse complement of the 5΄-A−11ACA-(6N)-UA/G+1-3΄ late promoter consensus, provided that uracils were present in positions –11 and –10. The ability of the AR9 nvRNAP for promoter-specific transcription of single-stranded DNA is, to our knowledge, unprecedented for a multisubunit RNAP. The AR9 DNA is very AU-rich (72.25 %) and may be present in partially single-stranded form in infected cells, especially during phage DNA replication, thus facilitating late promoter recognition.
The fact that AR9 nvRNAP can recognize its promoter consensus element in single-stranded form suggests that during transcription initiation from a double-stranded DNA, the consensus element is also recognized in a single-stranded form and then stabilized to ensure transcription bubble maintenance. Since no binding to or melting of thymine-only late promoter templates by nvRNAP is observed, 5-methyl groups of thymines at positions -11 and -10 of the template strand must interfere with the recognition by AR9 nvRNAP. On the other hand, substitution of –11 and –10 positions for non-consensus bases also abolishes transcription. Thus, nvRNAP may be specifically recognizing uracil residues in the consensus element. The mechanism by which AR9 nvRNAP recognizes uracils is a subject of ongoing investigation. It could resemble that of uracil-DNA glycosylase, a well-studied enzyme that efficiently distinguishes uracils in DNA from all other bases including thymines by base-flipping of uracil nucleotides and burying them in a binding pocket where other, more bulky bases do not fit (28).
AR9 nvRNAP lacking gp226 is unable to bind promoter DNA but is catalytically active. Therefore, gp226 may be directly responsible for promoter recognition. We have found a limited similarity of gp226 (∼aa170–aa255) with the region 2 of bacterial σ factors belonging to the σ70 class using HHpred program (Supplementary Figure S4A) (29, 30). Region 2 is the most conserved part of the σ70 class proteins that is involved in core binding and –10 promoter element recognition (4,8,10). Binding to the core proceeds through a conserved coiled-coil structure in the largest (β’) subunit (31). HHpred and sequence analysis indicates that the corresponding structure should be present in the AR9 gp270, a homolog of the N-terminal part of bacterial RNAP β’ subunit (Supplementary Figure S4B). Unlike RNAP holoenzymes containing σ70 class proteins, AR9 RNAP recognizes the template strand of promoter DNA. Thus, the function of sequence-specific recognition of promoters must reside in a region of gp226 with no homology to proteins of known function. Homologs of gp226 are encoded in all phiKZ-related phages, suggesting that all nvRNAPs may share a common mechanism of promoter recognition. Thus, gp226 and its homologs may constitute a new group of transcription initiation factors.
Footprinting experiments show that AR9 nvRNAP complexes on uracil-containing double-stranded templates appear to be similar to bacterial RNAP open complexes based on the extent of DNA protected from DNase I digestion and the extent of localized promoter melting. The presence of DNase I hypersensitive sites located with a ∼10 bp periodicity suggests that upstream DNA is wound around the AR9 nvRNAP, similarly to the situation in open promoter complexes formed by bacterial RNAP (10,32). However, in bacterial RNAP, the upstream DNA contacts are accomplished by the dimer of α subunits, which are absent from the AR9 nvRNAP. The unusual subunit composition of giant phages RNAPs and apparently new mechanisms of transcription initiation utilized by these enzymes make them attractive subjects for comparative analysis of transcription machinery mechanisms, structures and evolution.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Kira S. Makarova for noticing gp226 similarity to bacterial sigma factors and useful discussions.
FUNDING
This work was supported by Ministry of Education and Science of the Russian Federation [14.B25.31.0004], Russian Foundation for Basic Research [16-34-00805 mol_a] and Russian Science Foundation grant 15-15-10017 to Roman Kozlov. The work was carried out using scientific equipment of the Center of Shared Usage ‘The analytical center of nano- and biotechnologies of SPbPU’. Funding for open access charge: Skolkovo Institute of Science and Technology.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments