





Abstract
LuxR-type transcription factors control diverse physiological functions necessary for bacterial adaptation to environmental changes. In the intracellular pathogen Brucella, the LuxR homolog VjbR has been shown to regulate the expression of virulence factors acting at early stages of the intracellular infection and, directly or indirectly, hundreds of additional genes. However, the precise determination of VjbR direct targets has so far proved elusive. Here, we performed chromatin immunoprecipitation of VjbR followed by next-generation sequencing (ChIP-seq). We detected a large amount of VjbR-binding sites distributed across the Brucella genome and determined a markedly asymmetric binding consensus motif, an unusual feature among LuxR-type regulators. RNA-seq analysis performed under conditions mimicking the eukaryotic intracellular environment revealed that, among all loci associated to VjbR-binding, this regulator directly modulated the expression of only a subset of genes encoding functions consistent with an intracellular adaptation strategy for survival during the initial stages of the host cell infection. Other VjbR-binding events, however, showed to be dissociated from transcription and may require different environmental signals to produce a transcriptional output. Taken together, our results bring new insights into the extent and functionality of LuxR-type-related transcriptional networks.
INTRODUCTION
Brucella is a genus of Gram-negative, facultative intracellular bacteria responsible for brucellosis, a widely distributed zoonosis that affects a broad range of wild and domestic mammals (1). In livestock, this disease produces abortion in females and orchitis and sterility in males, thus compromising reproduction of the infected animals and generating severe economic losses worldwide (2). Brucellosis can be transmitted to humans through consumption of contaminated dairy products, inhalation of bacterial aerosols, or contact with wounds or mucosal tissues (1). The first manifestations of human brucellosis are characterized by acute febrile symptoms and, in the absence of antibiotic treatment, this disease eventually leads to a chronic phase characterized by persistent foci of infection and complications such as arthritis, endocarditis, neurological symptoms and even death.
As an intracellular pathogen, the success of Brucella to establish persistent infections depends on its ability to survive and proliferate within different cell types of the mammalian host. Accordingly, this genus has evolved different strategies to overcome host bactericidal mechanisms through subversion of the eukaryotic vacuolar trafficking (3,4). Upon internalization, Brucella is enclosed within a vacuole that rapidly acquires early endosome surface markers and establishes transient contacts with lysosomes, resulting in limited intraluminal content exchange and a consequent acidification of the vacuole (5). This process constitutes a decisive step for subsequent stages of the infection, since low pH is one of the signals that trigger expression of the VirB apparatus (6), a Type-IV Secretion System (T4SS) necessary for the biogenesis of an endoplasmic reticulum (ER)-like organelle permissive for the replication of Brucella (4). The VirB system translocates several effector proteins to the eukaryotic host (7–12), which affect host-cell pathways contributing to the establishment of the intracellular infection. Consistent with the pivotal role of VirB function in subverting host-intracellular trafficking, virB mutants are no longer able to sustain interactions with ER-derived membranes, fail to establish the intracellular replication niche, and are completely degraded in phagolysosomes (13,14).
Despite significant progress in identifying virulence factors and elucidating how Brucella exploits intracellular trafficking pathways, the genetic program orchestrating intracellular adaptation of this bacterium is still poorly understood. Recently, VjbR has been shown to play a major role in modulating expression of the virB operon and other Brucella virulence-associated genes such as flagellar components, many cell surface structures, and two VirB effectors (8,15,16). VjbR belongs to the LuxR-family, a widely-distributed group of transcription factors that control diverse physiological functions necessary for bacterial adaptation to changes in the environmental conditions. LuxR-type regulators participate in a mechanism called quorum sensing (QS), which regulates gene expression in a bacterial cell density-dependent manner through sensing of self-produced autoinducer signaling molecules of the type of acyl-homoserine lactones (AHL) in many Gram-negative bacteria (17). Noteworthy, in Brucella, instead of mediating collective responses, the LuxR-type regulator VjbR is required in the context of the confinement of a single bacterium isolated within a phagosome at early stages of intracellular infection.
Previous transcriptomic and/or proteomic analyses revealed that VjbR function is not restricted to the regulation of a small subset of virulence-related elements, but it also affects, directly or indirectly, the expression of hundreds of genes (18,19), pointing to a role for this LuxR-type transcription factor as a global regulator of Brucella gene expression. Nevertheless, overlap between these studies was 5–9% of the total lists of VjbR-dependent expressed genes, and promoter sequence analysis of the candidate target genes did not reveal a consensus binding motif. In an additional attempt to identify direct targets of VjbR, DNAse I Footprinting was performed on promoters containing a sequence previously observed at the center of the virB promoter VjbR-binding site, the only target DNA sequence characterized so far at the molecular level for this regulator (20). This sequence was shown to be 9 bp-long, asymmetric, and identical to half of the 20-bp palindromic binding site of MrtR, a LuxR-type transcription factor from the closely-related bacterial genus Mesorhizobium (21). However, the analysis of four different Brucella promoters containing the MrtR-like motif showed no binding of VjbR, suggesting that nucleotides flanking this 9-bp DNA sequence are critical for binding site recognition (20). Therefore, despite efforts by different groups, the precise identification of novel VjbR-binding targets has so far remained elusive.
In this work, we sought to determine the VjbR regulon through the identification of its direct targets and the consequent transcriptional output. To this end, we performed a direct assessment of VjbR activity by two complementary genome-wide technologies, ChIP-seq and RNA-seq, under conditions that trigger the expression of VjbR and mimic the environmental variables encountered by Brucella during the first stages of intracellular infection. The results presented here provide a comprehensive mapping of the VjbR binding across the genome of Brucella abortus and bring new insights into the VjbR-mediated mode of control of global gene expression of this intracellular pathogen.
MATERIALS AND METHODS
Strains, media and growth conditions
Brucella abortus 2308 wild-type or the isogenic deletion mutant B. abortus ΔvjbR strain were cultured in Tryptic soy broth (TSB) 5 mM nalidixic acid, or incubated in MM1 medium (22) pH 5.5 in the presence of 5 mM urocanic acid (Sigma-Aldrich) at 37°C in a rotary shaker (250 rpm).
ChIP-seq
In order to induce VjbR expression, B. abortus 2308 wild-type or B. abortus ΔvjbR control strain (OD600 = 0.05) were grown in TSB up to OD600 of 1.0, a condition where VjbR is not yet expressed in the wild-type strain (22). Subsequently, bacteria were harvested, resuspended in MM1 5 mM urocanic acid pH 5.5, and incubated at 37°C (250 rpm) for 3 h. Induction of the expression of both VjbR and one of its direct targets, VirB7, was corroborated by Western blot as previously described (22). After induction with urocanic acid, formaldehyde was added to a final concentration of 1% (vol/vol) and incubated for 15 min at room temperature with gentle agitation. Crosslinking was stopped by the addition of glycine to a final concentration of 125 mM, and bacteria were subsequently washed three times in ice-cold phosphate-buffered saline buffer. Bacterial pellets were resuspended in lysis buffer [50 mM Hepes pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% (vol/vol) Triton X-100, 0.1% (wt/vol) sodium deoxycholate (DOC), 0.1% (wt/vol) sodium dodecyl sulfate (SDS), complete protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride (PMSF)], and chromatin was fragmented using a sonicator Misonix XL-2000 with 12 pulses of 30 s at 1.5 min intervals in an ice bath. Bacterial debris was removed by centrifugation for 10 min at 14 000 rpm, and a fraction of the supernatant was stored as the input sample for IP assays. Protein G magnetic beads (Thermo Scientific), pre-washed with lysis buffer and blocked O.N. with 5 mg/ml BSA, were incubated with the fragmented chromatin and a polyclonal mouse anti-VjbR antiserum for 4 h at 4°C on a rotator. Beads were then washed twice with lysis buffer, once with wash buffer (500 mM NaCl lysis buffer), once with LiCl buffer [20 mM Tris–HCl pH 8.0, 0.25 M LiCl, 1 mM EDTA, 0.5% (wt/vol) NP-40, 0.5% (wt/vol) DOC, 1% (wt/vol) PMSF], and twice with buffer TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA). Elution and reverse crosslinking were carried out by incubation of the samples (IP or input) with elution buffer [50 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% (wt/vol) SDS, 1 mM dithiothreitol (DTT)] for 16–18 h at 65°C. Supernatants were treated with 10 mg/ml RNAse A (Sigma-Aldrich) for 1 h at 37°C, 0.2 mg/ml Proteinase K (Sigma-Aldrich) for 30 min at 55°C, and DNA fragments were purified using the Wizard kit (Promega). Both control input and immunoprecipitated DNA samples corresponding to three wild-type and one ΔvjbR mutant biological replicates were submitted to Illumina TruSeq library preparation and 50-bp single-end HiSeq 2000 sequencing.
RNA-seq
Brucella abortus 2308 or B. abortus ΔvjbR strains were cultured and induced with urocanic acid as described for ChIP-seq. After induction, bacteria were washed and total RNA was isolated using MasterPure RNA Purification Kit (Epicentre) and then treated with TURBO DNAse (Ambion – Life Technologies). RNA samples corresponding to two biological replicates from the wild-type or the isogenic ΔvjbR deletion mutant were submitted to Illumina TruSeq rRNA-depleted (bacteria) stranded library preparation and 50-bp single-end HiSeq2000 sequencing.
Molecular methods
Western blot was performed to analyze the expression of the VjbR, VirB7 and RibH1 proteins as previously described (22).
Occupancy of VjbR at selected regions of the B. abortus 2308 genome was performed on two independent ChIP assays of B. abortus 2308 or B. abortus ΔvjbR strains by quantification of immunoprecipitated fragments corresponding to sequences containing the virB promoter VjbR-binding site or the VjbR-binding sequences identified upstream of genes BAB2_0068 (virB1), BAB2_1099 (ftcR), BAB1_1775 (rpoH1), BAB1_1657 (dsbB), BAB1_1663, BAB1_1882, BAB1_1700, BAB1_0676, the araC-homolog BAB1_0677, and BAB2_0081. This was carried out by quantitative real-time PCR (qPCR) using dilutions of the immunoprecipitated DNA as template and the qPCR primers listed in Supplementary Table S4. As a control for normalization, we also quantified sequences of a fragment of the coding region of the housekeeping gene IF-1, which was not thought to bind VjbR, using primers previously described (22). Real-time quantitative PCR reactions (qPCR) and analysis were performed as previously described.
Validation of RNA-seq on four genes was performed by reverse transcription-qPCR (RT-qPCR). Total B. abortus 2308 or B. abortus ΔvjbR RNA from three independent experiments was isolated as described above. Subsequently, reverse transcription was performed using the first‐strand SuperScript III cDNA kit (Invitrogen) and random decamer primers (Invitrogen) in the presence of RNasin ribonuclease inhibitor (Promega). Transcript levels were measured in a Mx3005P quantitative-PCR system (Stratagene) using cDNAs as template, KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems), and the RT-qPCR primers listed in Supplementary Table S4. Transcript levels were normalized to that of the IF-1 houskeeping gene as described above.
Electrophoresis mobility shift assays (EMSA). DNA probes corresponding to intergenic sequences upstream of genes BAB2_0653, BAB1_0677 (araC), BAB1_1775 (rpoH1), BAB1_1550, BAB1_2037 or BAB2_1099 (ftcR), and one control probe lacking VjbR-binding motifs, were 32P-labeled by PCR as described previously (20), using B. abortus 2308 genomic DNA as template and the EMSA primers listed in Supplementary Table S4. The recombinant VjbR protein was expressed and purified as described previously (20). Binding reactions were performed by incubating VjbR with 5000 cpm of the different 32P-labeled probes in binding buffer (15 mM Tris–HCl [pH 8.0], 0.5 mM EDTA, 10 μg ml−1 bovine serum albumin [BSA], 1 mM dithiothreitol [DTT], 30 mM KCl, 80 mM NaCl, 6% glycerol, 50 μg ml−1 salmon sperm DNA). After incubation at room temperature for 30 min, the protein–DNA complexes were separated from the free probes on 4% (for the araC or BAB2_0653 probes) or 8% (for ftcR or BAB1_1550) nondenaturing polyacrylamide gels at a constant voltage of 220 V in a Protean II xi cell (BioRad), which contains a central cooling core to prevent overheating during electrophoresis. Subsequently, gels were vacuum dried, exposed on a phosphor screen, and scanned with a Storm 860 PhosphorImager (Molecular Dynamics). EMSA were carried out in duplicate.
Bioinformatic analysis
ChIP-seq: single-end 50-bp Illumina HiSeq 2000 sequencing of ChIP-seq samples provided 20–23 million reads per library. Removal of low-quality bases at the ends of reads (phred33<30) and clipping the first three bases in addition to Illumina adaptor sequences was carried out by Trimmomatic software v0.32 (23) using palindrome mode. A sliding window quality trimming was performed, cutting once the average quality of a window of four bases fell <30. Reads shorter than 32 bp after trimming were discarded. FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to assess quality of the reads before and after trimming. Subsequently, reads were aligned to the B. abortus 2308 genome (RefSeq accession numbers for chromosomes I and II: NC_007618.1 and NC_007624.1, respectively) using SAMtools and the Burrows-Wheeler Alignment software (BWA) (24,25), obtaining 84–86% uniquely mappable reads per library. Bigwig files were generated with custom bash scripts and BEDtools (26). Alignments were then visualized using the software Integrated Genome Viewer (IGV) (http://broadinstitute.org/igv). To determine the ChIP-seq enriched regions, read alignments were analyzed using the peak caller software MACS2 (P-value 1 × 10−5, false discovery rate 0.01) (27). BEDtools were used to merge and intersect peak intervals, obtaining 554 peaks present in at least one out of the three wild-type replicates, from which 468 (84.4%) were present in all three wild-type replicates, and 516 (93.1%) were present in at least two out of the three, these latter being used for further analyses. Peaks displaying low-affinity ChIP-seq were filtered by applying a cut-off value of 1.8, according to which the number of peaks in the control mutant strain was equivalent to 5% of those of the wild-type. Subsequently, custom python scripts were used to subtract low-confidence peaks, defined as those displaying ChIP-seq signals in the ΔvjbR mutant localized at a distance < 3 standard deviations from the wild-type average peak summit coordinate.
The VjbR-binding consensus motif was identified using the Multiple Em for Motif Elicitation (MEME) software (28) and 235 input sequences spanning 150 bp to each side of the bona fide VjbR ChIP-seq peak summits, using options -dna -mod zoops -maxw 16 -revcomp. Custom python scripts were used to discriminate intergenic and intragenic peaks, and to measure peak summit distances from the closest start codon of divergently transcribed genes. VjbR-binding motif occurrence on either ChIP-seq intervals or the complete B. abortus 2308 genome was determined using the Find Individual Motif Occurrences (FIMO) algorithm software (29) and the position weight matrix generated by MEME (28).
Analysis of the association between motif occurrence and effective binding of VjbR was performed as follows. From the total intergenic 5΄ spacer regions > 90 bp of the B. abortus 2308 genome, we used BEDtools and custom python scripts to generate a set of ChIP-seq-negative putative promoters. This was carried out by subtracting those intergenic 5΄ spacer regions overlapping any of the 516 VjbR ChIP-seq peaks present in at least two out of the three biological VjbR ChIP-seq replicates. 300 bp-long sequences were randomly extracted from the control set of ChIP-seq negative promoters following the same distribution of distances from the closest start codon as that of the 129 bona fide intergenic ChIP-seq peaks. Occurrence of the 16-bp VjbR-binding consensus motif was analyzed by FIMO in the 129 ChIP-seq negative promoters randomly extracted in 1000 iterations, plotted the results as a frequency histogram, and compared the results with the number obtained from the 129 bona fide intergenic ChIP-seq peaks.
RNA-seq: single-end 50-bp Illumina HiSeq 2000 sequencing of RNA-seq samples provided 17–21 million reads per library. Removal of low-quality reads and Illumina adapters, assessment of the quality of the reads, alignment of reads to the B. abortus 2308 genome, and visualization, was performed in the same way as for ChIP-seq, obtaining 80–82% uniquely mappable reads per library. Read counts were quantified using the software FeatureCounts (30) using the strand specific mode. Differential expression analysis was performed using the DESeq software (31). Hits displaying adjusted P-value <0.05 were informed as differentially expressed genes.
GO terms enrichment analysis
Using GO.db Bioconductor annotation data package in R language, all Gene Ontology (GO) terms and ancestors were retrieved and annotated for all genes of B. abortus 2308. For differentially expressed genes, an enrichment test was performed for the following categories: BP (biological process), MF (molecular function) and CC (cellular component). The enrichment factor (EF) was estimated as the ratio between the proportions of genes associated with a particular GO category present in the dataset under analysis, relative to the proportion of the number of genes in this category in the whole genome. p-values were calculated using the Fisher Exact Test and adjusted by the Benjamini–Hochberg method.
RESULTS
VjbR binds with high specificity to a large number of sites across the Brucella genome
Previous analyses of VjbR-mediated gene expression indicated that incubation of Brucella in rich bacteriological media can lead to markedly different outputs (18,19), probably due to the contribution of direct and indirect transcriptional effects highly sensitive to slight variations in the experimental conditions used (e.g. bacterial culture density or VjbR protein concentration in the bacterial cell). In this work, instead, we sought to determine the VjbR regulon under a restricted set of conditions mimicking the acidic, nutrient-poor environment that Brucella encounters within the host cell. Accordingly, we used as starting material mid-exponential bacteria grown in rich bacteriological medium up to an OD600 where the VjbR protein is not yet expressed. At this point, bacteria were shifted to a medium that meets the above-mentioned conditions and also triggers the expression of the VjbR protein (22). As determined by western blot, VjbR and one of the VirB proteins were induced in the B. abortus 2308 wild-type strain after 3 h of incubation, thus indicating de novo expression of both the regulator and its proven direct target, whereas no changes were observed in the isogenic deletion mutant B. abortus ΔvjbR (Figure 1). We then performed ChIP using a polyclonal antiserum against the native VjbR protein and analyzed the immunoprecipitated DNA fragments by genome-wide next-generation sequencing, obtaining 17–21 million uniquely-mapped reads per sample.
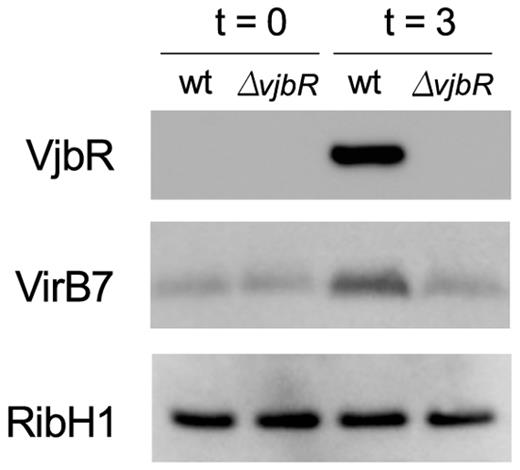
Induction of the native VjbR protein and one of its direct targets under conditions mimicking the intracellular environment.B. abortus 2308 or B. abortus ΔvjbR strains were grown in rich medium (TSB) up to OD600 ∼1.0. Subsequently, cultures were centrifuged and bacteria were resuspended in MM1 medium (22) pH 5.5 in the presence of 5 mM urocanic acid. At the indicated incubation times, samples were analyzed by western blot using anti-VjbR, anti-VirB7 polyclonal antibodies, or anti-RibH as a control for protein loading.
Alignment of reads to the B. abortus 2308 genome resulted in well-defined, highly reproducible ChIP-seq signals (Figure 2A), with 93% of peaks present in at least two out of the three biological replicates. Low-intensity ChIP-seq signals were removed by applying a fold-enrichment (FE) cut-off value of 1.8, with a resulting number of peaks in the B. abortus ΔvjbR mutant equivalent to 5% of those of the wild-type strain (Figure 2B). In this way, we obtained a list of 235 bona fide ChIP-seq peaks distributed across the Brucella genome with densities of 71.4 and 70.8 peaks/Mbp in chromosomes I and II, respectively. For each identified ChIP-seq peak summit, we determined the distance from the nearest gene based on RefSeq annotations for the B. abortus 2308 genome, obtaining a binding distribution that pointed to a preferential localization between 20 and 140 bp upstream the corresponding start codons (Figure 2C). Among these ChIP-seq signals, 129 (55%) displayed peak summits located at intergenic positions (Supplementary Table S1), indicating a high enrichment of VjbR-binding sites at promoter regions since 88% of the Brucella genome is composed of coding sequences. Finally, the observed ChIP-seq FE intensity values displayed a relatively high dynamic range, spanning from 1.8 to 6.78 (Figure 2D), which may be indicative of heterogeneity in VjbR-binding affinity for each site, differences in the number of VjbR molecules bound per promoter, or the presence of other DNA-binding proteins that hinder the access of the antibodies.
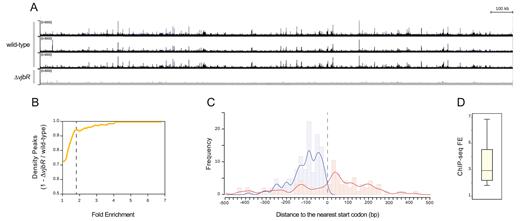
VjbR binds with high specificity to a large number of sites distributed across the B. abortus 2308 genome. (A) Representative image of the VjbR ChIP-seq read alignments to the B. abortus 2308 chromosome I visualized as ChIP-seq peaks using the Integrated Genome Viewer software. Peak height correlates with the number of reads from the wild-type strain B. abortus 2308 (black) or the isogenic deletion mutant B. abortus ΔvjbR control strain (gray) at the corresponding positions of the B. abortus 2308 genome. (B) Comparison of number of peaks between the wild-type and the B. abortus ΔvjbR control strain by a density plot of (1 – number of peaks ΔvjbR / wild-type) as a function of ChIP-seq FE. Dotted line indicates the FE cut-off value used to filter low-intensity peaks. (C) Histogram and fitting line of distance frequencies from peak summit position to the nearest start codon for the 235 bona fide ChIP-seq peaks localized at intergenic (blue) or coding regions (red). Dotted line indicates the relative position of the start codon. (D) Boxplot showing distribution of the observed ChIP-seq FE values.
VjbR presents an asymmetric 16-bp binding consensus motif
To determine the VjbR-binding motif, we analyzed sequences extending 150 bp to each side of the 235 peak summits using the MEME algorithm (28). Considering that LuxR homologs usually recognize 18–20 bp sequences with either perfect or imperfect dyad symmetry (32–34), we first used MEME parameters to search for palindromic motifs in the VjbR ChIP-seq intervals. However, using this restriction no motif was identified. Instead, relaxing symmetry constraints allowed us to determine an asymmetric 16-bp consensus sequence characterized by a 6-bp GC-rich region flanked by 8 bp- and 2 bp-long sequences with predominance of A and T (Figure 3A). As shown in Figure 3B, the MEME algorithm located the 16-bp motif in the virB promoter at the precise position of the VjbR-binding site previously identified by DNAse I Footprinting (20), thus indicating that determination of the VjbR-binding consensus motif was carried out successfully and validating the adopted strategy. Interestingly, unlike what we expected based on previous observations, the novel motif exhibited low similarity to the binding site of the closely related LuxR-type regulator MrtR, but displayed a better match to the AT-rich region that flanks the 5΄ end of the MrtR-binding-like motif of the virB promoter (Figure 3B).
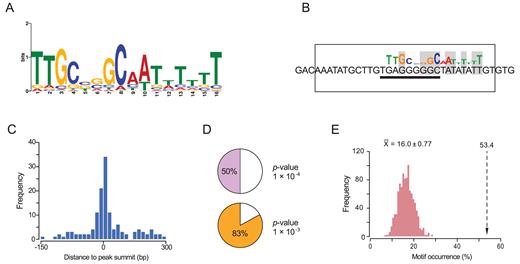
Determination of the VjbR-binding consensus motif. (A) VjbR DNA-binding motif generated by the MEME algorithm. (B) VjbR-binding motif determined by the MEME algorithm on sequences corresponding to the virB promoter (P-value 1.21 × 10−4) in the genomic coordinates ChrII:67,199–67,214 based on RefSeq annotations for B. abortus 2308. The open rectangle indicates the VjbR-binding site previously determined by DNAse I Footprinting in the virB promoter (20). Solid line indicates the position of a 9-bp motif identical to half-binding site of the LuxR-type regulator MrtR from Mesorhizobium (21). Nucleotides that match the VjbR-binding consensus motif are highlighted in gray. (C) Distance frequencies from the position of the VjbR-binding consensus motifs, identified by the MEME algorithm, to the corresponding ChIP-seq peak summits. (D) Occurrence of the VjbR-binding motif in the 235 ChIP-seq peaks determined by the FIMO algorithm with the indicated cut-off P-values. (E) Occurrence of the VjbR-binding motif determined by the FIMO (P-value 1 × 10−4) in the 129 intergenic VjbR ChIP-seq peaks (dotted lined arrow) or in 129 ChIP-seq-negative promoters randomly sampled in 1000 iterations (pink bars).
As shown in Figure 3C, the analysis performed with MEME identified the VjbR-binding consensus motif preferentially distributed in the close proximity of the peak summit positions, indicating high positional resolution of this ChIP-seq analysis. On the other hand, a scan of the novel motif using FIMO detected the presence of the consensus sequence in 50 or 83% of the identified ChIP-seq peak intervals, using cut-off p-values of 1 × 10−4 or 1 × 10−3, respectively (Figure 3D). As can be observed in Figure 3A, the VjbR-binding consensus sequence does not contain fully conserved nucleotide positions and presents four different low scoring sites, hence constituting a highly degenerate consensus motif. Consistent with this assessment, FIMO analysis detected occurrence of the motif in multiple positions throughout the B. abortus 2308 genome (cut-off P-value 1 × 10−4), even outside the VjbR ChIP-seq enriched regions. This prompted us to further assess the significance of the association between occurrence of VjbR-binding consensus sequences and effective binding of VjbR to DNA. To this end, we quantified the occurrence of VjbR-binding motifs either in the 129 intergenic ChIP-seq peaks, or in 1000 iterations of 129 randomly sampled ChIP-seq-negative promoters. As shown in Figure 3E, the motif occurrence in intergenic ChIP-seq peaks (53.4%) was significantly higher than in control VjbR ChIP-seq-negative promoters (average 16.0 ± 0.77%, empirical P-value < 0.001), thus indicating that sequence-specific recognition of the VjbR-binding consensus sequence plays an important role in the production of VjbR–DNA complexes in vivo.
In order to validate direct binding of VjbR to the sequences identified by ChIP-seq, we performed ChIP-qPCR on independent immunoprecipitation experiments to analyze sequences extending ±50 bp from the intergenic VjbR-binding motifs determined by MEME in selected ChIP-seq peaks. Using this procedure, occupancy of VjbR was confirmed in all analyzed sequences (Figure 4A), showing a direct correlation between ChIP-qPCR FE levels and the number of normalized sequencing reads mapping to peak summit positions (Figure 4B). To test the ability of VjbR to interact in vitro with the genomic regions identified by ChIP-seq, we performed electrophoretic mobility shift assays (EMSA) using a recombinant VjbR protein and DNA probes corresponding to different promoter sequences containing the novel VjbR-binding motif. We have previously observed that the VjbR–virB DNA complex has low stability under electrophoretic conditions, and that binding of VjbR to this region cannot be assessed by EMSA unless the affinity of the complex is enhanced by duplication of the native VjbR-binding site (20). Based on this previous evidence, in the present work we assessed the ability of VjbR-DNA complexes to withstand electrophoresis in promoters containing either one (BAB2_0653) or two VjbR-binding motifs (the araC homolog BAB1_0677, the rpoH1 homolog BAB1_1775, BAB1_1550, BAB1_2037 and the gene BAB2_1099 named ftcR). As shown in Figure 5A, VjbR specifically interacted in the nanomolar range with probes corresponding to the upstream regions of ftcR and BAB1_1550. VjbR was also able to interact with the upstream sequences of araC and BAB2_0653 but not with the control probe, although micromolar protein concentrations were required to observe these interactions (Figure 5B). Moreover, these latter protein–DNA complexes displayed a smeared pattern (Figure 5B), suggesting that binding of VjbR to both targets were less stable than those analyzed in Figure 5A. Furthermore, higher pore size polyacrylamide gels (4%) were required to prevent the retention of VjbR-araC or VjbR-BAB2_0653 promoter complexes in the wells during EMSA, suggesting that this regulator is able to form high-molecular weight multimers upon binding to DNA at high VjbR protein concentrations. On the other hand, EMSA analyses of the rpoH1 and BAB1_2037 upstream sequences showed no retarded bands at any of the assayed protein concentrations (data not shown). Taken together, these results demonstrate that VjbR is able to interact in vitro with four novel binding sites identified by ChIP-seq, although displaying different stability patterns. Moreover, the lack of signal in two of the selected targets assayed suggests that, similarly to that observed in the virB promoter, binding of VjbR to sequences upstream of rpoH1 and BAB1_2037 may not withstand the electrophoretic conditions of EMSA.
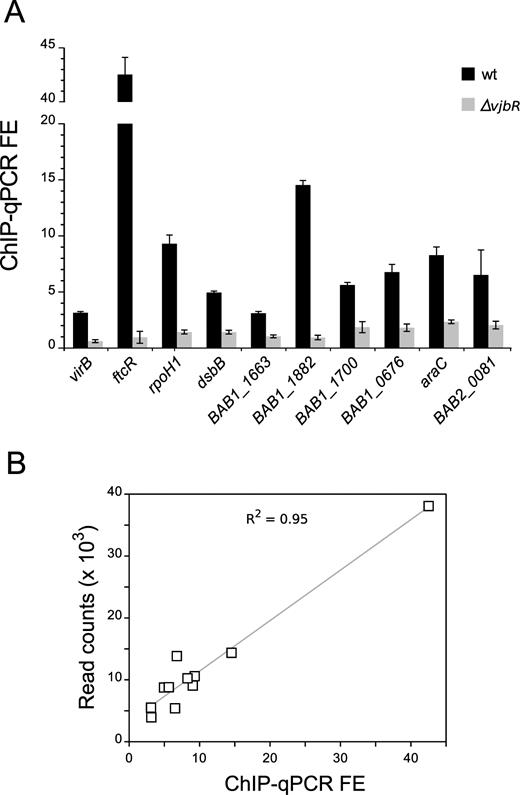
Validation of VjbR occupancy at selected peaks. (A) Independent ChIP assays performed on strains B. abortus 2308 or B. abortus ΔvjbR were analyzed using real-time qPCR amplifying fragments extending ∼50 bp to each side of the VjbR-binding motifs identified upstream of the indicated genes. Fold-enrichment (FE) represents the immunoprecipitated DNA:input DNA ratio for B. abortus 2308 (black bars) or B. abortus ΔvjbR (gray bars). (B) Correlation between ChIP-qPCR fold-enrichment data shown in (A) and ChIP-seq peak height (read counts) at the position of the VjbR-binding sequences in the wild-type strain B. abortus 2308.
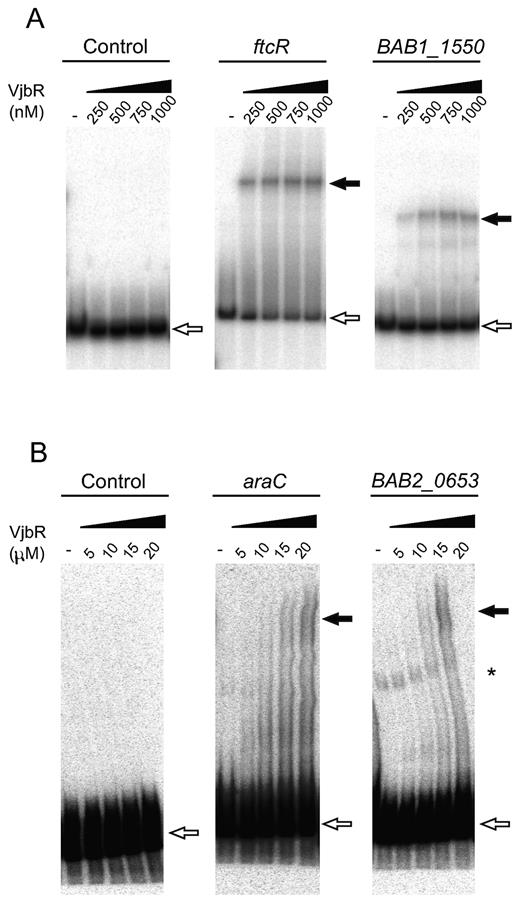
VjbR interacts in vitro with different targets identified by ChIP-seq. EMSA performed with VjbR recombinant protein at the indicated nanomolar (A) or micromolar (B) concentrations and radiolabeled DNA probes corresponding to the upstream sequences of the indicated genes, or with a control probe lacking VjbR-binding motifs. White arrows indicate the position of the free probes. Black arrows indicate the position of the different VjbR–DNA complexes. Asterisk indicates the position of an extra band probably derived from unspecific PCR products formed during labeling of probe BAB2_0653.
Examination of representative ChIP-seq peaks from the validated VjbR-binding sites showed that, in the virB promoter, both ChIP-seq peak height and ChIP-qPCR FE displayed values consistent with the low in vitro binding affinity of VjbR to this region (Figure 6A) (20). In agreement with previous works that reported direct binding of VjbR to sequences located between virB1 and virB2 (8–18), we also observed a ChIP-seq signal within this intergenic region. Yet, FIMO reported no occurrence of VjbR-binding consensus motifs within the virB1–virB2 intergenic ChIP-seq peak interval (Figure 6A), suggesting that recruitment of VjbR to this genomic region is not due to specific recognition of a conserved VjbR-binding consensus sequence but to a concerted action involving additional elements. Among all identified ChIP-seq peaks, the signal located upstream ftcR exhibited the highest ChIP-seq enrichment values, displaying read counts 32-fold higher than the background at the peak summit position and two VjbR-binding consensus motifs centered 47- and 90-bp upstream of the start codon (Figure 6B). Indeed, VjbR has been shown to induce the promoter of ftcR, which encodes a two-component transcriptional regulator that activates the expression of flagellar genes involved in persistence of Brucella infection in mice (35). Thus, in line with the previous study, our observation of a high-affinity VjbR ChIP-seq signal upstream of ftcR constitutes the first evidence of a direct interaction between this LuxR-type transcription factor and regulatory elements of flagellar gene expression. Other examples of VjbR high-affinity binding to intergenic regions are depicted in Figure 6C–E, which show ChIP-seq signals identified upstream of genes coding for two helix-turn-helix-containing proteins belonging to the AraC- and MerR-families (BAB1_0677 and BAB1_1201, respectively) and RpoH1 (BAB1_1775).
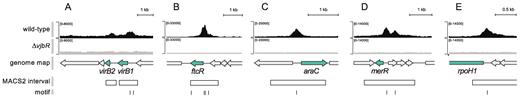
VjbR ChIP-seq peaks of selected genes. Representative images of ChIP-seq read alignments of the wild-type strain B. abortus 2308 (black) or the isogenic deletion mutant B. abortus ΔvjbR control strain (gray) at the corresponding positions of the genome of B. abortus 2308. Schematic representation of genomic regions containing the selected (green) and flanking genes (gray) of (A) virB1 and virB2; (B) ftcR; (C) the araC homolog BAB1_0677; (D) the merR homolog BAB1_1201; and (E) rpoH1. Open rectangles indicate position of the ChIP-seq intervals identified by the MACS2 peak caller software (27). Bars indicate position of the VjbR-binding motif identified by the FIMO algorithm within the ChIP-seq intervals using cut-off P-value 1 × 10−3 (A) or 1 × 10−4 (B–E).
Genome-wide effect of VjbR on Brucella gene expression
In order to assess the transcriptional effects of VjbR binding, we performed a genome-wide expression profiling of two biological replicates of the B. abortus 2308 wild-type strain or the isogenic ΔvjbR mutant under the same experimental conditions as those applied for ChIP-seq. As a result, our RNA-seq analysis yielded 14–17 million uniquely-mapped reads per sample with high reproducibility among biological replicates, which allowed us to identify 71 genes with statistically significant differential expression changes between the wild-type and the ΔvjbR mutant strain (adjusted P-value < 0.05). As assessed by integration of RNA-seq with ChIP-seq data, expression of 37 genes organized either as mono- or polycistronic transcripts was modulated by VjbR through binding to their promoter regions (Supplementary Table S2). 32 of these target genes exhibited intergenic VjbR ChIP-seq peaks at their promoter regions, whereas the remaining five displayed VjbR-mediated repression from VjbR-binding sites located at intragenic upstream sequences. In addition to these direct targets, 10 additional genes showed VjbR-mediated transcriptional effects together with a VjbR ChIP-seq peak in their promoter regions, but were nevertheless categorized as ‘low confidence’ direct VjbR-target genes owing to the presence of ChIP-seq signals in the mutant control strain or to low VjbR ChIP-seq FE values. Finally, we identified 24 differentially expressed genes displaying no association with VjbR ChIP-seq peaks, indicative of indirect effects of deletion of vjbR (Supplementary Table S2).
Among all VjbR direct targets, genes exhibiting the highest expression changes displayed VjbR-mediated transcriptional activation, including the virB operon (BAB2_0057-68), a gene encoding a ‘universal stress’-family protein containing an Usp domain (BAB2_1016), and an operon displaying homology to Class I NADH-ubiquinone oxidoreductase complex subunits (BAB2_0735 to BAB2_0738) (Figure 7A–C and Supplementary Figure S1). Additionally, we observed high levels of VjbR-mediated differential expression of a ‘low confidence’ bicistronic operon comprising btaE, a trimeric autotransporter adhesin involved in the attachment of Brucella to host cell structures (36). As shown in Figure 7D, the promoter region of btaE exhibited a VjbR ChIP-seq peak in both the wild-type and the mutant control strain. However, both ChIP-seq signals are separated by 87 ± 56 bp, and, therefore, the possibility that VjbR directly regulates expression of this bicistronic transcriptional unit cannot be ruled out.
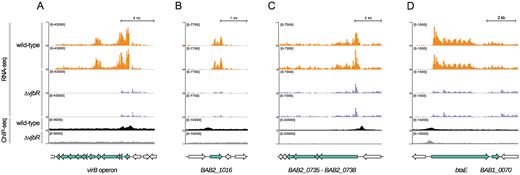
Integration of RNA-seq and VjbR ChIP-seq data of selected genes. Representative images of the alignment of RNA-seq and VjbR ChIP-seq reads of two biological replicates of the wild-type (orange and black, respectively) or the vjbR mutant control strain (blue and gray, respectively), and schematic representation of the genomic regions containing the selected (green) and flanking genes (gray) of (A) the virB operon, (B) BAB2_1016, (C) the operon containing genes BAB2_0738 to BAB2_0735, and (D) the bicistronic unit containing btaE and BAB1_0070.
To assess the possible functional significance associated to VjbR activity, we performed a gene ontology (GO) analysis of the differential expression data focusing on both the genes directly regulated by VjbR and also in the overall list of genes displaying either direct or indirect VjbR-mediated expression effects. As shown in Figure 8, the GO category ‘Biological Processes’ (BP) exhibited, as expected, a high enrichment in the GO term ‘pathogenesis’ owing to the well-known role of the 12 virB genes in Brucella virulence. Consistently with previous works that proposed a major role for VjbR in the control of membrane structures (15), ‘Cellular Compartment’ (CC) GO annotations showed a statistically significant 1.71-fold enrichment of membrane-associated proteins and an underrepresentation of cytoplasmic components of 0.12-fold (Figure 8). In the VjbR direct targets, it was observed a depletion of elements related to the term ‘catalytic activity’ in the ‘Molecular Function’ (MF) GO category (Figure 8), suggesting that under the assayed conditions the VjbR function is mainly involved in processes other than enzymatic reactions. We also observed a significant enrichment of the GO term ‘oxidative phosphorylation’ due to VjbR-mediated regulation of respiratory chain components such as cbb3-type terminal cytochrome c oxidase and a Class I NADH–ubiquinone oxidoreductase complex (Supplementary Table S2). Accordingly, ‘oxidoreductase activity’ acting on either NAD(P)H or heme groups with oxygen as acceptor are also significantly enriched in MF GO annotations due to the direct role of VjbR in regulation of the above-mentioned respiratory chain complexes. In addition, a third type of oxidoreductase activity showed significant enrichment as a result of differential expression of ribonucleotide reductases mapping to different locations in the B. abortus 2308 genome (BAB1_1063 and BAB2_0888-9), although in a VjbR-independent manner, probably owing to downstream effects of two direct targets of VjbR-mediated regulation (i.e. the putative regulatory proteins encoded by BAB1_1587 and BAB2_0310).
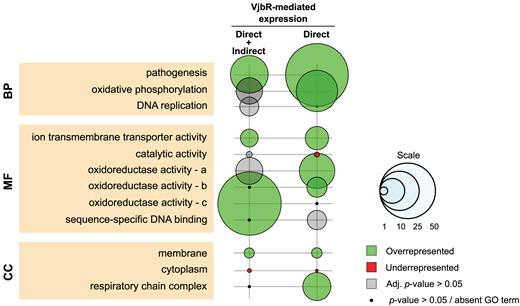
Gene Ontology (GO) enrichment analysis of differentially expressed genes. GO enrichment was evaluated at three different levels: Biological Processes (BP), Molecular Function (MF) and Cellular Component (CC) for either the total list of differentially expressed genes (n = 71) or those displaying bona fide direct VjbR-mediated regulation (n = 37). Bubble size correlates with enrichment factor values. Colored and gray bubbles represent Benjamini–Hochberg adjusted P-value <0.05 or >0.05, respectively. Black dots indicate non-corrected P-value >0.05 or absent GO term. a, oxidorreductase activity acting on heme groups; b, NAD(P)H; c, CH or CH2.
DISCUSSION
Since its first description as a central element for Brucella virulence, significant advances have been made in the study of VjbR and the mechanisms that modulate the regulatory activity of this LuxR-type transcription factor (18,19,37). Yet, determination of a VjbR-binding site motif and the precise identification of direct targets have so far remained elusive, which hampered progress in our understanding of the molecular bases of VjbR function and its role in Brucella global gene expression. To address these issues, here we carried out a genome-wide assessment of VjbR-binding under acidic, nutrient-limited conditions.
Our results revealed that in B. abortus VjbR indeed binds to a large number of genomic positions and displays a network of interactions with properties consistent with those used to define global regulators (38,39). Analysis of the identified DNA-binding consensus sequence showed that, unlike most well-studied LuxR-family transcription factors, the VjbR-binding motif displays a notable asymmetry, strongly marked by one half of the consensus sequence containing only AT base pairs, with a tract of six T nucleotides (Figure 3A). So far, crystallographic analyses of LuxR-type regulators showed symmetrical protein-DNA interactions according to the symmetry of the palindromic or pseudo-palindromic DNA-binding sequences in all well-studied examples (32,40–42). In view of the atypical features of the VjbR-binding motif, it will therefore be interesting to determine which amino acid residues are critical for establishing contacts with DNA, how the asymmetric VjbR-binding site structure impacts on the affinity and stability of the protein-DNA complexes, and why such an asymmetry had been selected over symmetry during the evolution of Brucella.
Determination of the VjbR-binding motif with the MEME algorithm showed high positional resolution relative to the observed ChIP-seq peaks (Figure 3C), and also achieved high precision in localizing the consensus sequence within a VjbR-binding site previously determined by DNAse I Footprinting (Figure 3B). Moreover, VjbR occupancy was confirmed in the virB promoter and in all newly identified VjbR-binding sequences analyzed by ChIP-qPCR (Figure 4), and by EMSA in several promoter regions (Figure 5). However, our study also revealed dissimilar VjbR binding patterns in different targets and, moreover, similarly to what previously observed in the virB promoter (20), binding of VjbR could not be assessed by EMSA in two of the analyzed promoter sequences. This indicates that despite the ability of VjbR to bind in vivo to all these sequences as assessed by ChIP-qPCR and/or ChIP-seq, some of these VjbR-DNA complexes resist electrophoresis in vitro (Figure 5A), whereas other targets such as rpoH1, BAB1_2037 and virB, do not. In turn, araC and BAB2_0653 showed an intermediate situation with a smeared EMSA pattern suggesting reduced resistance to the EMSA conditions (Figure 5B). It is worth noting that all of the analyzed targets contain VjbR-binding sequences with similar levels of conservation, and the number of binding motifs present in either of these promoters (one or two) did not seem to be a determinant of whether or not VjbR binding withstands the EMSA conditions in the analyzed probes. Accordingly, we hypothesize that DNA properties of the flanking sequences (e.g. intrinsic curvature and bendability) may play a role in the stability of the VjbR–DNA complexes in vitro during electrophoresis.
In addition to the data supporting the notion that VjbR interacts with its target promoters through sequence-specific DNA recognition, here we also identified ChIP-seq peaks at genomic regions that had no match with the VjbR-binding consensus motif. On the other hand, even though most of the highly conserved VjbR-binding sequences are found within VjbR ChIP-seq peak intervals, we still observed that 30% of the motifs displaying high conservation (P-values < 1 × 10−7) are located at genomic positions showing no VjbR ChIP-seq signals. Therefore, our results suggest that in addition to sequence-specific recognition, there must exist additional elements operating in the process of interaction of VjbR with its actual binding sites in vivo, either by assisting VjbR with the binding to regions where no binding motifs are observed, or by hampering the interaction with highly conserved VjbR-binding sequences at specific positions in the genome. In view of these observations, further research will be required to reach a comprehensive understanding of the mechanisms that dictate VjbR-binding site recognition and define its network of regulatory interactions.
Our results showed that under the experimental conditions tested here, VjbR modulated expression of 71 genes, 66% of which displayed direct VjbR-mediated regulation, as assessed by integration between ChIP-seq and RNA-seq data. GO term enrichment analysis of these targets indicated that the main biological processes directly modulated by VjbR not only encompass the well-studied virulence genes known to control intracellular trafficking and escape lysosome-mediated degradation, but also involve electron transport chain components. Analysis of RNA-seq data showed that VjbR positively regulates NADH-ubiquinone oxidoreductase genes while reducing the expression of the terminal cbb3-type oxidase (Supplementary Table S2), suggesting that VjbR function increases respiratory activity by induction of complex I though diminishing the electron flow towards downstream components of the branched respiratory chain of Brucella (Figure 9). Consistent with our observations, two previous transcriptomic and/or proteomic analyses of Brucella melitensis grown in rich medium also revealed VjbR-mediated upregulation of complex I and downregulation of cbb3- and aa3-type cytochrome c terminal oxidases, respectively, and to similar extents (18,19). In this way, different lines of evidence support the notion that VjbR redirects electron transfer towards ubiquinol oxidases, which may result in translocation of one proton per NADH oxidized instead of three (Figure 9). Such a VjbR-mediated redirection of the respiratory chain electron flow at the expense of lower coupling of ATP synthesis could be part of a mechanism for adaptation of the Brucella metabolism to the low oxygen concentration encountered within the host cell, since bd-type ubiquinol oxidases are proposed to have the highest oxygen-affinity among all cytochromes present in the Brucella genome. In agreement with this, previous studies showed that the bd-type ubiquinol oxidase is expressed within host macrophages and contributes to intracellular bacterial replication of Brucella suis (43). Noteworthy, in line with the two main biological processes significantly enriched by GO term analysis of RNA-seq data from our study, virB genes and respiratory chain components constitute the 77% and the 100% of the VjbR-dependent differentially expressed genes in common with the previous transcriptomic analyses by Weeks et al. (19) and Uzureau et al. (18), respectively (Supplementary Table S3). Thus, taken together these observations highlight the conserved role of VjbR on modulation of T4SS expression and respiratory chain components, the latter of which may be part of an adaptive response of Brucella to adjust the electron flow and energy metabolism under the harsh conditions encountered within the intracellular environment, probably by optimizing oxygen affinity or efficiency of proton-translocation.
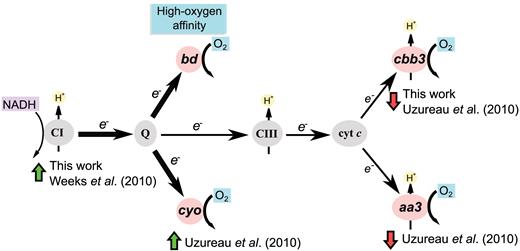
Organization of the branched respiratory chain of Brucella. CI, complex I NADH:ubiquinone oxidoreductase; Q, quinone pool; CIII, complex III; Cyt. c, Cytochrome c. Pink ovals represent the four terminal oxidases of Brucella. Black arrow thickness indicates electron flow intensity. Green and red arrows indicate positive or negative VjbR-dependent differential expression, respectively.
Integration of genome-wide binding of VjbR and RNA-seq data obtained under acidic and nutrient-poor conditions indicated a number of direct VjbR-mediated regulatory events substantially lower than the 129 intergenic ChIP-seq peaks identified across the Brucella genome. Among this repertoire of VjbR-binding sites, if we assume that VjbR-mediated regulation occurs in only one direction at divergent promoters and consider genes putatively co-expressed as operons, VjbR may potentially directly regulate a number of genes ranging from 183 to 199 of those listed in Supplementary Table S1. Thus, even hypothesizing the existence of a high amount of non-functional VjbR-binding sites, our observations allow us to propose that many of the observed non-productive VjbR target DNA-sequences may be able to produce transcriptional effects under conditions different from those studied here. This notion is supported by the observation that genes BAB1_0660 and BAB1_1355 encoding the outer membrane protein Omp2b and an acid-shock calcium binding protein, respectively, displayed both direct VjbR promoter binding and VjbR-dependent expression in one of the aforementioned microarray-based transcriptomic analyses of bacteria grown in rich medium (18), whereas in this work showed ChIP-seq peaks but no VjbR-mediated transcriptional effects. Thus, even though VjbR is able to bind to a given promoter under two different environmental conditions, only one of them provides the context necessary to achieve VjbR-mediated modulation of gene expression, probably through induction of co-activators required by VjbR to perform its regulatory function. Indeed, we have recently found that the virB genes and the trimeric autotransporter adhesin btaE, both displaying VjbR binding and VjbR-mediated activation, share three transcriptional regulators interacting with their respective promoter regions in addition to VjbR (44), one of them being HutC, an urocanic acid-inducible GntR regulator that participates in intracellular expression of the virB operon. Hence, based on the results from this and previous works, we propose that acidification and low-nutrient experimental conditions provide the stimuli necessary to trigger the expression of the VjbR protein, allowing the regulator to bind to all its target promoters but inducing the expression of only a subset of genes, whereas the rest of the direct targets of VjbR seem to require additional regulatory elements that are not present or active under the assayed conditions (Supplementary Figure S2).
The data presented in this work expanded our knowledge of the role that VjbR plays in the global control of Brucella gene expression and revealed the extent of the interaction network of an AHL-dependent member of the LuxR family at the genomic scale. On the basis of our observations, we propose a model where, under nutrient-deprived and acidic conditions, this regulator directly modulates expression of a subset of 47 genes including virulence determinants, components of the respiratory chain, efflux systems, elements predicted to participate in ion transport and adaptation to stress, hypothetical proteins, as well as transcriptional regulators probably responsible for the observed indirect VjbR-mediated effects (Supplementary Table S2). This number of productive interactions, which constitute only a fraction of the intergenic VjbR-binding events observed in this work (Supplementary Figure S2), results in modulation of bacterial functions consistent with the requirements this pathogen during the first stages of intracellular infection to rapidly escape lysosome-mediated degradation and adapt energy metabolism to the variables found within the intraphagosomal environment. The rest of the genes and operons adjacent to VjbR ChIP-seq peaks, instead, could experience direct VjbR-mediated regulation upon different environmental conditions. Consistent with our model, 24 of the VjbR-binding events that were non-productive under the conditions tested here are associated to genes which showed VjbR-mediated modulation in other studies (Supplementary Table S3), suggesting that the latter elements are direct targets of VjbR that may exert roles necessary for adaptation of Brucella at subsequent stages of the intracellular trafficking. Taken together, our observations point to a dissociation between VjbR-binding and control of transcription at specific promoters and highlight the ability of VjbR to generate different outputs depending on the environmental context.
ACCESSION NUMBER
All sequencing data have been deposited to the Gene Expression Omnibus (GEO) under accession number GSE95722.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Nicolas De Jay, Steven Hébert, Alain Bateman, Jordan Prince Tremblay, Maud Marques and Karine Choquet for helpful suggestions for the bioinformatics analyses, and Julieta Mateos for help with setting the conditions for immunoprecipitation.
FUNDING
National Scientific and Technical Research Council of Argentina (CONICET) [PIP 0336 to R.S.]; National Agency for the Promotion of Science and Technology of Argentina (ANPCyT) [PICT 0365 to R.S.]; Canada Foundation for Innovation; Natural Sciences and Engineering Research Council of Canada (NSERC) (to C.L.K.); Fonds de Recherche du Quebec - Santé (FRQS) (to C.L.K.). Funding for open access charge: ANPCyT [PICT 2012 0365 to R.S.].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments