





Abstract
Mitochondrial ribosomal RNAs (rRNAs) often display reduced size and deviant secondary structure, and sometimes are fragmented, as are their corresponding genes. Here we report a mitochondrial large subunit rRNA (mt-LSU rRNA) with unprecedented features. In the protist Diplonema, the rnl gene is split into two pieces (modules 1 and 2, 534- and 352-nt long) that are encoded by distinct mitochondrial chromosomes, yet the rRNA is continuous. To reconstruct the post-transcriptional maturation pathway of this rRNA, we have catalogued transcript intermediates by deep RNA sequencing and RT-PCR. Gene modules are transcribed separately. Subsequently, transcripts are end-processed, the module-1 transcript is polyuridylated and the module-2 transcript is polyadenylated. The two modules are joined via trans-splicing that retains at the junction ∼26 uridines, resulting in an extent of insertion RNA editing not observed before in any system. The A-tail of trans-spliced molecules is shorter than that of mono-module 2, and completely absent from mitoribosome-associated mt-LSU rRNA. We also characterize putative antisense transcripts. Antisense-mono-modules corroborate bi-directional transcription of chromosomes. Antisense-mt-LSU rRNA, if functional, has the potential of guiding concomitantly trans-splicing and editing of this rRNA. Together, these findings open a window on the investigation of complex regulatory networks that orchestrate multiple and biochemically diverse post-transcriptional events.
INTRODUCTION
Mitochondria are semi-autonomous organelles of the eukaryotic cell that contain not only a distinct genome—typically a multicopy, single type of circular-mapping chromosome—but also their own translation machinery. Although protein components of the mitoribosome are partly or completely encoded by the nuclear genome, synthesized in the cytosol and imported into mitochondria, the genes specifying the large subunit (LSU) and small subunit (SSU) ribosomal RNAs always reside on mitochondrial DNA (mtDNA) (1). Mitochondrial rRNAs (mt-rRNAs) are sometimes fragmented, extreme cases being dinoflagellates and apicomplexans (2–4). In Plasmodium the ∼20 gene pieces are spread across the genome on both DNA strands, are separately transcribed and then assembled into the ribosome, without covalently joining of the rRNA pieces (2). Further peculiarities observed in certain mt-rRNAs are homo-nucleotide appendages at their 3′ end, e.g. oligo(A) tails in Plasmodium (5) and short poly(U) tails in kinetoplastids (6).
Identifying mt-rRNA genes and accurate termini mapping in mitochondrial genome sequences can be challenging, particularly in taxa that are not closely related to model organisms and whose mtDNA has diverged far away from its bacterial ancestor. This applies in extremis to the unicellular protozoan (protist) group diplonemids, the sistergroup of kinetoplastids. Mitochondrial genes of Diplonema papillatum and its relatives are not only highly divergent but also systematically fragmented in a unique way. Genes consist of up to 11 pieces (modules) that are ∼80–530-nt-long, and each is encoded on a distinct circular chromosome of 6 kb (class A) or 7 kb (class B). Modules are transcribed separately and subsequently joined into continuous RNAs. With each chromosome containing only 1–6% coding sequence, the estimated genome size of Diplonema mtDNA is unusually large [∼600 kb; (7)].
In contrast to the eccentric genome structure, the gene complement of Diplonema mtDNA is rather conventional. Mitochondrial genes encode components of the respiratory chain, oxidative phosphorylation and mitoribosome, notably NADH dehydrogenase subunits 1, 4, 5, 7 and 8; apocytochrome b, cytochrome oxidase subunits 1–3, ATP synthase subunit 6 and LSU rRNA. The gene for mitochondrial SSU rRNA has not yet been identified (8). For rnl (encoding LSU rRNA), we only found a 352-nt long 3′-terminal portion that is otherwise well conserved. Incidentally, this RNA piece is the most highly expressed transcript in poly(A) libraries. However, the complete sequence and overall organization of rnl has remained unrecognized for many years, partly due to technical challenges in culturing sufficient cell material and isolating mitochondria from Diplonema, but also, as we know now, because of the intricate structure and biosynthesis of mt-LSU rRNA. We succeeded to resolve the puzzle by high-throughput RNA sequencing (RNA-Seq) and show here that maturation of Diplonema mt-LSU rRNA proceeds by multiple steps including extensive RNA editing. We also identify antisense RNA molecules that have the potential for guiding both trans-splicing and RNA editing of mt-LSU rRNA, but their function has yet to be demonstrated.
MATERIALS AND METHODS
Sequences deposited in public-domain databases
We have deposited in GenBank the genomic sequence of rnl-module 1 plus adjacent chromosome regions (accession no. KF633465) and the cDNA sequences of cytosolic 5.8S, 18S and 28S rRNA of D. papillatum (accession nos. KF633466-KF633468). The sequence of rnl-module 2 was deposited previously under the accession number JQ302963. A partial sequence of D. papillatum cytosolic 18S rRNA had been deposited before by others (GenBank accession no. AF119811).
Strain, culture and extraction of mtRNA
D. papillatum (ATCC 50162) was obtained from the American Type Culture Collection. The organism was cultivated axenically at 16–20°C in artificial seawater enriched with 1% fetal horse serum (Wisent) and 0.1% bacto tryptone. For extended large-scale cultivations, chloramphenicol (40 mg/L) was added to prevent bacterial contamination. To isolate mitochondria, cells were collected by centrifugation at 3000g for 10 min, washed once with ice-cold ST buffer [0.65 M sorbitol, 20 mM Tris (pH 7.5), 5 mM EDTA] and disrupted by nitrogen decompression at 600 psi (Parr Instrument Company) in the same buffer. Mitochondrial RNA and DNA were extracted from an organelle-enriched fraction isolated by differential and sucrose gradient centrifugation essentially as devised earlier (9). More specifically, intact cells and nuclei were removed by centrifugation at 3000g. The mitochondria-enriched fraction was obtained after centrifugation at 30 000g (20 min) followed by two consecutive separations on a discontinuous sucrose gradient [15, 25, 35, 45 and 60% sucrose supplemented with 20 mM Tris (pH 7.5) and 5 mM EDTA] at 130 000g (1 h). Mitochondria accumulated at the interface between the sucrose layers of 35 and 45% (and/or 25 and 35%). Mitoribosomes were enriched via separating a cell lysate by two consecutive kinetic centrifugations, the first on a step gradient (10–35% glycerol, in steps of 5%) at 250 000×g for 2 h and the second on a continuous gradient (10–40% glycerol) at 250 000g for 4 h. Fractions enriched in mt-LSU rRNA (as determined by agarose gel electrophoresis) were pooled. RNA was extracted by a home-made Trizol substitute (9). Residual DNA was removed from RNA preparations by either RNeasy (Qiagen) column purification or digestion with RNase-free DNase I (Fermentas), or TURBO DNase (Invitrogen) followed by phenol-chloroform extraction. Poly(A) RNA was enriched by a passage through oligo(dT)-cellulose (Amersham), after denaturation of the aqueous solution at 72°C for 2 min and subsequent chilling on ice.
Northern hybridization
DNase-treated RNA was separated electrophoretically in a MOPS/formaldehyde denaturing gel (1.2% agarose, 3% formaldehyde), side by side with the Riboruler High and Low Range RNA ladders (0.2 – 6.0 kb and 0.1 – 1.0 kb, Fermentas). As a size marker for smaller molecules, we used single-stranded DNA, which was obtained from denatured RT-PCR products of 130–440-nt-long rnl segments. This marker was visualized by hybridization to a radioactively labeled oligonucleotide (see later in text). Primers used for RT-PCR (and product sizes) are dp210 + dp211 (130 nt), dp72 + dp211 (240 nt), dp168 + dp169 (355 nt), dp210 + dp208 (440 nt) and dp72 + dp208 (560 nt). As size markers and positive controls for mono-modules, we used RNAs synthesized by in vitro transcription of PCR products amplified with primer pairs dp230 + dp216 (module 1) and dp232 + dp168 (module 2). Oligonucleotides used as primers and hybridization probes are listed in Supplementary Table S1. The electrophoretically separated nucleic acids were blotted on a nylon membrane (Zeta-Probe, BioRad) and fixed by baking the membrane at 80°C for 60 min. As hybridization probes, we used oligo-deoxynucleotides radio-labeled by T4 polynucleotide kinase in the presence of [γ-32P]ATP. For the detection of antisense transcripts, we used an oligoribonucleotide probe that was in vitro transcribed from PCR amplicons that in turn were produced with primer pairs dp225 + dp210 (antisense targeting) and dp226 + dp211 (sense-targeting control); for each primer pairs, one contained the T7 promoter in addition to gene-specific sequence. In vitro transcription with T7 RNA polymerase [New England BioLabs (NEB)] was performed in the presence of [α-32P]UTP, for internal labeling. Membranes were hybridized overnight at 55°C in either 5× saline sodium citrate (SSC) supplemented with 5× Denhardt’s solution (0.1% polyvinylpyrrolidone, 0.1% BSA, 0.1% Ficoll 400) and 0.5% sodium dodecyl sulfate (SDS) when oligonucleotide probes were used or the ULTRAhyb buffer (Ambion) when RNA probes were used. Subsequently, membranes were washed twice at 50°C in 2× SSC plus 0.1% SDS (oligonucleotide probes), or twice at 68°C in 0.1× SSC plus 0.1% SDS (RNA probes) and visualized using a phosphor-imaging screen scanned by a Personal Molecular Imager (Bio-Rad). Quantitative measurements of relative band intensities were conducted with the Image Lab 4.1 software (Bio-Rad).
CircRT-PCR and RT-PCR
DNase-treated RNA was incubated with tobacco acid phosphatase (TAP; Epicenter) and T4 polynucleotide kinase (PNK; NEB). For circRT-PCR experiment, we used an unmodified kinase that possesses 3′-phosphatase activity. RNA was diluted to 20 ng/μL and circularized using T4 RNA ligase (Roche). The first strand (cDNA) was generated with Powerscript reverse transcriptase of the Creator Smart cDNA library construction kit (Clonetech) or avian myeloblastosis virus (AMV) reverse transcriptase (Roche). PCR was performed with the Takara PCR kit (Bio Inc.), typically for 35 cycles. Generally, two gene-specific primers were used, but for certain RT-PCR experiments, amplification was conducted with only one gene-specific primer (for first-strand synthesis) plus the Smart IV primer that anneals with the overhanging G residues at the 5′ end extension of the first-strand DNA (10). Primer sequences are given in the Supplementary Table S1. For all RT-PCR experiments, a negative control was performed where no template RNA was added.
Cloning and sequencing of amplicons
Amplicon termini were rendered blunt with T7 DNA polymerase and the Klenow fragment of DNA polymerase I (NEB), agarose gel-purified, phosphorylated with T4 PNK (NEB) and ligated into the vector pBFL6cat, which is an in-house constructed, small pBlueScript derivative. Libraries of cDNA were cloned into pDNR-LIB (Clonetech). After transformation into Escherichia coli DH5α, plasmid DNA was extracted using the Qiagen 96-well mini-prep kit. Sequencing reactions were performed with the BigDye Terminator version 3.1 Cycle Sequencing Kit from Applied Biosystems and sequenced on an ABI 370 Analyzer.
High-throughput RNA sequencing
Total RNA and mitochondrial RNA-enriched samples from D. papillatum were depleted of cytosolic 5, 5.8, 18 and 28S rRNA using a series of 5′ end biotinylated oligonucleotides (IDT) complementary to these rRNAs. For oligonucleotide design, we used the 5S rRNA sequence published earlier by others (GenBank accession no. AY007785) and the 5.8, 18 and 28S rRNA sequences reported here. The amount of the overabundant mt-LSU rRNA in mitochondrial RNA preparations was reduced by an oligonucleotide (dp72-5biosg) targeting the rnl module 2 (for oligonucleotides, see Supplementary Table S1). After annealing, oligonucleotide:rRNA hybrids were removed by streptavidin-coated magnetic beads (MyOne C1 and/or M-270 Dynabeads; Invitrogen). The library PA was made from cytosolic rRNA-depleted total RNA enriched for poly(A) RNA (see earlier in text), and the libraries F1 and F2 from mitochondrial RNA, following the supplier-recommended protocol devised for strand-specific RNA-Seq libraries and using the ScriptSeq™ RNA-Seq Library Preparation Kit (Epicentre). The difference between the F1 and F2 libraries is that for F2 the RNA fragmentation step was omitted to minimize further fragmentation of short RNA molecules. The F1, F2 and PA libraries were constructed and paired-end-sequenced (2 × 101 nt; Illumina HiSeq 2000) at the commercial technology platform Macrogen (Korea). According to the service provider, spurious antisense reads are below 2% and typically at 1% with the methodology used. For the GG library, we used RNA extracted from a subcellular fraction enriched in mitoribosomes. The library was constructed using the TruSeq Stranded Total RNA Sample Prep kit (Illumina) following the suppliers instructions and paired-end sequenced (2 × 250 nt; Illumina MiSeq) at the Genome Quebec Innovation Center in Montreal.
RNA-Seq data analysis
From the libraries F1, F2 and PA, we obtained between 50 and 70 Mio raw fastq reads of 101-nt length, and from the library GG ∼3 Mio raw reads of 250-nt length (Supplementary Table S2). Reads corresponding to cytosolic rRNAs were filtered out using Geneious 5.6 (Biomatters, New Zealand) leaving 40% (F1), 33% (F2), 95% (PA) and 15% (GG) reads. Adapters were removed from the 5′ and 3′ termini of reads with cutadapt version 1.2.1 (http://journal.embnet.org/index.php/embnetjournal/article/view/200). As parameters, we used a sub-sequence of 12 nt at the 3′ end or 5′ end of the 5′ and 3′ adapters, respectively, to allow for partial adapter sequence in the reads. The error rate was set to 0.1. Cutadapt was also used for quality clipping with a quality threshold of 20. Reads <20 nt were discarded. Statistics for the cleaning steps of reads are compiled in Supplementary Table S2. The data set used for further analysis was built from paired reads; reads that lost their mate during filtering were discarded using an in-house script. As a reference on which to map the read pairs to, we constructed a set of theoretically possible reference transcript sequences, including the expected intermediary molecules from RNA processing, trans-splicing and RNA editing. Paired reads were mapped onto each of these reference transcripts using bowtie2 (http://bowtie-bio.sourceforge.net/index.shtml). Bowtie was executed independently on each reference transcript and for each sense (forward and reverse) using the corresponding –norc/–nofw option. Read pairs where only one mate maps to the reference transcript or which are discordant (i.e. not mapping to the same strand or where the forward mate maps downstream of the reverse mate) were discarded from the alignment. Finally, using in-house scripts, pairs were removed that do not overlap with any of the reference transcripts, have a mapping quality <30, or a number of deletions ≥3. From libraries F1 and F2, we removed read pairs representing insert sizes ≥165 nt that originate from spurious dp72-amplification products primed by residual, contaminating dp72, an oligonucleotide that was used during sample preparation for the removal of cytosolic rRNA. Output files in sam format were subsequently transformed into ‘.bam’ files with SAMtools version 1.4 (http://samtools.sourceforge.net/). Alignments were visualized with tablet version 1.13.05.17 available at URL http://bioinf.scri.ac.uk/tablet/ (11). The statistics for the length distribution of the poly(A) tail and the poly(U) tract were calculated using an in-house script, which filters fastq files or the ‘sam‘ alignment file, respectively, for reads that overlap the upstream and/or downstream modules by a minimum number of nucleotides (typically 10–12 nt) and which contain a minimum number of homopolymeric nucleotides (typically 4 nt). The exact parameters are given in the figure legends.
RNA secondary structure modeling
We searched for conserved primary sequence and secondary structure motifs of mitochondrial LSU rRNAs by using the phylogeny-based consensus model available at the Comparative RNA Web (http://www.rna.ccbb.utexas.edu) (12). Thermodynamic folding was predicted by RNAfold 2.0 (13). Identified conserved motifs served as anchors for manual folding of the entire sequence to fit the model. Conventional nomenclature for sequential numbering of secondary structure elements has been used [e.g. (14)]. The secondary structure was drawn with XRNA 1.1.12 (http://rna.ucsc.edu/rnacenter/xrna/xrna.html) and finalized using CorelDRAW X4.
RESULTS
Identification of mt-LSU rRNA and its gene in Diplonema
The 352-nt-long 3′-terminal portion of mt-LSU rRNA from Diplonema was early on recognized as a top candidate for an unidentified rRNA, due to its extremely high abundance (representing 1% of all ESTs) in cDNA libraries constructed from total poly(A) RNA [(7); GenBank record JQ302963]. This RNA species carries an A tail of >25 nt and as we show here, is a precursor transcript of mt-LSU rRNA (see section later in text). For identification of mt-LSU rRNA from Diplonema, neither BLAST nor Rfam searches, nor comparison with mitochondrial rRNA sequences from other taxa was successful. Counterparts from euglenozoan species (i.e. the euglenid Euglena gracilis and kinetoplastids) not only are as highly divergent as mt-LSU from Diplonema but also display an extremely dissimilar nucleotide composition (15–20% G+C-content in kinetoplastids and Euglena versus ∼50% in Diplonema).
Mature Diplonema mt-LSU rRNA was first detected by northern hybridization, using an oligonucleotide as a probe that is specific for the 3′-terminal rnl portion. In total RNA, this probe lights up a major band of ∼0.9 kb, together with a weaker band at 0.4 kb (Figure 1A, right panel). The same band pattern is seen when using the entire 3′-terminal piece as a probe (Supplementary Figure S1). The 0.9-kb band is most likely the mature mt-LSU rRNA, whereas the smaller one, present in >20-times lower steady-state concentration, corresponds to the polyadenylated 3′-terminal portion. A size of 0.9 kb may appear small for mt-LSU rRNA, but the kinetoplastid counterpart is not much longer [1.1 kb; GenBank acc. no. TRBKPGEN; (15)]. In poly(A) RNA, the RNA species of 0.4 kb is highly enriched, whereas that of 0.9 kb is nearly undetected [Figure 1A, lane ‘poly(A)’; Supplementary Figure S1], which is in accordance with evidence from cDNA sequencing. Apparently, mature mt-LSU rRNA has a shorter A tail than the 352-nt RNA species, so that only a small fraction of is pulled down during the poly(A) enrichment procedure.
![Mitochondrial LSU rRNA of Diplonema. (A) Northern blot hybridization. Lane 1, in vitro transcription product of rnl module 1 (540 nt); lane 4, in vitro transcription product of rnl module 2 (359 nt; synthetic RNAs are 6 and 7 nt longer than the corresponding modules); lanes 2, 3, 5 and 6, total RNA (∼5 μg); lanes 7 and 8, poly(A) RNA (∼0.5 μg) extracted from whole cells. RNA in lanes 2 and 5 is from one preparation; that in lanes 3 and 6 is from an independent preparation. Blotted RNA was probed with radioactively labeled oligonucleotides dp216 (lanes 1–3) and dp218 (lanes 4–8) that target module 1 and module 2 of rnl, respectively. Bands represent the mature mt-LSU rRNA (∼900 nt), mono-module 1 transcripts (∼550 nt; the weak band in lane 3 is clearly visible on the original image), mono-module 2 transcripts (∼450 nt) and presumptive end-processing intermediates of single-module transcripts. The size markers are indicated on the left. The signal ratio of mt-LSU rRNA versus mono-module 1 transcripts varies noticeably from one preparation to another; it is 100:1 in lane 2 and 60:1 in lane 3. The signal ratio of mt-LSU rRNA versus mono-module 2 transcripts (lanes 5 and 6; total RNA) is ∼20:1. This ratio is ∼1:5 to ∼1:17 in poly(A)-enriched RNA (lanes 7 and 8), a variation depending on the particular oligo(dT) pull-down experiment. Notably, the steady-state of mono-module 1 transcript is lower than that of mono-module 2. The same is seen in RNA-Seq experiments (see Figure 4). (B) Upper part, schematic sequence of mtLSU rRNA. The U-tract between modules 1 and 2 (black box) is not encoded by mtDNA, but added post-transcriptionally. Regions with which northern hybridization probes dp216 and dp218 anneal are indicated. Lower part, coding regions of mt-LSU rRNA on mitochondrial chromosomes. Modules 1 and 2 are contained in cassettes of B-class chromosomes, but oriented in opposite direction relative to the chromosome’s constant region [indicated as B(+) and B(−), see text]. Non-coding regions within the cassettes (‘unique flanking regions’) are shown in dark gray. The constant region of chromosomes (light gray) is ∼95% identical across all B-class chromosomes (7). The black part of the constant region is also present in A-class chromosomes (‘shared constant region’).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/42/4/10.1093_nar_gkt1152/3/m_gkt1152f1p.gif?Expires=1750282457&Signature=GCOjz3EUnr4MxLpIEuE~gjMoBvVfPy27u7oAPC3t60S-RDN0Ov0xWRjd6kIMKL0sh9aslEOwFS54ZdMetCCoBQvNR8VrhSK9iHDWndVIHhwKR6gS3FOlsPoGlqVI-9tOq9acRKI7-5AYNXeVU8rln1iMSvWyM177Zd6PYcD8fIR4NanjgkZB711X8H2Y7A6eUwdW6g4PkZGXL1MdEhVboFR6P1frMuOjc5E-hqsDLnaTOUZL3QmWL3MiudBtkcEe0vv0Jp-uVOIuXSCgsM75aOe683da5Gn9J-ZFnn3FG~htXvhuxdeCbonpdTcpfZMGTERSIbOcZ1EJvLmtSDr6Iw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mitochondrial LSU rRNA of Diplonema. (A) Northern blot hybridization. Lane 1, in vitro transcription product of rnl module 1 (540 nt); lane 4, in vitro transcription product of rnl module 2 (359 nt; synthetic RNAs are 6 and 7 nt longer than the corresponding modules); lanes 2, 3, 5 and 6, total RNA (∼5 μg); lanes 7 and 8, poly(A) RNA (∼0.5 μg) extracted from whole cells. RNA in lanes 2 and 5 is from one preparation; that in lanes 3 and 6 is from an independent preparation. Blotted RNA was probed with radioactively labeled oligonucleotides dp216 (lanes 1–3) and dp218 (lanes 4–8) that target module 1 and module 2 of rnl, respectively. Bands represent the mature mt-LSU rRNA (∼900 nt), mono-module 1 transcripts (∼550 nt; the weak band in lane 3 is clearly visible on the original image), mono-module 2 transcripts (∼450 nt) and presumptive end-processing intermediates of single-module transcripts. The size markers are indicated on the left. The signal ratio of mt-LSU rRNA versus mono-module 1 transcripts varies noticeably from one preparation to another; it is 100:1 in lane 2 and 60:1 in lane 3. The signal ratio of mt-LSU rRNA versus mono-module 2 transcripts (lanes 5 and 6; total RNA) is ∼20:1. This ratio is ∼1:5 to ∼1:17 in poly(A)-enriched RNA (lanes 7 and 8), a variation depending on the particular oligo(dT) pull-down experiment. Notably, the steady-state of mono-module 1 transcript is lower than that of mono-module 2. The same is seen in RNA-Seq experiments (see Figure 4). (B) Upper part, schematic sequence of mtLSU rRNA. The U-tract between modules 1 and 2 (black box) is not encoded by mtDNA, but added post-transcriptionally. Regions with which northern hybridization probes dp216 and dp218 anneal are indicated. Lower part, coding regions of mt-LSU rRNA on mitochondrial chromosomes. Modules 1 and 2 are contained in cassettes of B-class chromosomes, but oriented in opposite direction relative to the chromosome’s constant region [indicated as B(+) and B(−), see text]. Non-coding regions within the cassettes (‘unique flanking regions’) are shown in dark gray. The constant region of chromosomes (light gray) is ∼95% identical across all B-class chromosomes (7). The black part of the constant region is also present in A-class chromosomes (‘shared constant region’).
The 5′-terminal region of mt-LSU rRNA was identified by RT-PCR applied to circularized RNA (circRT-PCR) using a pair of ‘divergent’ primers annealing with the molecule’s 3′ end region (see ‘Methods’). Subsequent cloning and sequencing revealed a 534-nt-long stretch upstream of the 3′ end portion of rnl. As only two such clones were obtained (in multiple experiments), we confirmed their authenticity by northern hybridization. An oligonucleotide specific to the presumed 5′-terminal portion lights up the 0.9-kb product and in addition a faint 0.5-kb band that corresponds to the 5′-terminal portion alone (Figure 1A, left panel). RNA-Seq data (later in text) provided the ultimate confirmation for the 534-nt-long sequence being the 5′ moiety of mt-LSU rRNA in Diplonema.
The most remarkable sequence feature of Diplonema mt-LSU rRNA is a run of ∼26 uridines (Us) immediately upstream of its 3′ moiety (Figure 1B, upper part; Table 1 and Supplementary Figure S2). This homopolymer tract was confirmed independently by RT-PCR using a primer pair that anneals upstream and downstream of this tract (Supplementary Table S5 and Supplementary Figure S3). The observed U-tract length varies by about ±3, which is apparently due to experimental rather than biological variation (Supplementary Table S6); errors probably occur during PCR amplification or the sequencing reaction itself, as commercial RT-enzymes have high synthesis fidelity. We posit that this long U-tract is the reason why RT-PCR-based experiments yielded extremely low numbers of reads. This bias is observed also in RNA-Seq (see later in text).
Non-encoded U-tract length of mt-LSU rRNA and its precursor transcriptsa
| Transcript structure | Mean number of Us (minimum–maximum) | Major peak RNA-Seq (nt)b | |
|---|---|---|---|
| circRT-PCR (nt) | RNA-Seq (nt) | ||
| ∼m1.[U]n | 5 (3–7)c | n.d.d | n.d.d |
| ∼m1.[U]n.m2∼ | 26.6 (26–28)e | 25.1 (14–33)f | 26f |
| [U]n.m2∼ | / | n.d.d | n.d.d |
| Transcript structure | Mean number of Us (minimum–maximum) | Major peak RNA-Seq (nt)b | |
|---|---|---|---|
| circRT-PCR (nt) | RNA-Seq (nt) | ||
| ∼m1.[U]n | 5 (3–7)c | n.d.d | n.d.d |
| ∼m1.[U]n.m2∼ | 26.6 (26–28)e | 25.1 (14–33)f | 26f |
| [U]n.m2∼ | / | n.d.d | n.d.d |
am1, m2, rnl modules 1 and 2; [U]n, uridine-homopolymer of length n; m1.[U]n, module 1 with 3′-terminal U tract; m1.[U]n.m2, LSU-rRNA; [U]n.m2, module 2 with 5'-terminal U tract; and ∼, exact module terminus not determined. n.d., not identified; /, not observed.
bPeak positions of tract length distribution is taken from Supplementary Figure S2. Libraries F2, PA and GG display similar U-tract length as F1 shown here.
cFour clones (dp11056, dp11060, dp11084, dp11088).
dThis type of transcript could not be identified unambiguously.
eSeven clones (dp9540, dp10594, dp11008, dp11009, dp11012, dp11017, dp11064).
fNot-quality clipped individual reads from the F1 library.
Non-encoded U-tract length of mt-LSU rRNA and its precursor transcriptsa
| Transcript structure | Mean number of Us (minimum–maximum) | Major peak RNA-Seq (nt)b | |
|---|---|---|---|
| circRT-PCR (nt) | RNA-Seq (nt) | ||
| ∼m1.[U]n | 5 (3–7)c | n.d.d | n.d.d |
| ∼m1.[U]n.m2∼ | 26.6 (26–28)e | 25.1 (14–33)f | 26f |
| [U]n.m2∼ | / | n.d.d | n.d.d |
| Transcript structure | Mean number of Us (minimum–maximum) | Major peak RNA-Seq (nt)b | |
|---|---|---|---|
| circRT-PCR (nt) | RNA-Seq (nt) | ||
| ∼m1.[U]n | 5 (3–7)c | n.d.d | n.d.d |
| ∼m1.[U]n.m2∼ | 26.6 (26–28)e | 25.1 (14–33)f | 26f |
| [U]n.m2∼ | / | n.d.d | n.d.d |
am1, m2, rnl modules 1 and 2; [U]n, uridine-homopolymer of length n; m1.[U]n, module 1 with 3′-terminal U tract; m1.[U]n.m2, LSU-rRNA; [U]n.m2, module 2 with 5'-terminal U tract; and ∼, exact module terminus not determined. n.d., not identified; /, not observed.
bPeak positions of tract length distribution is taken from Supplementary Figure S2. Libraries F2, PA and GG display similar U-tract length as F1 shown here.
cFour clones (dp11056, dp11060, dp11084, dp11088).
dThis type of transcript could not be identified unambiguously.
eSeven clones (dp9540, dp10594, dp11008, dp11009, dp11012, dp11017, dp11064).
fNot-quality clipped individual reads from the F1 library.
The gene specifying Diplonema mt-LSU rRNA was pinpointed by mapping the rRNA sequence on the available mtDNA sequence, revealing two previously unannotated coding regions embedded in cassettes of separate B-class chromosomes (for a definition of ‘cassette’, see legend of Figure 1B). These coding regions are referred to as rnl modules 1 and 2 (Figure 1B, lower panel). With 534 bp length, rnl module 1 is the longest among all known gene modules in Diplonema mtDNA, whereas rnl module 2 (352 bp) is of average size. Gene module 2 of rnl lacks a 3′ terminal A-homopolymer stretch, which is obviously added by post-transcriptional polyadenylation. Also absent from both the module 1 and module 2-coding regions is a terminal T tract, otherwise present in the center of the mt-LSU rRNA cDNA sequence. The sequence of gene module 1 ends precisely upstream, whereas that of gene module 2 starts exactly downstream of the U tract in mt-LSU rRNA. Therefore, these non-encoded nucleotides must be added post-transcriptionally, resulting in U-insertion RNA editing. This is by far the longest stretch of non-encoded Us seen in Diplonema mitochondria and also the largest number of nucleotides added at a single editing site ever observed.
2D structure modeling of mt-LSU rRNA
The secondary (2°) structure of the 3′ moiety from Diplonema mt-LSU rRNA was modeled based on comparison with the mitochondrial consensus structure—the homologs from kinetoplastids and E. gracilis are too divergent for a meaningful comparison of covariant residues (Figure 2A). Only domains IV to VI [as defined for E. coli (Figure 2B)] are conventional, albeit reduced. Domain V encompasses the peptidyl-transferase center (PTC) and is the most conserved region of LSU rRNAs. As in many other reduced mt-LSU rRNAs, the Diplonema molecule lacks the helices H76-H79 that in E. coli bind the ribosomal protein L1 and H83-H86 that associate with 5S rRNA. Domain VI lacks major parts, and the sequence that connects H73 and H95 in most other mt-LSU rRNAs (12,16) is unusually short. Just a few of the universally conserved sequence motifs are readily recognizable in the Diplonema molecule, namely, those corresponding to the basis of helix H90 and its single-stranded junctions to H89 and H93, as well as the terminal loops of helices H80, H92 and H95 (the latter is also known as the α-sarcin/ricin loop). Nonetheless, domain V of Diplonema mt-LSU rRNA resembles bacterial 23S rRNA somewhat more closely than that of kinetoplastids, the latter lacking for example H97 (17–19).
![Putative secondary structure of the mt-LSU rRNA (3′ moiety) from Diplonema. (A) The structure was modeled according to the mitochondrial reference sequence and structure (http://www.rna.icmb.utexas.edu). Residues identical to the universal consensus sequence (12,16) are shown in bold. Domain IV is composed of the 3′ portion of module 1 (dark gray shading) and the post-transcriptionally added U-tract (black shading). Domains V and VI are encoded by module 2 (light gray shading). The thin dashed line marks helix 26a (see ‘Discussion’). Base pairing is indicated as thin lines, thick lines, dots and open circles corresponding to A:U, G:C, G:U and other base pairs, respectively. Residues are numbered according to nucleotide positions in rnl modules 1 (upstream of U-tract) and 2 (downstream of U-tract). The nucleotide pair U305:A314 in the module 2 corresponds to a conserved trans Watson-Crick/Hoogsteen pair in the E. coli structure. (B) The 2° structure of the 3′ moiety from Diplonema mt-LSU rRNA mapped onto the structure from E. coli LSU rRNA. Helices are numbered according to (14). H95, α-sarcin/ricin loop. Thick gray and black lines indicate the structure elements present in the Diplonema model [same shading as in (A)]. Triangles indicate breakpoints in the 3′ half of fragmented LSU rRNAs from apicomplexan (2,20,21) and dinoflagellate (3,4) mitochondria (light gray triangles), several green algal mitochondria (gray triangles; 22–25), and the kinetoplastid (26) and euglenid (27) cytosol (black triangles). It is noteworthy that among all known cases of discontinuous domain-IV LSU rRNA (apicomplexans and dinoflagellates), none is split in the 3′ half of H61.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/42/4/10.1093_nar_gkt1152/3/m_gkt1152f2p.gif?Expires=1750282457&Signature=JDVhMascZKboilftrO3WD4kIjAkoLcJnRzOBnzQbOfxF0LryxCxHXikngg7Y9kW8RH3Nffgmo6nu-vaYNtxv5Wau2tFAZq-V35EiyzKOZ2~hRsom0vEKxBgip81niXqbLNWn1Rwn404VdL2m71NEVHeVcJDAvVDB3GOxKar2BzD309xiSQ1oV7nf~48-rhNYu6zB2NB1buazymMnhG7nvUBQ9txbv4-e-pxQ75qTXa3sa5fv9i8hkpUckRcUrlbAuE3DLPLsS1mpwRJv8A-fwoyUrcILEIduKNJVdD-UbxVx0q9-m~GOTQmWSNLUJPufI9FM0t23fk5n7IIGZeGuNA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Putative secondary structure of the mt-LSU rRNA (3′ moiety) from Diplonema. (A) The structure was modeled according to the mitochondrial reference sequence and structure (http://www.rna.icmb.utexas.edu). Residues identical to the universal consensus sequence (12,16) are shown in bold. Domain IV is composed of the 3′ portion of module 1 (dark gray shading) and the post-transcriptionally added U-tract (black shading). Domains V and VI are encoded by module 2 (light gray shading). The thin dashed line marks helix 26a (see ‘Discussion’). Base pairing is indicated as thin lines, thick lines, dots and open circles corresponding to A:U, G:C, G:U and other base pairs, respectively. Residues are numbered according to nucleotide positions in rnl modules 1 (upstream of U-tract) and 2 (downstream of U-tract). The nucleotide pair U305:A314 in the module 2 corresponds to a conserved trans Watson-Crick/Hoogsteen pair in the E. coli structure. (B) The 2° structure of the 3′ moiety from Diplonema mt-LSU rRNA mapped onto the structure from E. coli LSU rRNA. Helices are numbered according to (14). H95, α-sarcin/ricin loop. Thick gray and black lines indicate the structure elements present in the Diplonema model [same shading as in (A)]. Triangles indicate breakpoints in the 3′ half of fragmented LSU rRNAs from apicomplexan (2,20,21) and dinoflagellate (3,4) mitochondria (light gray triangles), several green algal mitochondria (gray triangles; 22–25), and the kinetoplastid (26) and euglenid (27) cytosol (black triangles). It is noteworthy that among all known cases of discontinuous domain-IV LSU rRNA (apicomplexans and dinoflagellates), none is split in the 3′ half of H61.
Domain IV is most likely constituted by the 3′ third of the module 1 sequence. We recognize the conserved helices H69 and H71 with their surrounding single-stranded regions that are involved in the majority of inter-subunit contacts with ribosomal SSU and functionally important interactions with ribosome-bound tRNAs (28). Two other consensus helices of this domain lack a substantial peripheral portion in Diplonema as well as in kinetoplastids and several other taxa. The structure model places the poly(U) tract at the 3′ end of domain IV. Two 4-nt-long purine stretches upstream in module 1 might base-pair with poly(U) to form a helix analogous to H61. However, this region could also remain single-stranded as in the 2° model of kinetoplastid and nematode mt-LSU rRNAs (16,19).
Although we were able to reconstruct a reasonable 2° structure model of the 3′ half of mt-LSU rRNA from Diplonema, folding the 5′ half of this molecule (domains I-III) is challenging due to several reasons (but see Supplementary Figure S7). First, this part of the molecule is in general moderately conserved. In addition, comparative modeling was not feasible due to low sequence similarity between Diplonema mt-LSU rRNA and homologs with available 2° models. Finally, modeling based on thermodynamic folding leads to an excessive number of alternatives because the G+C rich (51%) sequence allows profuse base-pairing possibilities. As to length and structure, the 5′ half of mt-LSU rRNA from Diplonema is more reduced and shorter than that from kinetoplastids, yet comparably deviant as that from certain animals as detailed in the Discussion section.
Deep sequencing and RT-PCR analysis of the rnl transcript population
To capture rnl transcripts of Diplonema in a comprehensive way, we performed massively parallel sequencing (RNA-Seq) of three RNA samples (F1, F2, PA). Samples F1 and F2 were extracted from a subcellular fraction enriched in mitochondria; sample PA was enriched for poly(A) RNA. The applied RNA-Seq approach involved paired-end library construction by RNA fragmentation for F1 and PA (but not F2), random hexamer priming and strand-specific sequencing. Average fragment (insert) length is 300 nt, read length is 101 nt and read depth is ∼60 Mio reads per sample. Primer and quality trimming resulted in ∼100 Mio paired reads of ≥20-nt length for all three libraries together (60%). Of these, 1.066 Mio paired reads (1%) contain rnl sequences. A fourth small library (GG) was constructed with an RNA sample that was extracted from a mitoribosome-enriched subcellular fraction of Diplonema. Reads of this library were used to characterize the mitoribosome-associated LSU rRNA. Information on RNA-Seq data are compiled in Supplementary Tables S2 and Supplementary Data.
First, we mapped read pairs to the sequence of mt-LSU rRNA. Read coverage of the mitochondrial libraries is depicted in Supplementary Figure S4A. Detailed inspection of coverage showed that only 14 (quality-clipped) reads span completely the internal U-tract and include ≥10 nt of both adjacent modules, although ∼150 000 reads map to the module-1/module-2 junction region; the majority of U-tract containing reads maps to either the 3′ end of module 1 or the 5′ end of module 2 (Supplementary Table S4). This bias is due to low sequence quality in homopolymer tracts. More than 99.9% reads have quality values <20 from the 13th U-tract position on, so that all sequence beyond this position is removed by quality clipping during the read preprocessing step (Supplementary Figure S5). Therefore, we used the inferred ‘inserts’ (i.e. the interval inferred from paired-end reads) instead of reads for mapping onto mt-LSU rRNA (Figure 3; for logarithmic scale, see Supplementary Figure S4A) and most of the other analyses described later in text.
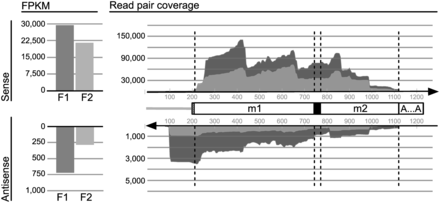
Coverage of Diplonema mt-LSU rRNA by RNA-Seq data. Mapping of inferred inserts from two mitochondrial libraries, F1 (dark gray) and F2 (light gray). Vertical scales, counts of inserts. Cartoon in the center, schematic representation of the virtual reference transcript to which inserts were mapped. Unfilled boxes labeled m1 and m2, rnl modules 1 and 2, respectively. Black box, poly(U) of ∼26 length added by RNA editing; dashed box upstream module 1, unique flanking region; gray line, transcribed constant region of B-class chromosomes (see Figure 1B). ‘A … A’, A-tail. It should be noted that inserts (and reads) cannot be mapped unambiguously beyond ∼80 nt upstream and downstream of modules because these regions are nearly identical in sequence with those from other modules residing on B-class chromosomes. Stacked-area chart on the right side, coverage by sense (upper area) and antisense (lower area) inserts, respectively. The bar charts to the left represent the total number of reads covering the corresponding area in the stacked-area chart. The scales for sense and antisense transcripts differ by a factor of 30. Sharp drop-off in antisense read coverage ∼100 nt upstream of rnl module 1 (a zone corresponding to the constant region of B-class chromosomes) reflecting a discrete 3′ end of antisense RNAs. Uneven read coverage along the sequence is probably due to sequence bias.
For targeted detection of long transcripts and accurate mapping of their termini, we conducted in addition RT-PCR using specific primers that anneal within module 1 or module 2 of rnl. In experiments with circularized RNA, primers point in divergent direction, otherwise they are oriented in convergent fashion.
Maturation intermediates of rnl transcripts
To characterize intermediates of mt-LSU rRNA, we mapped RNA-Seq inserts from the mitochondrial libraries F1 and F2 to three virtual reference transcript sequences, which represent the primary transcript of each individual module and a trans-spliced, edited and polyadenylated transcript. LSU rRNA precursors were also characterized by RT-PCR and circRT-PCR experiments.
End-processing intermediates are of two types, transcripts including an rnl module plus either both adjacent non-coding regions or a single adjacent region retained on either end (Figure 4). Fully processed module transcripts are seen as well (Table 2). Notably, not only fully processed modules but also end-processing intermediates engage in trans-splicing. For example, we detected a transcript with joined modules 1 and 2, whose 3′ end still has non-coding sequence attached. Mapping of RNA-Seq data to unprocessed reference sequences is shown in Supplementary Figures S4B and Supplementary Data.
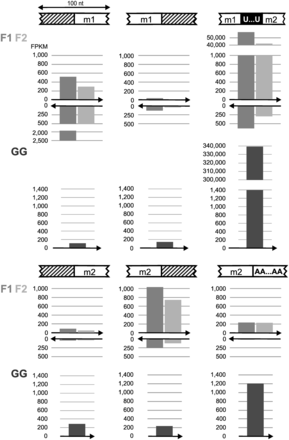
Maturation intermediates of rnl transcripts. Cartoons depict schematically the regions where maturation processes take place. White, hatched and black boxes indicate modules and the A tail, non-coding regions and the U-tract at the module junction, respectively. Bar charts beneath cartoons show the number of paired reads from the mitochondrial libraries F1 (medium gray) and F2 (light gray), and the mitoribosome library GG (dark gray) that map to the designated regions. The arrow below the bars specifies reads in sense (pointing to the right) and antisense (pointing to the left) direction. Counted reads suffice the following criteria: within a 100-nt-long region around the maturation site, reads (forward or reverse read of mapped read pairs) are required to cover at least 55 nt of this window, i.e. overlap boundaries (between modules and other regions) by at least 5 nt. The proportion of immature rnl transcripts in the library GG serves as a measure for mitoribosome enrichment.
End processing intermediates of rnl modules transcriptsa
| Module | Methodology | Number of clones/inserts representing intermediate type | |||
|---|---|---|---|---|---|
| —m— | —m∼ | ∼m— | ^m^ | ||
| Module 1 (≥534 nt) | circRT-PCR | / | 3b | 3c | / |
| RNA-Seq | /d | 941 | 55 | /d | |
| Module 2 (≥352 nt) | circRT-PCR | 3e | / | 2f | 2g |
| RNA-Seq | 4d | 167 | 2593 | /d | |
| Module | Methodology | Number of clones/inserts representing intermediate type | |||
|---|---|---|---|---|---|
| —m— | —m∼ | ∼m— | ^m^ | ||
| Module 1 (≥534 nt) | circRT-PCR | / | 3b | 3c | / |
| RNA-Seq | /d | 941 | 55 | /d | |
| Module 2 (≥352 nt) | circRT-PCR | 3e | / | 2f | 2g |
| RNA-Seq | 4d | 167 | 2593 | /d | |
aNumber of observed clones in RT-PCR experiments or inserts in RNA-Seq libraries F1 and F2 (latter data taken from Figure 4). Symbols and abbreviations used: —, non-coding adjacent region; m, rnl-module 1 or 2; ^m^, module end-processed at both termini; ∼m, m∼, nature of module’s 5′ end or 3′ end, respectively, is unknown (may be unprocessed or processed); /, not observed.
bThree clones (dp11008, dp11034, dp11059); length of non-coding regions is 324, 20 and 69 nt, respectively.
cThree clones (dp9411, dp9613, dp11051l); length of non-coding regions is 163, 22 and 3 nt.
dLow probability of observation, because the libraries have an insert size average of 300 nt.
eThree clones (dp9408, dp10574b, dp10586).
fTwo clones (dp9411, dp9613).
gTwo clones (dp10439rb, dp10526a).
End processing intermediates of rnl modules transcriptsa
| Module | Methodology | Number of clones/inserts representing intermediate type | |||
|---|---|---|---|---|---|
| —m— | —m∼ | ∼m— | ^m^ | ||
| Module 1 (≥534 nt) | circRT-PCR | / | 3b | 3c | / |
| RNA-Seq | /d | 941 | 55 | /d | |
| Module 2 (≥352 nt) | circRT-PCR | 3e | / | 2f | 2g |
| RNA-Seq | 4d | 167 | 2593 | /d | |
| Module | Methodology | Number of clones/inserts representing intermediate type | |||
|---|---|---|---|---|---|
| —m— | —m∼ | ∼m— | ^m^ | ||
| Module 1 (≥534 nt) | circRT-PCR | / | 3b | 3c | / |
| RNA-Seq | /d | 941 | 55 | /d | |
| Module 2 (≥352 nt) | circRT-PCR | 3e | / | 2f | 2g |
| RNA-Seq | 4d | 167 | 2593 | /d | |
aNumber of observed clones in RT-PCR experiments or inserts in RNA-Seq libraries F1 and F2 (latter data taken from Figure 4). Symbols and abbreviations used: —, non-coding adjacent region; m, rnl-module 1 or 2; ^m^, module end-processed at both termini; ∼m, m∼, nature of module’s 5′ end or 3′ end, respectively, is unknown (may be unprocessed or processed); /, not observed.
bThree clones (dp11008, dp11034, dp11059); length of non-coding regions is 324, 20 and 69 nt, respectively.
cThree clones (dp9411, dp9613, dp11051l); length of non-coding regions is 163, 22 and 3 nt.
dLow probability of observation, because the libraries have an insert size average of 300 nt.
eThree clones (dp9408, dp10574b, dp10586).
fTwo clones (dp9411, dp9613).
gTwo clones (dp10439rb, dp10526a).
RNA editing almost certainly takes place before trans-splicing, because neither RNA-Seq nor RT-PCR detected reads where the 3′ end of rnl module 1 is immediately upstream-adjacent to the 5′ end of module 2. Uridine residues are most likely added 3′ to module 1 and not 5′ to module 2 according to circRT-PCR experiments (Table 1). For Diplonema cox1, U-appendage editing of the module upstream of the editing site has been validated more rigorously. Only after 3′ dephosphorylation of RNAs did we observe upstream modules with Us appended at the 3′ end, but under no condition was the downstream module found with Us attached to its 5′ end (8).
RNA editing intermediates of rnl that have excess or deficit Us cannot be determined reliably, because sequences containing homopolymers are of low quality, especially those where the U-tract is at the 5′ end of the read (Supplementary Figure S5). At present, the two following editing scenarios remain indistinguishable: (i) uncontrolled addition of numerous Us and subsequent precise trimming as is the case for U-insertion editing of trypanosome mitochondria (29) and (ii) controlled addition of the exact number of nucleotides.
The A-tail of module 2-containing rnl transcripts displays substantial differences in length (Table 3). Mono-module 2 is polyadenylated by addition of up to 90 As, whereas trans-spliced transcripts have predominantly ∼20-nt-long A-tails, and mt-LSU rRNA incorporated in the mitoribosome has virtually no A-tail. These differences are seen consistently in all three experimental approaches used in this study. In northern hybridization, we observe different signal ratios of mono-module 2 versus mature rRNA. The ratio in total RNA is ∼1:20, but nearly inverse in the poly(A) RNA-enriched fraction (see Figure 1 and Supplementary Figure S1). In circRT-PCR experiments, A-tails of rnl mono-module 2 transcripts are up to ∼50-nt-long, whereas those of the trans-spliced transcript are not longer than 26 nt. Finally, A-tail size distributions in RNA-Seq data from total-cell poly(A) RNA exhibit a broad crest up to 80 nt, those from total mitochondrial RNA peak at ∼20 nt and the ones from mitoribosomal RNA have a dominant maximum at 0 nt (Table 3 and Supplementary Figure S6). The possible biological significance of this variation in A-tail length will be examined in the ‘Discussion’ section.
Poly(A) tail length of rnl transcriptsa
| Transcript (structure) | Poly(A) tail | |
|---|---|---|
| circRT-PCR: mean (minimum-maximum) length (nt) | RNA-Seq: mean length (major peak position) (nt)b | |
| rnl mono-module 2 (m2[A]n) | 24 (4–47)c | 46 (∼60) (PA)d |
| mature rRNA (m1.Us.m2[A]n) | 22 (19–26)e | 33 (∼20) (F1)f |
| 0 (0) (GG)g | ||
| Transcript (structure) | Poly(A) tail | |
|---|---|---|
| circRT-PCR: mean (minimum-maximum) length (nt) | RNA-Seq: mean length (major peak position) (nt)b | |
| rnl mono-module 2 (m2[A]n) | 24 (4–47)c | 46 (∼60) (PA)d |
| mature rRNA (m1.Us.m2[A]n) | 22 (19–26)e | 33 (∼20) (F1)f |
| 0 (0) (GG)g | ||
aSymbols used: m1, m2, rnl-modules 1 and 2; m1.Us.m2, mt-LSU rRNA sequence including (from 5′ to 3′) module 1, 20–30 Us, and module 2; [A]n, adenine homopolymer of length n. Transcripts length is ≥900 and ≥353 nt for m1.Us.m2[A]n, and m2[A]n, respectively.
bPeak positions of A-tail length distribution is taken from Supplementary Figure S6.
cFive clones (dp10594, dp11008, dp11009, dp11012, dp11017).
dLibrary PA was made from RNA that contains predominantly rnl mono-module 2 [m2:trans-spliced rRNA = ∼17:1 according to northern hybridization experiments; see Figure 1A, lane ‘poly(A)’].
eFifteen clones from the series dp104xx; e.g. dp10411r.
fLibrary F1 was made from RNA that contains predominantly mature mt-LSU rRNA (trans-spliced rRNA:m2 = ∼20:1 according to northern hybridization experiments; see Figure 1A, lane ‘total’, probe m2). Two mate pairs from this library span rnl modules 1 plus 2 (reads 1203:11003:25874 and 1216:19505:7846).
gLibrary GG was made from RNA that was extracted from a subcellular fractions enriched in mitoribosomes. Contamination with rnl transcripts not assembled in the ribosome is estimated at ≤0.1% (see Figure 4).
Poly(A) tail length of rnl transcriptsa
| Transcript (structure) | Poly(A) tail | |
|---|---|---|
| circRT-PCR: mean (minimum-maximum) length (nt) | RNA-Seq: mean length (major peak position) (nt)b | |
| rnl mono-module 2 (m2[A]n) | 24 (4–47)c | 46 (∼60) (PA)d |
| mature rRNA (m1.Us.m2[A]n) | 22 (19–26)e | 33 (∼20) (F1)f |
| 0 (0) (GG)g | ||
| Transcript (structure) | Poly(A) tail | |
|---|---|---|
| circRT-PCR: mean (minimum-maximum) length (nt) | RNA-Seq: mean length (major peak position) (nt)b | |
| rnl mono-module 2 (m2[A]n) | 24 (4–47)c | 46 (∼60) (PA)d |
| mature rRNA (m1.Us.m2[A]n) | 22 (19–26)e | 33 (∼20) (F1)f |
| 0 (0) (GG)g | ||
aSymbols used: m1, m2, rnl-modules 1 and 2; m1.Us.m2, mt-LSU rRNA sequence including (from 5′ to 3′) module 1, 20–30 Us, and module 2; [A]n, adenine homopolymer of length n. Transcripts length is ≥900 and ≥353 nt for m1.Us.m2[A]n, and m2[A]n, respectively.
bPeak positions of A-tail length distribution is taken from Supplementary Figure S6.
cFive clones (dp10594, dp11008, dp11009, dp11012, dp11017).
dLibrary PA was made from RNA that contains predominantly rnl mono-module 2 [m2:trans-spliced rRNA = ∼17:1 according to northern hybridization experiments; see Figure 1A, lane ‘poly(A)’].
eFifteen clones from the series dp104xx; e.g. dp10411r.
fLibrary F1 was made from RNA that contains predominantly mature mt-LSU rRNA (trans-spliced rRNA:m2 = ∼20:1 according to northern hybridization experiments; see Figure 1A, lane ‘total’, probe m2). Two mate pairs from this library span rnl modules 1 plus 2 (reads 1203:11003:25874 and 1216:19505:7846).
gLibrary GG was made from RNA that was extracted from a subcellular fractions enriched in mitoribosomes. Contamination with rnl transcripts not assembled in the ribosome is estimated at ≤0.1% (see Figure 4).
Antisense RNA covering module junction and editing site of mt-LSU rRNA
We posited earlier that trans-splicing and RNA editing of the mitochondrial protein-coding gene cox1 in Diplonema might be instructed by antisense RNAs. Preliminary evidence for antisense transcripts of a protein-coding gene came from targeted RT-PCR experiments (8). However, the yield of products was low and the informative sequence obtained (after subtraction of primer sequences) was only a few nucleotides long. Here we re-examine whether guiding antisense RNAs exist in Diplonema mitochondria by focusing on one of the most highly expressed mitochondrial genes, rnl, and by exploiting strand-specific RNA-Seq data.
Strikingly, putative antisense transcripts of mt-LSU RNA are detected at ∼2.5%, which is significantly above background (see ‘Materials and Methods’ section and Figure 3A, lower panel). The existence of such transcripts is also seen in RT-PCR experiments (Supplementary Figure S3 and Supplementary Table S5). Remarkably, antisense read coverage drops off sharply ∼100 nt upstream of module 1, a zone corresponding to the constant region of B-class chromosomes (Figure 3A). This drop-off reflects a discrete 3′ end of rnl antisense RNAs. The same phenomenon is seen in read mapping of antisense transcripts from cox1-module 1 (not shown), which is likewise a first module encoded on a B-class (+) chromosome (see Figure 1B). Whether the rnl-antisense 3′ terminus is generated by transcription termination or processing remains to be investigated.
In contrast to their 3′ end, the 5′ terminus of rnl antisense RNAs appears variable in the read coverage profile. We attempted to determine the length of these transcripts by northern experiments using either single-stranded oligo-deoxynucleotides or in vitro transcribed RNAs as a probe, but the signals were extremely weak (not shown). Antisense transcripts might be a heterodisperse assemblage of different length that do not form a homogenous band in gel electrophoresis; an already weak signal spread out instead of concentrated in a band would be difficult to detect by northern hybridization. Neither could we find the potential gene encoding the anti-mt-LSU RNA in the available ∼250 kb mtDNA nor in the currently draft assembly of nuclear DNA. It is possible that the gene was not found because it is encoded in a yet unsequenced genomic region, or alternatively, because there is no such gene as elaborated in the ‘Discussion’.
Putative antisense transcripts of unprocessed modules are also seen in RNA-Seq data. These RNAs apparently originate from bi-directional transcription of rnl-module-carrying chromosomes (Supplementary Figure S4B and Supplementary Data). Transcription in Diplonema mitochondria starts in the shared, constant region of chromosomes located opposite to modules (8). As modules are oriented in either sense relative to the shared region (as for example rnl modules 1 and 2 Figure 1B), the promoter(s) must be able to drive transcription of both strands.
DISCUSSION
Regulation of rnl gene expression in Diplonema mitochondria
Based on the observed types of rnl-transcript intermediates, two diametrically opposite maturation pathways of mt-LSU rRNA can be postulated. One interpretation of the results is that polyadenylation is a dead-end reaction, tagging molecules that failed to be trans-spliced or incorporated into the ribosome (Figure 5A). However, this view does not explain why only module 2 but not module 1 is polyadenylated. The other hypothesis, which we favour, considers that polyadenylation is crucial for mt-LSU maturation. We posit that module 2 is first polyadenylated and then deadenylated in two subsequent steps, with the particular A-tail length being the checkpoints for trans-splicing of modules 1 and 2, and then for assembly of the trans-splicing product into the ribosome (Figure 5B). This view would explain the difference in predominant A-tail length of mt-LSU rRNA from total mitochondrial RNA extractions (∼20 nt) versus mitoribosome-extracted RNA (∼0 nt) as follows. The former RNA preparation may contain mainly rRNA that is not incorporated into the ribosome. Still, we cannot fully exclude technical variation because different protocols were used for constructing the two libraries.
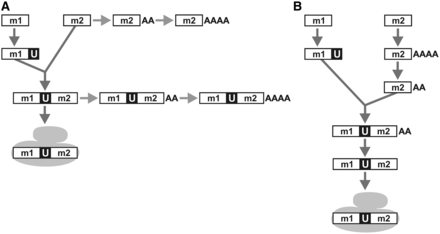
Maturation process of mt-LSU rRNA in Diplonema mitochondria. For clarity, the cartoon disregards end-processing of module 1 and module 2 precursor transcripts. m1, m2, rnl module 1 and 2, respectively. U, post-transcriptionally-added U tract. AA, AAAA, poly(A) tails of ∼20 nt or ∼40–90 nt length, respectively. The gray-filled shape symbolizes the mitoribosome. (A) Hypothetical pathway where polyadenylated rnl-transcripts represent dead ends instead of maturation intermediates. (B) Alternative pathway (preferred hypothesis) where the polyadenylation status plays a key role in mt-LSU rRNA maturation: a poly(A)tail length of ∼20 As signals a check point for trans-splicing, and absence of an A-tail from the trans-spliced product is a requirement for incorporation of the transcript into the mitoribosome.
The various biochemical reactions involved in the expression of Diplonema mt-LSU rRNA, module-end processing, adenylation, uridylation, trans-splicing and potentially A-tail trimming of the molecule’s 3′ end, must be catalyzed by an assortment of activities (ribonuclease, polymerase and ligase), as well as trans-factors that guide trans-splicing and editing. Traditionally, multi-step biochemical pathways are pictured as a cascade of catalytic steps, where the product of a given reaction is the substrate for the subsequent step. However, in Diplonema mitochondria, most transcript maturation-steps proceed independently from one another in the sense that the reaction at one extremity of the transcript is not influenced by the nature of the other extremity. This excludes a strictly linear, assembly line-like maturation pathway in this system. Parallelization, thought to accelerate this multi-step process, might be achieved by a molecular machine that combines all activities in one (‘processo-edito-spliceosome’), i.e. one that would properly position guiding factors relative to its catalytic domains and allow that the two extremities of a given transcript are sculpted in an independent fashion and in no particular order. The only steps where the nature of the ‘other’ end seems to matter in mt-LSU rRNA maturation of Diplonema is polyadenylation or deadenylation, which, according to the two above pathway hypotheses, appear to be the ‘rubbish’ or ‘quality’ stamps of molecules.
In contrast to the here proposed integrated multi-functional complex in Diplonema mitochondria, the current view of kinetoplastids mitochondrial (m)RNA maturation postulates the sequential action of two major complexes, each having dedicated functions. The RNA editing core complex conducts cleavage of pre-mRNA at the editing site, removal or addition of Us and resealing of the transcript, whereas the mitochondrial RNA-binding complex 1 recruits guide RNAs and interfaces with gRNA processing and mRNA tailing [reviewed in (30)].
Antisense transcripts
We detected two types of antisense RNAs, anti-rnl-mono-modules and anti-mt-LSU rRNA transcripts. Anti-mono-module transcripts most likely arise by bidirectional transcription of chromosomes, as the promoter(s) in the shared region must accommodate modules encoded on the plus and the minus strand [see Figure 1B and (8)]. The observed higher steady-state concentration of the rnl sense transcript could be achieved by either an elevated transcription rate in sense direction or faster degradation of antisense transcripts. Either scenario calls for controlled strand-dependent transcript regulation, whose nature is yet to be unraveled.
The origin of anti-mt-LSU rRNA is less obvious, as a corresponding gene has not been detected. Either the gene is encoded in yet unsequenced portions of the mitochondrial or nuclear genomes or alternatively, no such gene exists in Diplonema. The antisense RNA might be transcribed from mature mt-LSU rRNA and inherited epigenetically from generation to generation. Antisense transcription templated by mt-LSU rRNA would require an RNA-dependent RNA polymerase (RdRp). As this activity has broad taxonomic distribution (31–35), the Diplonema nuclear genome might well encode a mitochondrion-targeted enzyme. Epigenetic inheritance of RNAs has precedents as well, for example, in ciliates (36) and C. elegans (37), where RNAs transmitted to daughter cells are involved in genome rearrangement and antiviral response, respectively.
Diplonema mt-LSU rRNA is extraordinarily short and derived
With only ∼910 nt, mt-LSU rRNA of Diplonema is among the smallest known, but still longer than that of certain nematodes, bryophytes and rotifers [529–729 nt; (38–41)]. It is the module-1 portion (534 nt) that is substantially shorter in Diplonema (and even more in the aforementioned animals) compared with counterparts from other euglenozoans and heteroloboseans [∼730 nt in kinetoplastids (e.g. GenBank accession no. NC_000894), >800 nt in Euglena (42), and 1485 nt in Naegleria (GenBank accession no. AF288092)].
As stated in the ‘Results’ section, folding the Diplonema rnl sequence into the consensus 5′-half 2° structure of mt-LSU rRNA is challenging. The problems include low conservation, absence of comparative data from close relatives and the possibility to build numerous alternative structures with this G+C-rich sequence, making selection of the single most likely model difficult. For illustration, one of the multiple equally probable structure models is shown in Supplementary Figure S7. With the availability of mt-LSU rRNA sequences from other diplonemids, it should become feasible to model confidently this portion of the molecule. Finally, it is conceivable albeit not likely, that a separate 5′ mt-LSU rRNA piece exists. Whereas the mitoribosome-enriched fraction analysed here contains a highly abundant 350-nt molecule (not shown), this RNA species lacks 2° structure motifs typical for mt-LSU rRNA, but instead displays remote similarity to phylogenetically conserved mt-SSU rRNA signatures [helices h18, h44 and h45; numbering as in (28)]. Whether this molecule represents indeed mt-SSU RNA is currently uncertain, because its 5′ tier consists virtually exclusively of Gs and Ts impeding meaningful secondary structure modeling, and its length is much shorter than ever reported for this rRNA. These issues could be re-examined rigorously once a protocol is available for isolating pure mitoribosomes from Diplonema and by sequencing a mitoribosomal library prepared specifically for small RNAs.
The 3′ half is the most conserved portion of all mt-LSU rRNAs. The corresponding 2° structure of Diplonema mt-LSU rRNA was modeled based on comparison with the mitochondrial consensus structure—the homologs from kinetoplastids and E. gracilis are too divergent for a meaningful comparison of covariant residues. Overall, the fold of domains V and IV is less deviant in Diplonema than in kinetoplastids, where the PTC-abutting helices H89 and H91 are considerably truncated. The absent masses of these two helices appear to be the cause of the positionally shifted α-sarcin/ricin loop (H95) toward the PTC (19), seen in the cryo-electron microscopy map of the mitoribosome from Leishmania tarentolae. We posit that the extremely short single-stranded segment between helices H73 and H95 in Diplonema mt-LSU rRNA induces an even more pronounced overall shift of H95 together with H89 and H91 and stronger domain V/IV compaction in the mitoribosome.
Role of extensive U-‘insertion’ editing in mt-LSU rRNA from Diplonema
To our knowledge, LSU rRNA of Diplonema mitochondria is the only example of a massively edited rRNA and represents the most extensive editing ever observed at a single site. Other cases of rRNA editing include sparse nucleotide insertion or substitutions that mostly restore secondary structure elements and conserved sequence motifs (43,44). In kinetoplastids, mt-rRNAs are virtually never edited. Eukaryotic cytosolic rRNAs are chemically modified [guided by small nucleolar RNAs; ref. (45)], but sensu stricto RNA editing has not been described for these molecules.
The region occupied by the U-tract in our model of Diplonema mt-LSU rRNA corresponds to the 3′ half of H61 in the E. coli structure, a helix that plays an important role in the ribosome. The part of this helix abutting H64 ensures correct positioning of the SSU/LSU-connecting domain IV (14,16,28), whereas the part adjacent to H72 is deeply embedded in the ribosome (as are H72 and H73).
According to a most recent 2° structure model of LSU rRNA (46), the segment corresponding to the six 3′ terminal nucleotides in the U-tract together with the three first nucleotides in module 2 constitute the 3′ half of the newly proposed helix H26a. The corresponding 5′ half of this helix is a stretch traditionally modeled as single-strand connecting H26 and H47 in domain II. Helix 26a is thought to be a pivotal structural element of the proposed core domain 0, to which the traditional domains I-VI would be rooted. With the U-tract not only substituting the 3′ half of H61 but also being part of H26a, RNA editing of Diplonema mt-LSU rRNA would be function-critical.
ACCESSION NUMBERS
GenBank KF633465, KF633466, KF633467, KF633468.
FUNDING
Canadian Institute for Health Research [CIHR, grant MOP-79309; to G.B.]; Ph. D. scholarship from the Programme Canadien de Bourses de la Francophonie (PCBF; to G.N.K.). Funding for open access charge: Canadian Institute for Health Research.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors acknowledge M. Aoulad-Aissa for excellent technical assistance and thank M.W. Gray (Dalhousie University, Halifax, Canada) for help in modeling the rRNA secondary structure and B.F. Lang (Université de Montreal) for reading the article. G.B. designed and supervised the study. G.N.K. conducted RT-PCR. Both G.N.K. and M.V prepared RNA samples for RNA-Seq. Isolation of mitochondria and mitoribosomes, secondary structure analyses and cytosolic rRNA depletion was performed by M.V. Preliminary RNA-Seq data analysis was conducted by G.B. and M.V. S.M. performed the detailed data analyses including read mapping and statistics. All authors contributed to the final manuscript version.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments