





Abstract
Deregulation of mini-chromosome maintenance (MCM) proteins is associated with genomic instability and cancer. MCM complexes are recruited to replication origins for genome duplication. Paradoxically, MCM proteins are in excess than the number of origins and are associated with chromatin regions away from the origins during G1 and S phases. Here, we report an unusually wide left-handed filament structure for an archaeal MCM, as determined by X-ray and electron microscopy. The crystal structure reveals that an α-helix bundle formed between two neighboring subunits plays a critical role in filament formation. The filament has a remarkably strong electro-positive surface spiraling along the inner filament channel for DNA binding. We show that this MCM filament binding to DNA causes dramatic DNA topology change. This newly identified function of MCM to change DNA topology may imply a wider functional role for MCM in DNA metabolisms beyond helicase function. Finally, using yeast genetics, we show that the inter-subunit interactions, important for MCM filament formation, play a role for cell growth and survival.
INTRODUCTION
All organisms must duplicate their genome to provide each new cell with a full complement of genetic information prior to mitotic division. When a cell enters S-phase, double-stranded genomic DNA is separated into single strands to be copied into sister chromatids. This process is tightly regulated and highly coordinated to ensure the high-fidelity replication of the genome.
Eukaryotic and archaeal chromosomal DNA replication are initiated by the stepwise assembly of pre-replicative complexes (pre-RCs), composed of the origin recognition complex (ORC), Cdc6 and mini-chromosome maintenance (MCM) complexes ( 1 , 2 ). These components cooperatively catalyze the initiation of replication at the origin. Upon origin firing, the MCM ring complex acts as an ATP-dependent DNA helicase to unwind the genome, opening double-stranded DNA (dsDNA) into single-stranded DNA (ssDNA) templates ( 3 ).
Eukaryotes express six essential homologous MCM proteins (MCM2-7) that form hexamers and double hexamers in vitro ( 4–6 ). Archaeal genomes also encode MCM genes with sequence and structural homology to eukaryotic MCM2-7 ( 7 ). However, many archaea express only a single MCM subunit, which forms homo-oligomeric complexes with the same function as the eukaryotic MCM complexes ( 8–10 ).
In G1 and leading up to S-phase of the eukaryotic cell cycle, multiple MCM2-7 hetero-hexamers are recruited to each pre-RC at the origin and spread to nearby chromatin ( 11–13 ). Mutations that limit pre-RC to only a single iteration of MCM recruitment are not viable, which suggests that the recruitment of many MCM proteins is a requirement for proper pre-RC function ( 11 , 14–17 ). This is puzzling as only one or two MCM hexameric rings are sufficient to unwind DNA ( 8 , 18 , 19 ).
How MCM functions when bound to chromatin prior to and during initiation has been the subject of much interest, though results often raise more questions than answers. The chromatin-bound MCM complexes are categorized into two biochemically distinguishable subgroups. One is the salt-stable ‘loaded’ complexes that are bound tightly to the origin, likely locked onto DNA as hexamer or a double hexamer as the active helicase form in vivo ( 4 , 6 ). The other is the salt-sensitive ‘associated’ complexes and not specifically located at origins, which accounts for the majority of the MCM proteins. These MCM proteins distal from the origin may have different biological function(s) ( 12 , 20–22 ). MCM proteins have been detected associating with large regions of unreplicated chromatin during G1 and early S-phase ( 13 ). Despite the large number of MCM proteins in the nucleus, reduction in MCM gene dosage causes genome instability, demonstrating the importance of maintaining MCM protein level for cell survival ( 23–26 ). These peculiarities are part of what has been termed the ‘MCM paradox’ [reviewed in ( 27 ) and references therein].
In this study, we report a crystal structure of the near full-length MCM from the archaeon Sulfolobus solfataricus (sso) assembled as a wide helical filament with a large channel along the filament axis. We also have verified the formation of the same filament structure in solution using electron microscopy (EM). The crystal structure revealed a structural element critical for filament formation, which has been confirmed by structure-guided mutagenesis and EM studies. Furthermore, we used yeast genetics to show that this structural element, important for filament formation, is critical for cell growth and survival.
METHODS
ssoMCM cloning and MCM purification
Wild-type (WT) full-length ssoMCM containing residues 1–686 are cloned as His-tagged fusion proteins in vectors pGEX-6P-1. Mutants of MCM were made using either Quikchange or PIPE ( 28 ) and expressed as previously described ( 9 ). Full-length WT and mutant MCM proteins were purified from Escherichia coli grown at 25°C for 18 h. Cells were lysed by French-press and centrifuged at 10 000 rpm for 1 h. Supernatant was passed over nickel resin and washed with 10 column volumes of high-salt Buffer A (1 M NaCl, 50 mM HEPES, pH 7.5). MCM was eluted with five column volumes of Buffer A supplemented with 250 mM imidazole. Eluted MCM fractions were diluted or dialyzed to 100 mM NaCl and passed over a Resource Q column. MCM was eluted from the Resource Q by a 10-column volume salt gradient from 50 to 500 mM NaCl. Resource Q fractions were diluted to 100 mM NaCl and passed over a heparin column. MCM was eluted from the column with a 50–1000 mM NaCl linear gradient. Heparin fractions were collected, concentrated to 1 ml in 250 mM NaCl, 20 mM HEPES, pH 7.5, 2 mM DTT and then further purified by Superdex 200 column chromatography. Superdex 200 fractions were concentrated to 30–50 mg/ml and flash-frozen in liquid nitrogen.
Crystallization and structure determination
To assemble a homogeneous complex, MCM (50 mg/ml) was initially incubated with a 61-bp dsDNA with a 3′-T overhangs on each strand and dialyzed from 2 M NaCl to 50 mM overnight at 4°C for crystallization trays. DNA strands for annealing into the 61-bp dsDNA (strand1:tagctattagagcttggtttaattatacaaactcaatatttttcttttttccttcctttat, strand2:tatcgataatctcgaaccaaattaatatgtttgagttataaaaagaaaaaagaaggaaat) were purified using a MonoQ column (GE), annealed overnight and further purified on a Superdex 200 gel filtration column (GE). Hanging-drop crystal trays were set up at 4°C. Small but long needle crystals grew to dimensions of ∼30 × 40 × 200 µm at 4°C in 2 µl drops with ratios of 1–1.5 µl MCM to 1–0.5 µl of crystallization buffer (7.5% isopropanol, 420 mM NaSO 4 and 20 mM HEPES, pH 6.75). Crystals were harvested, cryoprotected in 420 mM LiSO 4 , 25% PEG 400, 25 mM HEPES, pH 6.75 and flash-frozen in liquid nitrogen. Data were collected at APS beamline GMCA/CAT 23-ID (and 19-ID), using the 5-µm microbeam vectored over the length of the crystal needles.
Diffraction spots were detected up to 3.8 Å with 30-s exposure at 23-ID. However, a 30-s exposure time essentially kills the crystal diffraction. As a result, we used a combination of 5-s exposure time, microbeam, translations every three to five exposures along the needle crystals and multiple crystals to collect data sufficient for obtaining the highest resolution data set to 4.29 Å. Later on, a different crystallization condition (0.1M HEPES, pH7.0, 18% MPD) yielded a crystal form that has the same space group and same filament structure, but are more resistant to radiation damage up to 120-s exposure time in 23-ID-D and we are able to extend the resolution of the structure to 4.1 Å ( Supplementary Table S1 ). The crystal is in space group P21, with five subunits per asymmetric unit.
The structure was determined by molecular replacement using 3F9V with loops trimmed off as a search model using the Phaser program in the Phenix suite ( 29 ). R -free flags (5%) were set at this point and communicated between Phenix and CNS as needed. The initial model was rebuilt as a polyalanine structure in Coot with reiterative rounds of solvent flipping and solvent flattening using CNS, density modification and 5-fold non-crystallographic symmetry (NCS) averaging from the CCP4 suite or CNS. Once the main chain was properly placed, side chains were added and refined using CNS 1.3 deformable elastic network (DEN) low-resolution refinement strategies ( 30 ). The reference model for DEN restraints was a hybrid of the N-terminal ssoMCM and homology model of mkaMCM threaded through the structure model from 3F9V. The DEN refinement improved the phases at this resolution, as evident by the improved density and correct side-chain positioning, which is the case for the refinement of another large complex structure with 5.0 Å data ( 31 ). After DEN refinement, the model was further rebuilt by reiterative rounds of density modification in CNS and model building in Coot using B-factor sharpening and Density Modification in CNS ( 32 ). Further model improvements were made using phenix.refine in Phenix version 1.7.2-863 ( 29 ). The 5-fold NCS restraints were imposed at all stages. Final refinements with geometry restraints were done using Refmac and phenix.refine imposing secondary structure restraints, TLS restraints, Ramachandaran restraints and 5-fold NCS to a final model R -work/ R -free 34.38/35.25 and Ramachandaran statistics 85.3% in the most favored and 0.8% outliers. Identical set of reflections were used for R -free at all stages of refinement ( Supplementary Table S1 ).
Once the model was refined satisfactorily, validation and final statistics were acquired using Molprobity server ( http://molprobity.biochem.duke.edu/ ) and phenix.validate ( 29 ) ( Supplementary Figure S1d ). Our structure fell into the 83rd percentile among structures from 3.25 to 4.36 Å resolution with 0 bad bonds, 0 bad angles and excellent statistics among structures of similar resolution. N-terminal residues 1–6 are not included in the structure as no density was seen. Density for the flexible C-terminal 88 residue wing-helix domain of ssoMCM was also not visible and thus the final model contains residues 7–598 for each of the five subunits in the asymmetric unit or half of one helical turn. Although broken density was observed within the central channel, we were unable to build a model for dsDNA into the filament.
Transmission electron microscopy and electron tomography
Negatively stained samples were prepared by placing a small drop (∼4 μl) of sample solution onto a glow-discharged carbon-coated copper grid. For transmission electron microscopy (TEM) and electron tomography (ET), 200-mesh and 100/400 slotted grids were used, respectively. After a period of 1 min at room temperature, the sample was blotted and a drop of 2.5% uranyl acetate solution was immediately placed on the grid. After staining for 1 min the drop was blotted off, the grid was washed four times with the same stain solution and then allowed to air dry.
The stained samples were visualized with an FEI Tecnai F20 transmission electron microscope with an accelerating voltage of 200 kV. The samples were imaged at 50 000–100 000× with an underfocus value of 3 μm. Tomography tilt series were taken using the FEI Batch Tomography software with a tilt range from −70° to +70°. The tilt series were recorded on a 16-megapixel TVIPS CCD camera.
Alignment of the tilt series was performed using the ‘etomo’ tomography processing software from the Imod package ( 33 ). The steps included removing X-ray hot spots, rough alignment by cross-correlation and fine alignment by patch tracking. The aligned tilt series were then used to make three-dimensional (3D) reconstructions using GPU-based SIRT (simultaneous iterative reconstruction technique) reconstruction implemented in ‘Inspect3D’ (FEI). Slices from the 3D tomography maps were displayed using the ‘slicer’ tool within the ‘3dmod’ program of the Imod package. Amira (Visage Imaging GmbH, http://www.amira.com/ ) was used to segment and to create volume renderings of the 3D density maps of the filaments.
Consurf, sequence alignments and electrostatics analysis
Conserved region alignments and coloring were done through the consurf server ( http://consurftest.tau.ac.il/ ) ( 34 , 35 ). Multiple protein alignments were done with the ClustalW server ( http://www.ebi.ac.uk/Tools/msa/clustalw2/ ) ( 36 , 37 ). Electrostatics of MCM filament structure were calculated using the APBS plug-in as part of Pymol 1.4 ( 38 ).
Assays for DNA binding, helicase activity and oligomerization
A range of concentrations of purified MCM protein was incubated with 0.2 µM 61-bp dsDNA in binding buffer (10 mM Tris pH 8.0, 50 mM NaCl) at room temperature for 30 min. 10 µl reactions with 5% glycerol were electrophoresed (in 0.5% agarose, 0.5× TBE) at 90 V for 40 min. After electrophoresis, gels were stained in ethidium bromide and visualized under UV light. The DNA binding was quantified using Quantity One software. Helicase assays were performed as previously described ( 39 ).
For oligomerization assay, purified MCM and MCM mutants were dialyzed in a buffer containing 10 mM HEPES, pH 7.5, 50 mM NaCl and 2 mM DTT. 500 µg protein in 100 µl was analyzed by gel filtration chromatography on an analytical Superose 6 column at 4°C in a buffer containing 10 mM HEPES, pH 7.5, 250 mM and 2 mM DTT.
DNA topology footprints
MCM was dialyzed into a buffer containing 50 mM NaCl and 10 mM Tris 8 and diluted to 6 mg/ml. A 15-µl reaction solution containing 500 ng of plasmid DNA (pBR233, New England Biolabs) and MCM was incubated at room temperature for 30 min. About 5 U of E. coli Topoisomerase I were added to the reaction and incubated for 3 h at 37°C. A quantity of 25 mM EDTA and 5% SDS were added to stop the reaction which was then deproteinated by addition of proteinase K. Samples were run on a 1% agarose gel either with or without 1.4 µg/ml of the intercalator chloroquine added.
Yeast plasmid and mutation construction for yeast genetics
Nucleotide changes to introduce the point mutation and the internal deletion were created using the Phusion site-directed mutagenesis kit (New England Biolabs) following the manufacturer’s instructions. The constructs were sequenced (Laragen and Genewiz) to confirm the presence of the mutation and to confirm that PCR mutagenesis did not introduce additional mutations.
Yeast strains and manipulations
Fission yeast strains used for the study were grown in yeast extract plus supplements or in Edinburgh minimal medium (EMM) with appropriate supplements. In this work, WT strain refers to FY 261 (h+ can1-1 leu1-32 ade6-M216 ura4-D18) and mcm4 ts refers to FY 784 (h+ cdc21-M68 ura4-D18 leu1-32 ade6-M210 can1-1).
Yeast plasmids used for this work are derived from REP82X and contain a ura4+ marker, the weakest nmt promoter and a HA tag at the C-terminus. pNR29 served as the WT (positive control), whereas pSLF372 served as the vector only (negative control). Transformations were carried out by electroporation and candidates were selected on EMM media lacking uracil, which also contained 15 µm thiamine for full repression of the nmt promoter. Plates were allowed to grow at 25°C for ∼7 days following the transformation or until colonies were visible.
Once colonies were present, complementation analyses of WT and mcm4 temperature-sensitive strains were carried out by streaking six independent colonies from each transformation on EMM media lacking uracil, supplemented with 15 µM thiamine and incubated at 25, 32 and 36°C for 7 days. Plates were scanned on Days 3, 5 and 7. The represented figures are following 5 days of incubation at the designated temperatures.
RESULTS
MCM filament crystal structure
Using a full-length MCM protein from the archeaon ssoMCM, crystals were obtained in the presence of 61-bp dsDNA, but not in the absence of dsDNA, suggesting dsDNA is an integral part of the crystal, which is also confirmed from agarose gel analysis of the solubilized crystals. The crystal structure is an unusually wide left-handed filament ( Figure 1 a), with 10 subunits per helical turn. Each asymmetric unit (asu) contains five MCM subunits. Despite the presence of DNA in the crystals, we can only detect non-featured extra density along the filament channel that could be accounted by the bound DNA with some freedom of rotation/sliding in each asu. Although full-length MCM protein was used in crystallization, density of a small wing helix-like domain at the C-terminus was not visible, and thus the final model contains MCM protein residues 7–598 and is missing residues 599–686.

Overall features of the MCM filament structure. ( a ) Surface representation of the crystal structure of a filament of full-length ssoMCM. The left-handed filament contains 10 subunits per turn. ( b ) Top–down view through the filament central channel, with dimensions indicated. ( c ) View of a MCM monomer (ribbon in green) in the filament. Regions playing important roles in making contacts in the filament are labeled. ( d ) Monomer structure with a neighboring subunit in the filament shown in gray. The division of the N-domain and C-domain and the subdomains (A, B, C, N–C linkers, etc.) are indicated. Notable structural features are labeled: Nt-hp; PS1 (pre-sensor 1), EXT (external hairpin), PH3-lp (pre-helix 3 loop), P-hel (P-loop helix), P-lp (P-loop), H2I (helix-2 insert), ACL (allosteric communication loop). The ATP binding pocket is marked with a red asterisk.
Even though the resolution of the diffraction data goes to 4.1 Å, the recently developed refinement methods [DEN refinement ( 30 ), also see ‘Methods’ section] worked well to enable the confident placement of side chains in the electron density map ( Supplementary Figure S1a , Supplementary Data and Supplementary Data ), with excellent geometry and statistics ( Table 1 ). The filament structure reveals an outer diameter of ∼175 Å and a large inner channel opening of 90 Å ( Figure 1 a and b). The structure shows a narrow filament groove. Parallel to the narrow groove is a furrow on the filament outer surface ( Figure 1 a) with side channels formed between neighboring subunits which connect to the interior of the filament central channel ( Figure 1 a).
X-ray refinement statistics
| Resolution (Å) | 4.1 |
| Rwork / Rfree | 33.14/34.13 |
| Number of atoms | 23 590 |
| Protein | 23 585 |
| Ligand/ion | 5 |
| Water | |
| B-factors | |
| Protein | 197.65 |
| Ligand/ion | |
| Water | |
| RMSD | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.768 |
| Resolution (Å) | 4.1 |
| Rwork / Rfree | 33.14/34.13 |
| Number of atoms | 23 590 |
| Protein | 23 585 |
| Ligand/ion | 5 |
| Water | |
| B-factors | |
| Protein | 197.65 |
| Ligand/ion | |
| Water | |
| RMSD | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.768 |
X-ray refinement statistics
| Resolution (Å) | 4.1 |
| Rwork / Rfree | 33.14/34.13 |
| Number of atoms | 23 590 |
| Protein | 23 585 |
| Ligand/ion | 5 |
| Water | |
| B-factors | |
| Protein | 197.65 |
| Ligand/ion | |
| Water | |
| RMSD | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.768 |
| Resolution (Å) | 4.1 |
| Rwork / Rfree | 33.14/34.13 |
| Number of atoms | 23 590 |
| Protein | 23 585 |
| Ligand/ion | 5 |
| Water | |
| B-factors | |
| Protein | 197.65 |
| Ligand/ion | |
| Water | |
| RMSD | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.768 |
To exclude the possibility that the filaments are a crystallographic artifact, we used ET to detect the oligomeric state of MCM bound to dsDNA in solution. The same type of filaments were observed when MCM protein was pre-incubated with naked dsDNA in solution but not when incubated with ssDNA or in the absence of DNA ( Figure 2 a). The 3D ET reconstructions of filaments revealed that the filament groove and pitch, dimensions and handedness, matches those of the crystal structure well, as shown in Supplementary Movie S1 .

ET and TEM imaging of MCM–dsDNA filament. ( a ) Side-by-side comparison of the MCM filament crystal structure and ET reconstruction. The crystal structure is filtered to 8 Å (left) and a central slice from the ET reconstruction is shown to the right. Note that the shadow in the EM image makes it to appear wider. The superimposition of the crystal structure over the ET reconstruction image shows a well-matched groove dimension and handedness ( Supplementary Movie S1 ). ( b ) An ET reconstruction of MCM filament showing DNA protruding from the ends (also Supplementary Movie S2 for views of different sections). ( c ) Electron micrograph of WT MCM filament on 1000-bp linear dsDNA, forming a filament with 175 Å thickness, but a small fraction of it (bracket) showing filament with similar thickness obtained from a mutant shown in panel d. Approximately 1/100 of the wide filaments from WT MCM contains such a small portion of thin filament. ( d ) Electron micrograph of α5-linker mutant on 1000-bp linear dsDNA, exclusively forming a filament with 125 Å thickness. Unlike the WT MCM, this mutant cannot induce supercoiling of plasmid DNA (also compare Supplementary Figure 2a with c and Supplementary Figure 4a with Supplementary Data ). Black scale bars are 500 Å.
A prior EM study reported a very thin right-handed helical filament of a different archaeal MCM (mtMCM) ( 10 ). We were unable to detect any thin right-handed filament forms by EM using either mtMCM or ssoMCM, in varying buffer conditions, with or without DNA or added nucleotides ( 40 ). However, when we were revising this article, a left-handed lock washer structure (with filament arrangement) for MCM2-7 was published ( 41 ), providing an example for a left-handed structure for an initiator protein, such as MCM.
The structure of the MCM monomer in the filament is shown in Figure 1 d, which consists of two separable domains, an N-terminal domain (containing A, B and C subdomains) and the C-terminal domain [containing AAA+, α–helix 5 (α5) and α-subdomains], joined by a long linker (N–C linker). Despite a similar overall core fold to the previously published monomeric structure of ssoMCM [3F9V ( 9 )], the structure in the context of the filament has some obvious conformational differences, as reflected by an RMSD of 2.5 Å 2 for the superimposition of the two structures. Compared with the previous structure, the C-terminal domain is rotated ∼17° about the N–C linker and swung away from the side facing the central channel. Within the C-terminal domain, an α5 helix ( Figure 3 a) rotates dramatically (90° rotation) to take a different position and orientation, which has significant consequences in filament assembly. At the N-terminus, the zinc-bearing B subdomain also has some positional shifts, revealing a structural flexibility about the β-sheet bridging the B- and C-subdomains ( 8 , 9 ). Within the N-terminal half, another noteworthy difference from the previous ssoMCM structure is that the long N-terminal hairpin (Nt-hp) from the filament has a large shift to point in a different direction ( Figure 4 a), which alters the surface charge features dramatically, generating a spiraling charged surface differing from that of a hexamer with a horizontal changed surface within a ring ( Figure 4 b). The new conformations of these structural elements appear to be important for forming contacts in filament formation, as discussed below.

The helix α5 rotation and its interaction with the α-subdomain of a neighboring subunit in the filament structure. ( a ) C-domain structural alignment between the filament subunit (fila-ssoMCM, green) and the previously determined monomer structure (3F9V, mon-ssoMCM, yellow). Fila-MCM helix-5 (α5) is rotated 90° (red) relative to the mon-ssoMCM α5. ( b ) α5 (red) of one subunit (mon2) docks on the α-subdomain (blue) of a neighboring subunit (mon1), forming a four-helix bundle between mon1 and mon2 in the filament structure. The EXT hairpin (cyan) is just above the 4-helix bundle. ( c ) The side chains (in stick model) of some of the critical residues at the interface between α5 and α-subdomain as well as on the α5-linker for regulating filament formation ( Figure 2 d and text).
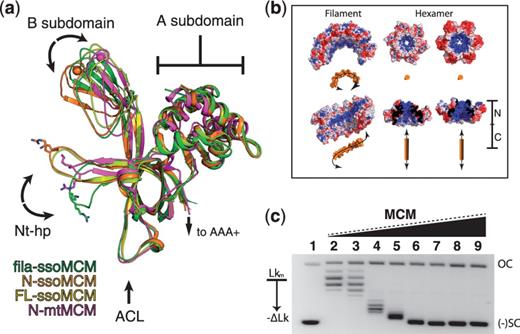
The N-terminal domain structural alignment and the DNA topology change induced by MCM. ( a ) Alignment of MCM N-terminal structures, showing the conformational change for the Nt-hp in the filament structure (green), with the Nt-hp pointing to a different direction to make contact with a neighboring subunit. Monomeric mon-ssoMCM in yellow (3F9V), N-ssoMCM in orange (2VL6), N-mtMCM in purple (1LTL). ( b ) Electrostatic patterning of ssoMCM filament (five subunits) compared with electrostatics of a hexamers of the N-terminal ssoMCM. The Nt-hp conformation in the filament structure reconstitutes an electro-positive surface (blue surface) to follow the spiral path in the filament. The orange objects represent the DNA with the expected DNA binding orientation on the filament or the hexamer. ( c ) DNA topology footprint with MCM, showing more negative supercoiling of DNA induced by increasing amount of MCM. Lane 1: negatively supercoiled pBR233 plasmid; lane 2: relaxed (nicked by topoisomerase) plasmid DNA and lanes 3–9: relaxed plasmid DNA incubated with increasing concentrations (1–10 µM) of MCM. OC, open circle; (−) SC, negative supercoiling; Lk, linker number.
dsDNA binds within the MCM filament channel
As only broken and not featured extra density was seen within the filament channel, which probably is due to the lack of fixed positioning of DNA, it was not possible to build the model for the 61-bp dsDNA in the co-crystal. Using ET to examine the MCM protein incubated with 1000-bp dsDNA, however, the dsDNA was detected with the same form of MCM filament, with dsDNA clearly visible protruding from both ends of the filament channel in EM tomographs ( Figure 2 b). The different Z-sections of DNA-bound filament are revealed in Supplementary Movie S2 , which shows that the filament features match those of the crystal structure.
Electrostatic analysis of the crystal structure revealed a remarkably charged inner surface that is highly electro-positive and forms a long, continuous ‘blue’ (electro-positive) strip along the filament inner channel surface ( Figure 5 a–d), immediately suggesting a role in DNA binding. Three pairs of residues, K246/R247 of one subunit, and R379/K381 and K408/R410 of an adjacent subunit, cluster on the filament channel interface to form this electro-positive strip in the filament ( Figure 5 c and d). To confirm that this electro-positive surface of the filament binds DNA, we mutated each residue pair to Ala and assayed their DNA-binding activity. As predicted, K408A/R410A mutant completely abolished dsDNA binding ( Figure 5 e). R379A/K381A and K246A/R247A mutants individually caused slight decrease in dsDNA affinity, and when combined dsDNA binding was completely abolished ( Figure 5 e). These results confirm the DNA-binding role for this spiral electro-positive strip on the filament channel surface. Examination of these mutants by EM revealed no filament formation, further illustrating the need for binding DNA for the filament formation.

The strong electro-positive ‘strip’ along the helical filament inner surface for DNA binding. ( a ) Spiral positively charged electrostatic pattern (the blue strip) along the inner wall of the helical MCM filament. ( b ) End view of the filament looking down the filament central axis, showing the spiral electrostatic pattern (the blue strip) down the central channel. ( c ) A half turn of the filament (five subunits in one asymmetric unit or asu), showing the electrostatic potential surface at −1 (red mesh) and 1 (blue surface) Kt/e. ( d ) Arrangement of the six positively charge residues (or three pairs) on the electro-positive strip. ( e ) Mutational effects of residues on the positively charged strip for dsDNA binding. ( f ) Top–down view along the filament channel, showing the periodicity (26 Å) of positively charged residues of each subunit and the length (153 Å) of the electro-positive surface along the filament in one asu (five subunits), which match the groove periodicity (24.6 Å) and the length (150 Å) of a 61-bp DNA in A-form, as shown in panel g. ( g ) The 61-bp A-form dsDNA modeled on to the electro-positive strip over a path of five subunits of the filament, showing charged protein surface and matching periodicity of groove spacing of the A-form DNA. ND, none detected.
MCM filament induces negative DNA supercoils
Knowing that the left-handed MCM filament can bind linear dsDNA in packed crystal and in solution, we subsequently used EM to examine MCM binding to closed circular plasmid dsDNA. The result revealed that the plasmid DNA coated by WT MCM filaments became heavily supercoiled ( Supplementary Figure S2a ), indicating MCM can induce topological changes to DNA.
To further confirm that MCM generates and stabilizes negative supercoils, we used a topology footprinting assay to observe changes in DNA topology as described previously ( 42 , 43 ). In this assay, we added Topoisomerase I to plasmid DNA bound by various amount of MCM to nick the circular DNA backbone and relieve topological stress of supercoils introduced by MCM binding, allowing DNA to relax into the lowest energy topoisomer that was then stabilized by ligation. Proteins were then degraded by proteinase K to isolate the stabilized circular DNA with the altered linking number. The results demonstrated that MCM generated the negatively supercoiled topoisomers of plasmid DNA in a dose-dependent manner ( Figure 4 c).
To confirm that the observed DNA topology change induced by MCM is indeed negatively supercoiled, the same footprinting reactions were performed, but the reactions were analyzed on agarose gels containing chloroquine, as chloroquine can bind dsDNA and change the relative mobility of topoisomers depending on the negative or positive supercoiling, allowing a differentiation between negative and positive supercoiling ( 43 ). The result showed that presence of chloroquine on the agarose gel shifted the MCM-induced supercoiled DNA toward the sample wells relative to the chloroquine-free gels (compare Supplementary Figure 4b and Supplementary Data ), confirming that changes in DNA topology induced by MCM in the assay were due to a negative change in linking number (−ΔLk).
A closer examination of the filament crystal structure revealed that the previously mentioned positively charged residue pairs (such as K246/R247) critical for DNA binding are spaced periodically ∼26 Å apart along the left-handed electro-positive strip ( Figure 5 f), which is similar to the groove periodicity of A-form DNA (24.6 Å/turn, ∼150 Å in length for a 61-bp DNA), but very different from that of B-form DNA (34.5 Å/turn, ∼210 Å in length for a 61-bp DNA). Although we cannot confirm that A-form DNA is indeed present due to a lack of defined DNA electron density in the crystal structure, we used molecular modeling to position the 61-bp DNA on the electro-positive strip of the filament, which would require a transition from B-form to A-form to fit both the periodicity of the 26 Å and the 153 Å unit length per asu ( Figure 5 f and g). The transition from B-form DNA to A-form requires untwisting (or net negative twist) of the right-handed duplex DNA, loosening the double helix. Such a local loosening of the right-handed double-helix introduced through binding to the left-handed MCM filament will generate supercoiling of a circular plasmid DNA in order to compensate for the local untwisting of the duplex, thus providing a structural basis for the observed supercoiling of DNA induced by MCM.
MCM helix α5 regulates oligomerization
Spatial alignment of the AAA+ subdomains with the previously solved ssoMCM monomer structure ( 9 ) revealed a 90° rotation of α5 ( Figure 3 a). This rotation brings α5 close to the three helices of the α-subdomain of a neighboring subunit, allowing α5 to form an inter-subunit four helix bundle with the α-subdomain in the filament structure ( Figure 3 b and c). Linkers flanking α5 (α5-linkers) contain Gly and Pro, which can provide the flexibility as well as rigidity of the linkers to enable α5 to have the large rotations necessary for interacting with the α-subdomain of another subunit in the filament form. Additionally, some residues at the binding interface between α5 and α-subdomain helices are highly conserved among archaeal and eukaryotic MCMs ( Supplementary Figure S5 ). Thus, we predicted that not only the Gly/Pro residues of the α5-linkers but also the conserved residues (such as F540, Supplementary Figure S7a ) at the interface between α5 and α-subdomain are important for proper inter-subunit interactions for filament formation. To test this, we created mutants of the conserved residues on the interface between α5 and α-subdomain (F540A mutant) and of the Gly and Pro residues on the α5-linker (G485P/G501P mutant). By gel filtration chromatography, the G485P/G501P and F540A mutant proteins eluted as smaller oligomers (with a molecular weight similar to a trimer) than the WT oligomeric form (a hexamer form) in 250 mM NaCl buffer ( Supplementary Figure S6a ), clearly indicating compromised subunit association, even though not a complete disruption.
We also examined whether these mutations would affect filament formation. By EM examination, we found that F540A mutant could not form any filaments, but formed only closed or open ring circular structures consisting of six to eight subunits. Surprisingly, when tested for helicase activity, F540A still showed significant amount of unwinding, with ∼50–60% of WT ( Supplementary Figure S7b and Supplementary Data ). As for the G485P/G501P mutant on the α5 - linker, EM study revealed that this mutant exclusively formed thinner filaments with diameters of ∼125 Å ( Figure 2 d), 50 Å narrower than the 175 Å filaments observed in WT MCM. After surveying the WT filaments extensively, we occasionally found very small portion of the large filaments of the WT (about once out of 100 filaments) to contain small portion of the ∼125 Å narrow filament (bracketed portion in Figure 2 c).
Unlike the wide filament that can spiral DNA along its helical electro-positive strip, the narrow filament formed by G485P/G501P mutant would only have sufficient space in the central channel to allow dsDNA to thread straight through the filament, generating little or no untwisting of the bound double helix, which would predict little or no DNA supercoiling to be induced by this mutant MCM protein. Indeed, EM observation of this mutant bound to circular plasmid DNA revealed very little supercoiling ( Supplementary Figure S2c ). This result is also corroborated by the DNA topology footprint assay demonstrating that little supercoiling was induced by the α5-linker mutant (G485P/G501P) ( Supplementary Figure S4a ).
Testing the biological relevance of α5–α subdomain interactions in vivo
We employed yeast genetics to test the biological relevance of the α5–α subdomain interactions important for filament formation using yeast Schizosaccharomyces pombe cells. As mentioned previously, the α-subdomain residue F540 at the interface with α5 is highly conserved as F/Y in MCM4 from several eukaryotic organisms ( Supplementary Figure S7a ), suggesting a conserved role for the equivalent F540 in other organisms in regulating α5 interactions for oligomerization. Previously, we showed that the F540A ssoMCM mutant could no longer form filament structure, but still retained 50–60% WT helicase activity ( Supplementary Figure 7b and Supplementary Data ). Therefore, we made the F540A equivalent mutation in S. pombe , which is Y751A on MCM4, to test the phenotype of this mutation in vivo (see ‘Methods’ section). We constructed plasmids containing mcm4 + WT gene or mcm4-Y751A mutant under control of a weakened thiamine-regulated nmt1 + promoter and transformed these into yeast strains with either WT mcm4 + or a temperature-sensitive allele mcm4ts which is not functional at 36°C.
Transformants were isolated in the presence of thiamine, which represses the promoter. WT cells expressing mcm4-Y751A mutant were able to form colonies at all temperatures and at all levels of expression. However, mcm4-Y751A was not able to complement mcm4ts at 36°C, indicating that the mutant MCM4 is not functional ( Supplementary Table S2 and Supplementary Figure S8 ). Interestingly, mcm4-Y751A is toxic in mcm4ts even at permissive temperatures when overproduced (minus-thiamine), as shown by very small colony size or failure to form colonies, compared with WT mcm4 + or the vector control ( Supplementary Figure S8 ). The cells are elongated, indicating a cell cycle delay or arrest. This phenotype indicates that an already attenuated protein ( mcm4ts , in this case) is outcompeted by a dominant lethal mutation ( mcm4-Y751A ) and suggests that the mutant may form non-functional assemblies with cellular MCM. This in vivo result using the equivalent F540A mutant suggests that the α5–α subdomain interactions are critical for cell survival.
In addition to the mutation of the conserve residue (equivalent of F540) on the α subdomain at the interface with α5 discussed above, we also want to examine the biological effect of mutating α5inin in yeast. As G485/G501 residues are poorly conserved in the α5 linker region among archaeal and eukaryotic MCMs, we generated a mutant by deleting the α5-helix (residues 690–704 of MCM4 from S. pombe ) to test the in vivo effect. When the α5-deletion mutant was expressed from the native mcm4 promoter in a plasmid, the mutant was not able to produce colonies following the transformation into the temperature-sensitive strain ( mcm4-ts , FY784 strain) under permissive temperature conditions, although the same α5 plasmid was able to successfully produce colonies in the WT strain (FY261 strain) at all three temperatures tested (data not shown). This indicates that the deletion mutant exhibits a synthetic dosage lethality phenotype.
When performing the same transformation experiments with the mutant derivatives expressed by the weakest nmt1 (‘81X’) promoter, we were able to successfully obtain colonies following transformation from both mutation types with WT and temperature-sensitive ( mcm4 ts) strains under low expression conditions (+thiamine), although the α5-deletion mutant still showed relatively smaller colonies in the mcm4ts host . Upon induction of the promoter (-thiamine), mcm4ts cells expressing either mutant were no longer able to produce colonies, again indicating a toxic phenotype for α5-deletion mutant ( Supplementary Figure S8 ).
These in vivo yeast genetics data obtained with the mutants designed to disrupting the α5 and α-domain interactions for MCM4 of S. pombe , together with the in vitro structural and biochemical data, suggest an important biological role for the interactions between the α5 and α-domain observed in the filament structure.
DISCUSSION
We described here an unusually wide filament structure of an archaeal MCM that exists both in crystals and solution. The formation of this large, left-handed filament requires the presence of dsDNA. Remarkably, MCM filament formed on DNA is capable of drastically changing DNA topology. This topology change is resulted from untwisting the bound dsDNA, changing the local helical parameters of the duplex from B-form to a looser isoform similar to A-form DNA. Furthermore, we identified the structure elements critical for filament formation through in vitro biochemical and EM studies and showed that mutation on these structural elements impacted MCM4 function and cell survival through in vivo genetics study in S. pombe yeast.
One possible functional implication of the new structural and biochemical data described here could be that MCM helical complexes bind and untwist DNA at origins to facilitate strand separation. Similarly, bacterial replication factor DnaA has been reported to ‘screw’ dsDNA within a right-handed helical filament, in a comparable manner to RecA, which is thought to catalyze origin-DNA melting and replication initiation ( 44 ). AAA+ family enzymes, including MCM, share a core fold similar to that of the RecA family of proteins and perform similar functions in DNA remodeling. The pitch and angle of RecA filament subunits can adjust in response to ligands such that a change in filament morphology occurs, and RecA and Rad51 (a eukaryotic RecA homolog) form both right- and left-handed helical filaments around DNA to facilitate disruption of the duplex ( 45 , 46 ).
The role for MCM filaments in replication initiation would fit well with the current understanding of pre-RC architecture. The central channel of this unusually wide filament is large enough to support a replication bubble and allow an MCM ring to clamp down on newly exposed ssDNA. This would also allow the filament to protect ssDNA from prolonged exposure to the cellular environment. In this case, the ORC could act as a nucleation point for filament growth ( 12 ). A modest conformational change via the α5 (and possibly other structural elements) would allow a transition of MCM from the filament form to the hexamer from that is the active helicase conformation. The in vitro reconstituted MCM double hexamer on dsDNA shows no DNA melting ( 6 ). One of the possible scenarios is that MCM initially loaded to the pre-RC as a double hexamer would transition to a filament to initiate origin melting, excluding the lagging strand and then closing back into a ring. However, the process in this scenario seems inefficient. Alternatively, the filament and double hexamer may exist together around origins, partitioning the role of melting and unwinding between the two structural forms of MCM. In any case, other replication factors are certainly involved in helicase activation. GINS and Cdc45 are reported to promote a switch from an open ‘lock washer’ of MCM2-7 to a planar ring ( 47 ). Given our data, it is possible that GINS and Cdc45 could also assume a role in switching MCM from a filament to a ring at melted origin DNA. It is noted that in addition to the previous right-handed ‘lock washer’ structure, a left-handed ‘lock washer’ form of MCM2-7 (with arrangement similar to a filament) was recently reported ( 41 ).
Notably, the MCM proteins that associate with unreplicated chromatin regions away from the origins (the MCM paradox) remain largely uncharacterized. So far, two chromatin-bound MCM populations have been described: an ‘associated’ population that can be removed by high salt, and a ‘loaded’ population that was shown to be origin-DNA-bound hexamers/double hexamers ( 13 , 20–23 ). As the MCM filaments described here could only be formed in relatively low salt concentration, this salt-sensitive nature of the filament form suggests that they may be categorized as the ‘associated’ population. Most studies focus on pre-RC assimilated MCM complexes at replication origins; however, a larger portion of MCM proteins is distributed over a wide range of unreplicated chromatin and distal to replication origins ( 13 ). It is an intriguing question whether the filament form of MCM partially accounts for those MCMs associated with chromatin regions away from the origins, as mentioned in the MCM paradox.
MCM’s capability of altering DNA topology suggests a possible mechanism for MCM proteins to regulate the diverse array of biological processes in addition to replication, such as chromatin remodeling and transcriptional regulation. Evidence suggests that MCM proteins (likely the origin distal MCM population) are involved in additional functions outside genome replication, such as transcription, chromatin remodeling and tumor suppression [reviewed by Forsburg ( 48 )]. It is conceivable that the ability of MCM filament formation on DNA to cause dramatic supercoiling of distal dsDNA would likely influence chromosome structure, which should impact the regulation of gene expression and other aspects of DNA metabolism. A more detailed structure/function understanding paired with further in vivo data will be required to resolve these exciting possibilities.
ACCESSION NUMBERS
The atomic model reported here has been deposited to the Protein Data Bank under the accession number 4FDG.
FUNDING
Funding for open access charge: NIH [GM080338 and AI055926 to X.S.C., GM071940 to Z.H.Z. and GM GM059321 to S.F.].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank the staff of USC NanoBiophysics core and the staff at synchrotron beamlines 23ID and 19ID at Argonne National laboratory, and 5.0.2, 8.2.1, 8.3.1 beamlines at Berkeley’s ALS for assistance with data collection. We are grateful to Pavel Alfone, Nat Echols of PHENIX group and Axel Brunger for early access to the program DEN (imbedded in CNS 1.3) designed for low-resolution refinement, Lauren Holden and Jared Peace for critical discussion and manuscript proofing and Lawrence Lee for helpful discussion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments