





Abstract
A complex of the three (αεθ) core subunits and the β2 sliding clamp is responsible for DNA synthesis by Pol III, the Escherichia coli chromosomal DNA replicase. The 1.7 Å crystal structure of a complex between the PHP domain of α (polymerase) and the C-terminal segment of ε (proofreading exonuclease) subunits shows that ε is attached to α at a site far from the polymerase active site. Both α and ε contain clamp-binding motifs (CBMs) that interact simultaneously with β2 in the polymerization mode of DNA replication by Pol III. Strengthening of both CBMs enables isolation of stable αεθ:β2 complexes. Nuclear magnetic resonance experiments with reconstituted αεθ:β2 demonstrate retention of high mobility of a segment of 22 residues in the linker that connects the exonuclease domain of ε with its α-binding segment. In spite of this, small-angle X-ray scattering data show that the isolated complex with strengthened CBMs has a compact, but still flexible, structure. Photo-crosslinking with p-benzoyl-L-phenylalanine incorporated at different sites in the α-PHP domain confirm the conformational variability of the tether. Structural models of the αεθ:β2 replicase complex with primer-template DNA combine all available structural data.
INTRODUCTION
The replicative DNA polymerases that synthesize the bulk of chromosomal DNA invariably contain two active sites. Primer DNA is extended processively by incorporation of nucleotides at the polymerase site, while mismatched nucleotides that are incorporated infrequently are removed at the 3′–5′ (proofreading) exonuclease site. In all proofreading polymerases, the two sites are spatially separated, so a mechanism is required to transfer the primer-template DNA from one site to the other when the polymerase needs to transit between the polymerization and proofreading modes (1).
The 17-subunit DNA polymerase III holoenzyme (Pol III HE) is the chromosomal replicase in Escherichia coli, and is composed of 10 different proteins (1). The three-subunit catalytic core contains one each of the α (1160 residues; 130 kDa), ε (243 residues; 27 kDa) and θ (8.8 kDa) subunits encoded by the dnaE, dnaQ and holE genes, respectively. The α subunit contains the polymerase active site (2,3), the ε subunit is responsible for the 3′–5′ proofreading exonuclease activity (4) and the θ subunit has no identified enzymatic activity (5). The αεθ core complex is active alone as a proofreading DNA polymerase, and co-purification of these three subunits demonstrates their tight physical association (6,7). Direct interactions between ε and α (8) and ε and θ (5) have been demonstrated, but no interaction has been detected between α and θ.
The αεθ core complex of E. coli DNA Pol III has proven unsuitable for X-ray crystallography, probably because crystallization is impeded by the highly flexible polypeptide linker connecting the globular N-terminal domain of ε with its C-terminal peptide that binds to α (9). The complex is also not amenable to detailed NMR studies because of its high molecular mass. However, 3D structures of subdomains of the complex have been determined by X-ray crystallography and in solution by NMR spectroscopy.
Two crystal structures of α have been reported: of a C-terminally truncated version (α917) of E. coli α (10) and of full-length Thermus aquaticus (Taq) α (11). Crystal structures are also available of the N-terminal globular domain of E. coli ε (ε186; Figure 1A), both alone (12) and in complex with HOT, the phage P1 homolog of θ (13,14). In addition, the structure of the ε186:θ complex was determined by NMR spectroscopy (15,16). Full-length ε includes an additional C-terminal segment of 57 residues, in the following referred to as εCTS (Figure 1A). Residues near the C terminus of the εCTS are known to be responsible for binding of α (9,17,18). The binding site of ε has been shown to be within the first 320 residues of α (19), which includes the N-terminal 270-residue PHP domain (20) (Figure 1C and D). Residues 190–212 of the εCTS had earlier been predicted to comprise an interdomain ‘Q-linker’ sequence (21). Solution NMR showed that residues in the εCTS are flexible and that those around the Q-linker (residues between Thr183 and Thr201, at least) remain so even in the context of the 165 kDa αεθ core complex. Moreover, the ε186 domain does not interact, even weakly, with α (9).
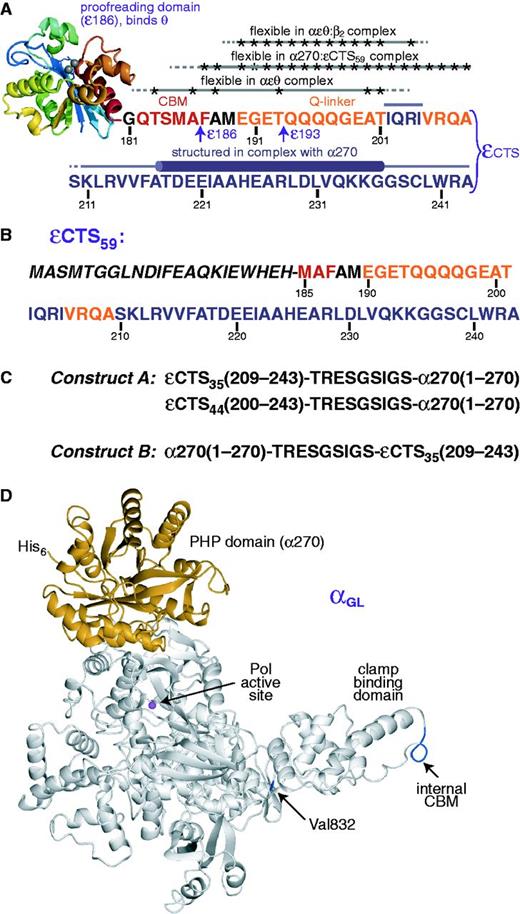
Protein constructs used in the present work. (A) The ε subunit is the proofreading 3′–5′ exonuclease of Pol III. It binds tightly to the α subunit via its C-terminal segment (εCTS). The globular domain of ε, ε186 (12) binds to the θ subunit (15,16). The εCTS comprises residues 181–243, including the β CBM at residues 182–187 (in red); the quadruple-mutant T183L/M185L/ A186P/F187L (εL) has a strengthened CBM (25). The α-binding site lies in the C-terminal part of the εCTS. At least 19 residues between ε186 and the α-binding site (i.e., between Thr183 and Thr201) remain flexible in the αεθ complex; Ala and Thr residues for which flexibility was established are indicated by asterisks (9). We refer to the segment comprising residues 190–209 (in orange) as the Q-linker (21). The present work reports: (i) that residues between Phe187 and Ala209 of εCTS (indicated by asterisks) remain flexible in the complex of εCTS59 and the PHP domain of α, α270 (Supplementary Table S3); (ii) the 3D structure of the complex between the εCTS (in purple) and the PHP domain of α (Figure 2), showing that part of the α-binding segment of ε forms a helix (residues 218–237) upon binding; and (iii) that residues between Glu190 and Arg204 (indicated by asterisks) remain flexible in a stabilized mutant version of the αεθ:β2 complex (Figure 4). The C-terminal residues of the ε186 and ε193 constructs are indicated. (B) Amino acid sequence of the εCTS59 construct. Residues 185–243 of ε are labeled with the sequence numbers of full-length ε. The preceding residues in italics are not part of ε; they comprise a T7 gene 10 tag (resulting in the N-terminal peptide MASMTG) for improved cell-free expression yields and a biotinylation site. (C) The first 270 residues of α (α270) contain the PHP domain. To determine the 3D structure of the εCTS in complex with α270, εCTS35 (residues 209–243) or εCTS44 (residues 200–243) was fused to either the N-terminus (constructs A; εCTS35–α270 and εCTS44–α270) or the C-terminus (construct B; α270–εCTS35) of α270. The amino acid sequence of the nine-residue linker is similar to one that had been determined to be flexible in another context (27,28). (D) Purification of the αεθ:β2 complex is difficult due to the limited affinity of β2 for the αεθ core. Increased affinity between α and β2 (for SAXS measurements) was achieved by changing residues 920–924 (internal CBM) from QADMF to QLDLF (26) to produce a mutant we refer to as αL (25), and then (for NMR measurements) introducing a further Val832Gly mutation (25,54) to yield αGL; αGL also contains an N-terminal His6 tag for purification. The figure shows the sites of Val832 and the internal CBM plotted in blue on the structure of the α subunit from T. aquaticus (11). The PHP domain is shown in orange and the Mg2+ ion in the active site in magenta.
The homodimeric β2 sliding clamp is responsible for processivity of DNA synthesis by the bacterial replisome. The β2 dimer forms a donut-shaped structure (22) that encircles (23) and slides on double-stranded (ds) DNA. Each protomer of β2 has a binding site for penta- or hexa-peptide clamp-binding motifs (CBMs) that are found in many proteins, including the δ subunit of the seven-subunit Pol III clamp loader and all five E. coli DNA polymerases (I–V) (24), among others (25). The interaction of CBMs with the β2 sliding clamp provides a specific way of recruiting requisite enzymes to 3′ ends of primer-template DNAs, and the Pol III α subunit has two CBMs: one is at the C-terminus and may be involved in polymerase recycling during lagging-strand replication; the other is an internal site that ensures processivity of the replicase (25,26).
In the present work, we identified the exact site and mode of binding of ε on α by determining the crystal structures of constructs where residues 209–243 and 200–243 of εCTS were fused to the N-terminus of the PHP domain of α (residues 1–270, referred to as α270) via a nine-residue linker that had been shown in another context to be flexible (27,28). A fortuitous PCR-generated mutation in α270, Leu21Pro, enabled crystallization. As the binding site of ε on α turned out to be far from the active site of the polymerase, we further investigated the tether between the N-terminal proofreading domain of ε and the C-terminal α-binding peptide. Building a model of the αεθ:β2 complex with primer-template DNA using this new information and published crystal and NMR structures of the various protein components indicates that the tether is sufficiently long to bring the exonuclease domain of ε closer to the active site of the α polymerase subunit when proofreading is required. The model positions two CBMs on the separate subunits of the β2 clamp, one being the internal CBM of α and the other the weakly binding CBM just beyond the C-terminus of the exonuclease domain of ε (25). Mutations of both CBMs for tighter binding to β produced an αεθ:β2 complex that was stable enough to be isolated chromatographically and used to collect small-angle X-ray scattering (SAXS) data that are consistent with the model. NMR measurements showed that the tether in the εCTS is nevertheless still flexible in a similar complex. In agreement with the model, p-benzoyl-L-phenylalanine (Bpa) residues site-specifically incorporated in α270 were found to afford photo-crosslinking to the εCTS, in particular at sites located closer to the active site of the polymerase. The remote attachment site of ε on α via a long flexible tether suggests that the mechanism for transition between polymerization and proofreading modes in Pol III is fundamentally different from those in other polymerases whose structures in both modes are known or can be reliably modeled (29,30).
MATERIALS AND METHODS
The 15N- and 15N/13C-labeled amino acids and a mixture of 15N/13C-labeled amino acids were from Cambridge Isotope Laboratories (Andover, MA, USA). p-Benzoyl-L-phenylalanine (Bpa) was from Peptech (Burlington, MA, USA). All other standard reagents required for cell-free protein synthesis were as described previously (31,32). New plasmids for overproduction of proteins or their cell-free synthesis were derivatives of the T7 promoter vectors pETMCSI, pETMCSII or pETMSCIII (33) and were constructed by standard methods, usually involving restriction digestion of PCR products and their insertion between corresponding sites in appropriate vectors (see Supplementary Methods for full details). Inserts in all plasmids were confirmed by nucleotide sequence determination.
In vivo protein expression and purification
The Pol III subunits α, θ (9), αL and εL (25) and the β2 sliding clamp (34) were purified as described. The αLεLθ core complex was isolated essentially as described for wild-type core (25,35). 15N-εL, 15N,13C-ε186 and 15N,13C-ε193 were expressed in vivo in M9 minimal medium containing 15NH4Cl and/or 13C-glucose; 15N,13C-ε186, 15N,13C-ε193 and their complexes with θ were purified essentially as described for ε186 and the ε186:θ complex, respectively (36). The εCTS–α270 fusion proteins (constructs A; Figure 1C) and His6-αGL (Figure 1D) were expressed in vivo and purified as described in Supplementary Methods. BpaRS was as described (31). Protein concentrations were determined spectrophotometrically using calculated values (37) of ε280.
Isotope labeling of α270 and α270 or εCTS in the α270:εCTS complex
Plasmid pKO1367 was used at a concentration of 16 µg ml−1 for cell-free synthesis of α270-His6 in 0.6 ml reaction mixtures at 30°C overnight. Five 15N-labeled α270-His6 samples were prepared following the combinatorial labeling scheme and reaction conditions described previously (32,38–40). The soluble fraction of α270-His6 was purified using ProPur IMAC Mini Ni-spin columns (Nalgene Nunc, USA), and the purified protein was dialysed against 2l of NMR buffer (20 mM Tris.HCl, pH 7.0, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol) and concentrated to a final volume of about 0.2 ml using Millipore Ultra-4 centrifugal filters (MWCO 10 kDa). D2O was added to a final concentration of 10% (v/v) prior to NMR measurements. In the same way, five samples of combinatorially 13C- and uniformly 15N-labeled α270-His6 were made by cell-free synthesis, using the requisite mixtures of isotope labeled amino acids (41). For improved sensitivity in the NMR experiments, two 0.6 ml reactions were pooled for each sample. Uniformly 13C/15N-labeled α270-His6 was made by cell-free synthesis in two 0.6 ml reactions, using a mixture of labeled amino acids as described (42).
Plasmid pKO1422 was used at 16 µg ml−1 for cell-free synthesis of εCTS (Figure 1B). All samples of the α270-His6:εCTS complex were purified and prepared for NMR as described above for α270-His6. One set of five samples contained combinatorially 15N-labeled α270 in complex with unlabeled εCTS; they were made by cell-free synthesis of εCTS in the presence of the combinatorially 15N-labeled α270-His6 samples described above. A second set of five samples contained combinatorially 15N-labeled εCTS in complex with unlabeled α270-His6; the εCTS was produced by cell-free synthesis in the presence of unlabeled α270-His6, which had itself been synthesized in a separate cell-free reaction. A third set of five samples contained combinatorially 13C- and uniformly 15N-labeled εCTS in the presence of separately purified and unlabeled α270-His6. Cell-free synthesis of these five samples used two 0.6 ml reaction mixtures.
NMR spectroscopy
All NMR spectra were recorded at 25°C using Bruker 600 and 800 MHz NMR spectrometers equipped with cryoprobes, using 200 µl solutions in 3 mm sample tubes. 15N-HSQC spectra used t1max = 32 ms, t2max = 102 ms and total recording times of 1–13 h. 2D HN(CO) spectra and 3D HN(CO)CA and HNCA spectra were recorded in 20–24 h per spectrum. D2O was added to all samples to a final concentration of 10% (v/v) prior to NMR measurements.
Crystallography
The εCTS35–α270(L21P) and εCTS44–α270(L21P) proteins were concentrated to 9.5–10 mg ml−1, respectively, by precipitation with ammonium sulfate (0.35 g ml−1); the pellets were dissolved in and extensively dialysed against 10 mM Tris.HCl (pH 7.6), 1 mM EDTA, 1 mM dithiothreiotol, 0.1 M NaCl. The crystals used for data collection were grown at 4°C in sitting drops with 4.5 µl of protein mixed with an equal volume of reservoir solution of 0.1 M Tris (pH 8.4), 0.2 M MgCl2, 3 mM tris(carboxyethyl)phosphine (TCEP), 16% (w/v) PEG 3350. Rectangular prisms 300–400 µm in length appeared within 3–4 days. They were cryoprotected by two transfers (5 min each) in reservoir solution supplemented with 15% (w/v) PEG 400 before being frozen for data collection at 100 K. X-ray data were collected on Beamline MX1 [εCTS35–α270(L21P)] or MX2 [εCTS44–α270(L21P)] at wavelengths of 0.96858 and 0.95369 Å, respectively.
The structure of εCTS35–α270(L21P) was solved at 1.7 Å resolution by molecular replacement, using the corresponding domain from the reported structure of E. coli α917 (10) as starting model to calculate phase information. The structure of εCTS44–α270(L21P) was subsequently solved at 2.15 Å resolution using the refined structure of εCTS35–α270(L21P) as starting model. Final models were obtained following cycles of refinement using REFMAC (43) and manually building using COOT (44). Data collection and refinement statistics are given in Supplementary Table S1.
DNA templates for site-directed Bpa mutants of α270
For site-specific incorporation of the unnatural amino acid Bpa into α270, amber stop codons were engineered at the corresponding sites of the dnaE(1–270) gene; primers used are listed in Supplementary Methods. The first five amber mutations were created by the Phusion site-directed mutagenesis kit (Finnzymes, Finland), and the genes were inserted between the NdeI and EcoRI sites of the T7 promoter vector pRSET-6b (45). The resulting plasmids pKO1481–1485 have the codons of Pro4, Asp25, Asp75, Gln106 and Lys229, respectively, replaced by amber codons and were used as DNA templates in cell-free synthesis reactions. Linear templates for cell-free synthesis of additional amber mutants (codons for Arg175, Tyr234 and Gln237) were generated by strand overlap PCR as described (46) using Vent DNA polymerase with outside primers and pairs of mutagenic primers (see Supplementary Methods). The PCR products were separately purified from an agarose gel using NucleoSpin Extract II kits (Macherey-Nagel, Germany). T7 promoter and terminator sequences were appended in two further separate PCR reactions (50 µl each) (46) with a mixture of 20–30 ng of purified PCR products from the previous step. Mixing of two sets of primer pairs in approximately equimolar ratio, removal of the residual primers by the NucleoSpin kit, denaturation at 95°C (5 min) and reannealing at room temperature (5 min) yielded DNA with complementary 8-nucleotide overhangs suitable for cyclization by the intrinsic ligase activity of the cell-free extract.
Complexes of Bpa mutants of α270 and the εCTS
Cell-free reactions were as described (31,32), with added Bpa (1 mM) and BpaRS (4–15 µM), as required. Plasmid templates pSH1017, pKO1367 and pKO1422 were used at 16 µg ml−1 for production of ε, α270-His6 and εCTS59 (Figure 1B), respectively. The reannealed amber mutant PCR products described above were used as template at ∼10 µg ml−1. The protein complex between α270-His6 (Bpa mutants or wild-type) and the εCTS were made by simultaneous cell-free synthesis of the two proteins in the same reaction mixtures. The α270-His6:ε:θ was produced by making ε in the presence of purified α270-His6 and θ or by co-synthesis of α270-His6 and ε in the presence of separately purified θ. The complex of α270 with εCTS-Bpa-His6 was made by cell-free co-synthesis of these partner proteins in the same reaction mixture. The reaction mixtures were then clarified by centrifugation (100 000 g, 1 h) at 4°C. The supernatants were loaded onto ProPur IMAC Mini Ni-spin columns and the complexes were partially purified by virtue of the C-terminal His6-tag of α270. The purified complexes were concentrated to ∼0.1 ml using Millipore Ultra-4 centrifugal filters (MWCO 10 kDa), replacing the buffer with 10 mM sodium phosphate (pH 6.8), 100 mM NaCl, 1 mM dithiothreitol for photo-crosslinking experiments.
Photo-crosslinking and LC-MS/MS analysis of crosslinked adducts
The isolated wild-type and Bpa-containing protein complexes (5–8 mg ml−1) were irradiated at 312 nm for 1 min using a Mini UV transilluminator system BTS 20 M (GAS700X) (UVItec, UK) and subsequently analysed by SDS-PAGE. The photo-crosslinked α270:ε or α270:εCTS adducts were analysed by LC-ESI-ion trap mass spectrometry/mass spectrometry (LC-MS/MS) using a described protocol for in-gel trypsin digestion of gel-fractionated proteins (47). The solution containing the tryptic peptides that diffused from the gel pieces was desalted using C18 Zip-tips (Millipore), dried in a desiccator and dissolved in 20 µl of 15% acetonitrile/1% formic acid for LC-MS/MS analysis using an Agilent 6530 Accurate Mass Q-TOF LC/MS.
Preparation of the His6-αGL:15N-εL:θ complex and titration with β2
A mixture of 11.0 mg of purified His6-αGL, 2.7 mg θ and 5.6 mg 15N-εL was treated at 0°C for 1 h, then dialysed against 50 mM HEPES-KOH (pH 7.5), 300 mM NaCl, 20 mM imidazole, 5% (v/v) glycerol (buffer A). The sample (10 ml) was separated from excess θ and εL on a 5 ml Ni-NTA column in buffer A (eluted in a linear 20–500 mM imidazole gradient). The His6-αGL:15N-εL:θ complex was concentrated to 100 µM in NMR buffer using Millipore Ultra-15 centrifugal filters (MWCO 10 kDa) and stored at −80°C. Sample purity was assessed by 15% SDS-PAGE. 15N-HSQC spectra were recorded before and after addition of concentrated β2 (separately dialysed in NMR buffer) to 50, 100, 150, 200, 300 and 400 µM (as dimer).
The His6-αGL:15N-εL:θ:β2 complex was separately isolated from a mixture of 100 µM His6-αGL:15N-εL:θ and 400 µM β2 by gel filtration (Supplementary Figure S1). The protein complex was concentrated to about 40 µM in NMR buffer using Millipore Ultra-4 centrifugal filters (MWCO 10 kDa) and stored at −80°C.
NMR titration of 15N,13C-ε:θ complexes with β2
Complexes of 15N,13C-ε186 and 15N,13C-ε193 with purified unlabeled θ were dialysed into NMR buffer and concentrated using Amicon Ultra-4 centrifugal filters (MWCO 10 kDa, Millipore). Resonances in the 15N-HSQC spectra of ε186 and ε193 in the two complexes with θ were assigned by reference to previous assignments of ε186 in ε186:θ (BioMagRes database entry: bmrb6184), our ε assignments in the αεθ complex (9) and new experimental data for residues Ala188, Gln182–Ala186 and Thr193 obtained from 2D HN(CO) spectra and 3D HN(CO)CA and HNCA spectra. Phe187 was assigned through combinatorial labeling with 15N and 15N/13C to identify the Ala186–Phe187 dipeptide. The assignment of resonances in the CBM of ε193 (Gln-Thr-Ser-Met-Ala-Phe) were confirmed using cell-free residue-specific 15N-labeling of wild-type and two CBM mutants of ε193, that is, εL193 (CBM: QLSLPL) and εQ193 (CBM: ATSMAF) (25), with these six amino acids, and of εL with 15N-Leu, all in the presence of excess unlabeled θ.
NMR titration of the 15N,13C-ε186: θ complex (100 µM) with β2 was made by recording of 15N-HSQC spectra before and after progressive addition of concentrated purified β2 to 100, 200, 300 and 400 µM, whereas the 15N,13C-ε193:θ complex (34 µM) was similarly titrated with 34 and 68 µM β2. Spectra were also recorded of the 15N-Gln, Thr, Ser, Met, Ala, Phe labeled ε193:unlabeled θ (27 µM) sample with and without added β2 at 30 µM.
Small-angle X-ray scattering
A mixture of the αLεLθ core (1 mg) and β2 (2 mg) was dialysed into buffer C (50 mM Tris.HCl pH 7.6, 1 mM EDTA, 1 mM dithiothreitol, 10% v/v glycerol) containing 100 mM NaCl. The stoichiometric αLεLθ:β2 complex was separated from excess β2 by anion exchange chromatography on a MonoQ 5/50 GL column (GE Healthcare) using a gradient of 0.1–1.0 M NaCl in buffer C, concentrated to 2.15 mg ml−1 using an Amicon Ultra 0.5 ml centrifugal concentrator (Millipore) and stored frozen at −80°C. SDS-PAGE was used to confirm the presence of all four subunits.
Scattering data were recorded on the SAXS/WAXS beamline at the Australian Synchrotron. The complex was analysed by size-exclusion chromatography-coupled small-angle X-ray scattering (SEC-SAXS). The complex was dialysed into 50 mM Tris.HCl pH 8.0, 0.1 M NaCl, 1 mM EDTA, 1 mM TCEP, 5% (v/v) glycerol and 70 µl were injected at 0.5 ml min−1 onto a Wyatt WTC-030S5 SEC column (7.8 × 300 mm) equilibrated at 12°C in the same buffer. A280 of the eluate was monitored immediately prior to its passage through a quartz capillary that was illuminated by a collimated 11 keV X-ray beam, λ = 1.127 Å. Scattering from the sample was measured by a Pilatus 1 M detector (Dectris, Switzerland) that recorded 2D scattering images in 2 s exposures from a position 3349 mm behind the sample. For all the frames used for the data analysis, no protein damage induced by X-rays was observed. Scattering from the eluate was stable as averaged from 10 exposures prior to and after elution of protein, to give the buffer scattering. Following radial averaging and buffer subtraction, the radii of gyration, Rg, of five exposure bins were determined by Guinier analysis using AUTORG (48) and plotted against elution volume. Sample scattering was averaged across the region of Rg stability, which corresponded to the main UV absorption peak in the elution profile and encompassed 20 exposures. The scattering pattern was truncated within the range 0.012 ≤ Q ≤ 0.16 Å−1. The theoretical SAXS patterns, radii of gyration and envelope volumes of various atomic models were calculated using CRYSOL (49), for comparison with experimental results.
RESULTS
Cell-free but not in vivo expression of the PHP domain of α yields correctly folded protein
As full-length E. coli α is prone to proteolysis and is unsuitable for crystallization (10), we studied the interaction between domains of α and ε. It had been shown that the N-terminal 320-residue fragment of α binds ε (19), but the PHP domain of α defined subsequently ends already at residue 270 (10). However, a soluble construct comprising the N-terminal 270 residues (α270) produced in vivo appeared to be unfolded as indicated by the 15N-HSQC NMR spectrum of a 15N-labeled sample (Supplementary Figure S2A). In contrast, samples made by cell-free protein synthesis routinely showed a chemical shift dispersion characteristic of a well-structured globular domain (Supplementary Figure S2B). Therefore, we subsequently produced all samples of α270 by cell-free synthesis.
Coarse mapping of the interface between α270 and the εCTS by NMR
Expression of a construct comprising the flexible 59 C-terminal residues of ε preceded by a 22-residue tag (εCTS59; Figure 1B) in the presence of 15N-labeled α270 led to a soluble complex that retained the overall chemical shift dispersion of α270 with some significant chemical shift changes, as expected for specific binding (Supplementary Figure S3A).
We also used NMR spectroscopy to map the binding site of the εCTS on α270. As the stability and concentration of α270 samples was insufficient for conventional triple-resonance NMR experiments, combinatorial labeling was used to obtain resonance assignments (38,39). Five samples were prepared, in which different residues of α270 were labeled with 15N in different combinations (Supplementary Figure S4A and Supplementary Data), allowing the residue-type identification of the 15N-HSQC cross-peaks. In addition, five samples were prepared with combinatorial 13C-labeling and uniform 15N-labeling. 2D HN(CO) NMR spectra of these samples provided the residue-type information of the preceding amino acid for each 15N-HSQC cross-peak (Supplementary Figures S3B and Supplementary Data) (50). In combination, the 10 samples provided sequence-specific resonance assignments for 50 15N-HSQC cross-peaks arising from amino acid pairs that are unique in the sequence of α270 (Supplementary Table S2).
A second set of five combinatorially 15N-labeled samples of α270 was prepared in complex with unlabeled εCTS59 and 15N-HSQC spectra were recorded (data not shown). Significant chemical shift changes occurred throughout the α270 domain, but the two largest were observed for amides within 15 Å of its N- and C-terminal ends (Supplementary Figure S3). This indicated that in contrast to α270, a fusion construct of it with the εCTS could be a stable, well-folded protein.
The 15N-HSQC spectrum of the 15N-εCTS59 construct in complex with unlabeled α270 displayed many narrow lines, indicating that much of it is highly mobile and not tightly interacting with α270. For resonance assignments, εCTS59 in the complex with unlabeled α270 was combinatorially 15N-labeled (Supplementary Figure S5A) and, in a second set of five samples, labeled combinatorially with 13C and uniformly with 15N. 3D HNCA and HN(CO)CA experiments with the second set were used to assign most of the flexible amino acid residues in the εCTS59 construct. Like 2D HN(CO) experiments, the 3D HN(CO)CA spectra of the combinatorially labeled samples identified for each 15N-HSQC cross-peak the amino acid-type of the preceding residue. In addition, the HN(CO)CA spectra delivered its Cα chemical shift which, together with the HNCA spectrum, provided more secure resonance assignments than could have been obtained from a single uniformly 15N/13C labeled sample. The final resonance assignments corresponded to the segment from Phe187 to Ala209. In addition, two-thirds of the residues of the non-native N-terminal 22-residue tag of the εCTS59 construct were assigned in the complex (Supplementary Table S3). The narrow line widths and random coil chemical shifts of these residues indicate high flexibility as expected. Most notably, no signals could be observed for the C-terminal segment (Ser210 to Ala243) of the εCTS, except for the amides of Glu221 and Gly237, indicating immobilization by tight association with α270. The delineation of the flexible residues in the εCTS was used to design fusion constructs of the εCTS with α270.
Crystal structures of intramolecular α270–εCTS complexes
The α270 domain was fused to the εCTS (residues 209–243) via a nine-residue linker, where the εCTS was N-terminal of α270 in construct A and C-terminal of α270 in construct B (Figure 1C). To assess the impact of the fusion on the structural integrity of α270, we compared the 15N-HSQC spectra of selectively 15N-alanine labeled samples of constructs A and B with corresponding spectra of the similarly 15N-alanine labeled non-covalent α270:εCTS59 complex. All samples were readily produced in soluble form by cell-free synthesis. The NMR spectrum of construct A was much more similar to the spectrum of the α270:εCTS59 complex than the spectrum of construct B (Supplementary Figure S6A and Supplementary Data), suggesting that construct A is a stable intramolecular α270:εCTS complex.
Both constructs were expressed in vivo, and could be purified in soluble form for use in crystallization trials, but neither yielded crystals. Crystals were obtained, however, for a variant of construct A that contained a fortuitous point mutation in the α270 domain, Leu21Pro. Some of the cross-peaks were broad or missing from the 15N-HSQC spectrum of the 15N-alanine labeled mutant protein, suggesting conformational exchange broadening of some of the NMR signals (Supplementary Figure S6C and Supplementary Data). The crystal structure, however, shows no evidence of conformational heterogeneity, which may be explained by crystal contacts involving Pro21 leading to preferential crystallization of a particular conformer; in all four molecules in the asymmetric unit, Pro21 makes a crystal contact with an alanine residue.
The 1.70 Å crystal structure of the α(Leu21Pro) mutant of construct A was solved by molecular replacement using the α270 domain from the structure of α917 (PDB: 2HQA) (10) as starting model (Supplementary Table S1). The four molecules in the unit cell show a maximum Cα RMSD of 0.284 Å in pairwise alignments, and in all four the structure is fully ordered from Lys211 of εCTS (numbered throughout as in full-length ε) through the linker region and the entire α-PHP domain to the final residue, Thr270. In one of the monomers, the backbone and side chain Cβ of Ser210 is also ordered, and in another Lys211 has alternate side chain conformations.
The refined structure of the εCTS35–α270 protein shows the εCTS assuming an extended structure across one face of α270, followed by an α-helix. The C-terminal residues that follow are located in a pocket formed by α270 and the α-helix of the εCTS (Figure 2). The εCTS portion is fully structured and in contact with α between residues Lys211 and the C-terminus of ε; electrostatic and H-bonded contacts between α and ε are listed in Supplementary Table S4, and these are complemented by a much larger number of hydrophobic interactions. Although it is engaged in crystal contacts, the linker peptide connecting the εCTS with the α270 domain is solvent exposed, so that the structure of the complex is unlikely to be affected by its length or conformation. The mutated residue Pro21 of α270 is far removed from the εCTS binding region on the opposite face of the PHP domain (Figure 2A) that is in closer proximity to the polymerase active site in the α917 structure (10).

1.7 Å crystal structure of construct A (εCTS35–α270) with the Leu21Pro mutation in α270. In the ribbon diagram in (A), εCTS35 and α270 are in green and cyan, respectively, while the nine-residue linker between the εCTS and α270 is in yellow. The location of residue 21 (in magenta) and the N- and C-termini of α270 (αM1, αT270) are indicated. The εCTS35 region is fully structured from Lys211–Ala243, and forms an α-helical segment between Thr218 and Gly237. The additional contacts of residues Ser210 and Ile202–Ile205 of εCTS with α270 in the 2.15 Å structure of εCTS44–α270(L21P) are also indicated. (B) A view in the same orientation with α270 in space-filling representation (gray) and side chains of selected residues of εCTS (green) shown as sticks (green). Residues Ser210 to Ala217 of εCTS form an extended structure that lies in a groove in α270. A list of H-bonding and electrostatic contacts between residues in α and ε is given in Supplementary Table S4.
To confirm the NMR data that εCTS in complex with α270 is indeed unstructured in the linker preceding Ala209, we made a longer type A fusion construct commencing at Ala200 (i.e., εCTS44–α270), crystallized it under similar conditions, and solved its structure at 2.15 Å (Supplementary Table S1). In two of the four chains in the asymmetric unit, residues preceding Lys211 were still disordered, but in one of the other two, weak electron density was interpreted as the tetrapeptide segment Ile202–Ile205 that has additional interactions with the region around His183 and Asp252 of α270 (Figure 2 and Supplementary Table S4), and Ser210 was also fully ordered. Because the Val206–Ala209 segment is disordered, we are unable to tell if this εCTS tetrapeptide derives from the same molecule to which it is bound, or from a neighboring molecule in the crystal lattice, and the NMR data show that this segment is inherently flexible in solution. Although it seems probable that these additional interactions are rather transient in the context of the full αεθ core complex, they nevertheless indicate where in space the flexible segment of ε (between Ala188 and Ala209) is likely to reside, at least in the closed form of the αεθ:β2 complex during DNA synthesis (25).
The PHP domain of α in the crystal structures of construct A and in α917 (10) is fully conserved structurally, except around the site of the Leu21Pro mutation, with a backbone RMSD of 0.532 Å over 253 Cα atoms. This allows straightforward modeling of the εCTS onto the structure of α917. As discussed in detail below (see also Supplementary Movie S1), the C-terminus of ε binds to α in a position that would place the extended peptide segment immediately preceding the C-terminal helix of ε and the exonuclease active site far from that of the polymerase. Its unusually remote location raises questions about how the N-terminal exonuclease domain of ε gains access to a mismatched primer terminus when proofreading is required. Thus, we sought to obtain further information about the location in the complex with α of the linker peptide that extends in ε from the CBM (i.e., from Ala188) to the structured part in the crystal structures above.
Photo-crosslinking to localize the flexible peptide segment of the εCTS on α270
In agreement with the NMR evidence for high mobility of residues prior to Ala209, the crystal structures of εCTS in the two type A constructs showed consistent electron density only from Lys211 onwards. To explore the location of the flexible residues of the Q-linker, we introduced the unnatural amino acid p-benzoyl-L-phenylalanine (Bpa) at different sites in α270, using the orthogonal Methanococcus jannaschii system developed by P.G. Schultz and co-workers, where the site of Bpa incorporation is encoded by an amber stop codon (51). The purification of the mutants was facilitated by producing the protein in a cell-free system, which relied on purified plasmid DNA with amber stop codons, purified Bpa-tRNA synthetase and a total tRNA preparation that contained the amber suppressor tRNA (31). Most Bpa mutants were produced in yields of up to 1.5 mg ml−1 in 7 h without evidence of truncation at the amber stop codon (Supplementary Figure S7A). Full-length proteins were readily purified using a Ni-NTA spin column, as all α270 mutants carried a C-terminal His6-tag. The production of the Ala25Bpa mutant, which was initially expressed in low yields, was improved dramatically by using the optimized (52) amber suppressor tRNAopt and doubling the amount of total tRNA (31).
Cell-free expression of ε in the presence of the purified Bpa mutants of α270 and of separately purified θ produced stable soluble complexes that could be purified as shown previously for wild-type α (9). Similarly, soluble complexes of the α270 Bpa mutants with the εCTS59 construct were obtained by cell-free co-expression of the α270 mutants and of εCTS59 (Supplementary Figure S7B and Supplementary Data).
UV irradiation (312 nm, 1 min) effects photo-crosslinking of Bpa to nearby residues (<3 Å) (53). SDS-PAGE revealed crosslinking with full-length ε when Bpa was located in positions 19, 21, 23, 25 and 229 of α270, but no crosslinks were observed with Bpa at positions 4, 75 and 106 (Supplementary Figure S7B and data not shown). Mass spectrometric analysis of in-gel tryptic digests confirmed that the crosslinks were with the εCTS rather than the globular N-terminal domain of ε. Additional experiments were carried out with the εCTS59 construct to eliminate the need to exclude binding to the N-terminal domain of ε. Bpa mutants at positions 175, 229, 234 and 237 displayed crosslinks with εCTS59 (Figure 3; Supplementary Figure S7C). The wide distribution of crosslinking sites across the surface of α270 confirms the NMR observation of high flexibility in the linker segment of ε before the C-terminal α270-binding region. Most interestingly, the Lys229Bpa mutant readily crosslinked with a peptide segment preceding Gln196 in εCTS59 (Supplementary Figure S8), although Lys229 is located on the opposite face of the PHP domain compared to the binding site of the C-terminus of ε. Therefore, the Q-linker region of the εCTS readily wraps around the PHP domain of α but is not poised for specific binding interactions with the PHP domain.
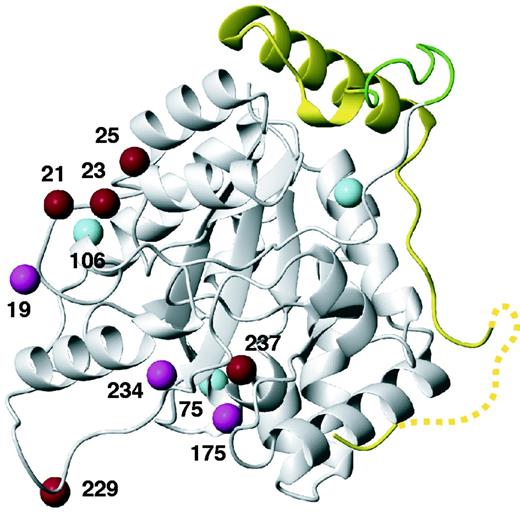
Sites in α270 where Bpa residues were introduced for photo-crosslinking with ε to detect proximity to residues for which no structural information was obtained by the crystal structure in Figure 2. The location of the εCTS determined by the crystal structure is shown in yellow, with the nine-residue linker peptide in green. The Cβ atoms of residues at sites leading to efficient, less efficient or no crosslinking are highlighted in red, magenta and cyan, respectively.
Photo-crosslinking experiments between α270 and the εCTS were also conducted with an εCTS construct that was extended at its C-terminus by Bpa-His6. MS analysis of a tryptic in-gel digest of the cross-linked complex revealed linkage to the segment of residues Ala31–Lys52 in α (data not shown). This result is in agreement with the crystal structure, which positions the peptide linker between the εCTS and α270 within 11 Å of the peptide identified by MS.
The εCTS Q-linker remains flexible in the αεθ:β2 complex
The ε subunit harbors a CBM immediately following the exonuclease domain (i.e., residues 182–187) (25), and α also contains a CBM between residues 920 and 924 (24). Although each binding interaction is individually weak, the cooperativity of binding of α to one subunit of the β2 dimer and of ε to the other maintains the integrity of the αεθ:β2 replicase complex with DNA during highly processive DNA replication (25). The questions remain whether such a binding arrangement is compatible with the available structural information on the replisome subunits and whether the εCTS can accommodate this arrangement. To investigate the structural confinement of the εCTS in the αεθ:β2 complex, we studied the NMR spectrum of the αGLεLθ:β2 complex, where αGL and εL are mutants of α and ε with improved binding affinities to β2 (Figure 1A and D); in addition to strengthening mutations in the CBM (as in αL) (25,26), αGL also contains an additional mutation (Val832Gly; spq-2) (25,54) that by itself strengthens binding of αεθ to β2 (S.J. and Thitima Urathamakul, unpublished). For example, a peptide with an optimized CBM as in εL interacts about 500-fold more strongly with β2 than the wild-type CBM of ε (25), while the αL mutations (A921L, M923L) in the CBM of α strengthen binding to β2 120-fold (26). For NMR measurements, αGLεLθ was made with uniformly 15N-labeled εL and mixed with a 3-fold excess of β2; the stoichiometric αGLεLθ:β2 complex was stable enough to be isolated by gel filtration (Supplementary Figure S1).
The molecular mass of the αGLεLθ and αGLεLθ:β2 complexes is so high (165 and 245 kDa, respectively) that only highly mobile peptide segments can generate cross-peaks in 15N-HSQC spectra. Remarkably, almost all the cross-peaks that could be observed for the αGLεLθ complex (Figure 4A and Supplementary Figure S5B) could also be observed in the presence of β2 (Figure 4B and C), although with generally decreased intensity as expected for the slower overall tumbling rate of the αGLεLθ:β2 complex (Supplementary Figure S9). The peaks did not arise from complexes with sub-stoichiometric amounts of β2, as they were observed even in the presence of an excess of β2. Assignments for many of these resonances (residues Glu190–Thr201 and Arg204) were obtained by comparison with spectra of α270 in complex with 15N-labeled εCTS (Figure 4B), and indicate that the Q-linker is clearly still mobile when the CBM of ε is tied to the β2 clamp.
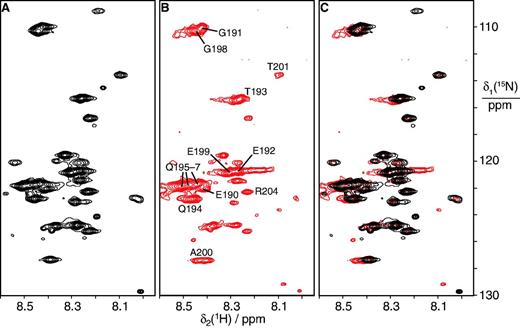
A large segment of the εCTS remains flexible in the αGLεLθ:β2 complex. The mutant subunits αGL and εL were used in the complex to avoid dissociation of β2 during purification using the N-terminal His6 tag on αGL. (A) 15N-HSQC spectrum of the αGLεLθ complex with 15N-labeled εL. Only amides from mobile residues are observable in the 165 kDa complex. (B) 15N-HSQC spectrum of the αGLεLθ:β2 complex with 15N-labeled ε. Resonance assignments obtained by comparison with spectra of α270 in complex with 15N-labeled εCTS are indicated. Most if not all of the observable peaks can be attributed to the εCTS. The same set of amides from mobile residues is observable in the purified 245 kDa complex as in (A). The spectrum was recorded using a 0.1 mM solution of the complex at 25°C. (C) Superimposition of the spectra in (A) and (B) demonstrates that most chemical shifts remain conserved.
The ε subunit interacts with β2 only through the CBM
Identification of the role of the CBM just following the structured domain of ε in DNA replication (25), when combined with the structure of the εCTS in complex with the α-PHP domain (Figure 2) enables us to position the proofreader between the β2 clamp and PHP domain of α in the αεθ:β2:DNA complex in the polymerization mode of DNA synthesis (25). We have previously shown by NMR that ε186 does not interact, even weakly, with α (9), and we now asked if ε contains a second site for interaction with β2 that orients it precisely in the αεθ:β2 complex. To do this, we made a new truncated version of ε we call ε193 (residues 2–193), that contains all of the structured exonuclease domain and the CBM (Figure 1A). We first used cell-free synthesis to prepare, in the presence of excess unlabeled θ, a sample of ε193 (27 µM) that was 15N-labeled only with amino acids that comprise the CBM (Gln, Thr, Ser, Met, Ala and Phe), and assigned these residues in the 15N-HSQC spectrum of the whole ε193 protein as described in Materials and Methods section. Addition of β2 to 30 µM led to disappearance of signals corresponding to all residues of the CBM (Gln182–Phe187), but no significant changes to the spectrum of the structured proofreading domain or of Ala188 and Thr193 in the region beyond the CBM (Figure 5). These data are the first to directly show the interaction of the CBM in ε with β2 at single-residue resolution.
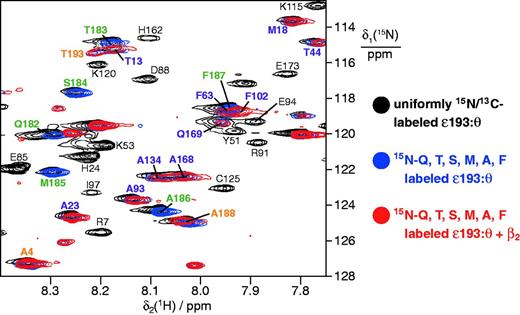
Isotope labeled ε193 in complex with purified unlabeled θ interacts with β2 only through the CBM. Superimposition of 15N-HSQC spectra of uniformly in vivo15N,13C-labeled ε193:unlabeled θ (black spectrum, selected resonance assignments in black) and of ε193:θ (27 µM) labeled specifically in ε193 with 15N-glutamine, threonine, serine, methionine, alanine and phenylalanine in the absence (blue spectrum) and presence (red spectrum) of β2 (30 µM). Resonances were assigned as described in Materials and Methods. Cross-peaks were observed for 52 of 59 Gln, Thr, Ser, Met, Ala and Phe residues in the structured ε186 domain, and all were unaffected by addition of β2 (selected signals labeled in purple); those of Ser2 and Thr3 in the disordered N-terminus could not be assigned, while Ala100, Thr128, Ser144, Ala164 and Thr179 had low intensity even in the absence of β2. Signals in the CBM that broaden beyond recognition in the presence of β2 (red spectrum; i.e., Gln182, Thr183, Ser184, Met185, Ala186, Phe187) are labeled in green, while assignments for flexible residues at the N- and C-termini (Ala4, Ala188 and Thr193) that are unaffected by β2 are labeled in orange.
We also isolated the complex of uniformly in vivo15N,13C-labeled ε193 with unlabeled θ, and recorded its 15N-HSQC spectra (at 34 µM) in the absence and presence of β2 at 34 and 68 µM (data not shown). Once again, the only cross-peaks broadened beyond detection in the ε193 spectrum were those in the region of the CBM; peaks throughout the remainder of the spectrum did not shift and were only broadened at the highest concentration of β2, consistent with the exonuclease domain being freely mobile in the complex with β2, except in the CBM that interacts directly with the clamp. In further support of the conclusion that ε contains no site of interaction with β2 beyond the CBM, we were unable to detect any significant changes in the 15N-HSQC spectrum of 15N,13C-ε186:θ (100 µM) on addition of up to 400 µM β2.
Structural modeling of the Pol III replicase complex in the polymerization mode
The 3D structures of many components of the Pol III replicase complex are known from different bacterial sources, including three crystal structures of Pol III α: of E. coli α917 (10), and of full-length Taq α alone (11) and in complex with primer-template DNA (55). The DNA-free protein structures are remarkably similar and reveal an open state that closes on binding primer-template DNA (discussed in 25). In addition, the crystal structures of an E. coli β2:dsDNA complex (23), ε186 (12) and the ε186:HOT complex (13,14) are known, as well as the NMR structure of the ε186:θ complex (16).
Combining these atomic-resolution structures with the present structure of the α270:εCTS complex and the identification of CBMs in α (24,26) and ε (25), we initially built a compact model of the replicase complex in the polymerization mode with the ε186 and θ domains in available space between the β clamp and the β-binding domain of α (Figure 6A, Supplementary Movie S1 and Supplementary Pymol Session File S1; model building is described in Supplementary Methods). This model fulfils all the known restraints, including the current results that the Q-linker region in ε is flexible and at least transiently close to Lys229 in α (Figure 3), and that residues 202–205 of ε are transiently close to His183 and Asp252 of α (Figure 2). In the model, the CBMs of α and ε bind to different subunits of β2 and the exonuclease domain of ε readily approaches the DNA, while the conformational space available to the Q-linker of ε is sufficiently large to allow high mobility (Figure 6A). Since we have been unable to detect an additional point of contact of ε with either α (9) or β (above), it may be that either (i) the globular ε186 domain remains mobile in the complex (it can still rotate in its position in this model without clashing with β2 or α) or it is held in a fixed position through transient electrostatic contacts with the double-stranded portion of the primer-template DNA. Its precise position could potentially be defined by further crosslinking studies, but we note that as with our Bpa data, all crosslinking methods are inherently unsuitable for precise definition of positions of components of intrinsically dynamic complexes; they demonstrate where subunits can be, not where they necessarily are.
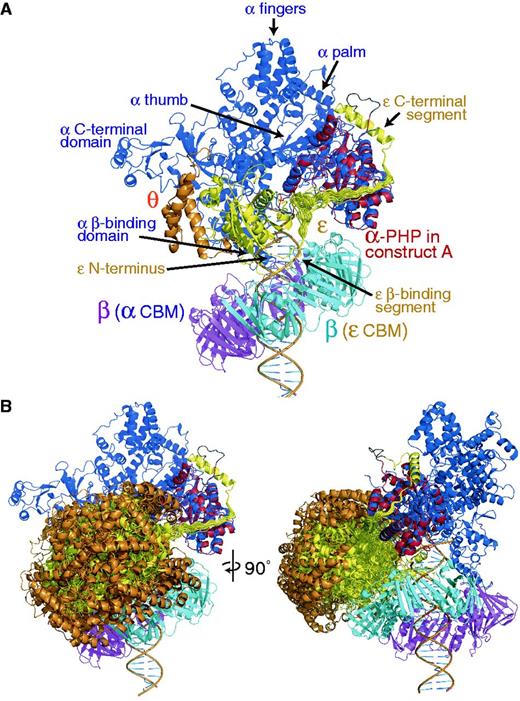
Models of the αεθ:β2 complex with primer-template DNA in the polymerization mode. Color coding: α (blue), with the PHP domain of the crystal structure superimposed in red, ε (yellow), θ (orange) and β2 (subunit in cyan contacting the CBM of ε and that in magenta contacting the internal CBM of α). (A) Compact structure of a form of the ‘closed’ complex (25) with εθ sandwiched between the β clamp and the PHP domain of α. Multiple conformations are displayed for the 22-residue linker segment connecting the α-bound portion of the εCTS with the globular exonuclease domain of ε (ε186) that was positioned to bring its CBM in proximity to the protein-binding groove of β. All conformations of the linker segments are sterically allowed, explaining the high mobility observed in this segment experimentally. (B) It is possible to rotate ε out of the complex into a more open structure while maintaining its contacts with the PHP domain of α and β2. Multiple (other) sterically allowed exonuclease domain (εθ) conformations are displayed; these represent a subset of the structures used to back calculate scattering curves in Figure 7. The view on the left is the same as in (A); that on the right is rotated 90° as indicated.
A third possibility is that the exonuclease domain remains much more freely mobile in the complex during DNA synthesis, and is reoriented to an appropriate position during proofreading. The structured region of ε186 ends at Gly180 and Gln182, the first residue of the CBM, is bound in the protein-binding groove of β2. Although the closeness of these residues restricts the space that can be occupied by εθ, it is still possible for εθ to rotate away from α:β2 to produce less compact and more mobile structures.
Assessment of structural models using SAXS data
The αLεLθ:β2 complex, with both the CBMs in α and ε strengthened, has been observed to be stable by ESI-MS under native conditions (25), and as with the corresponding complex containing αGL, it can be isolated chromatographically (see Supplementary Methods). To assess whether the αL subunit in this stabilized replicase complex has a closed structure similar to that in our model (Figure 6A) even in the absence of primer-template DNA, we collected real-time gel-filtration SAXS data on the αLεLθ:β2 complex at a synchrotron source (Figure 7) and compared it with predicted scattering curves for various structural models.
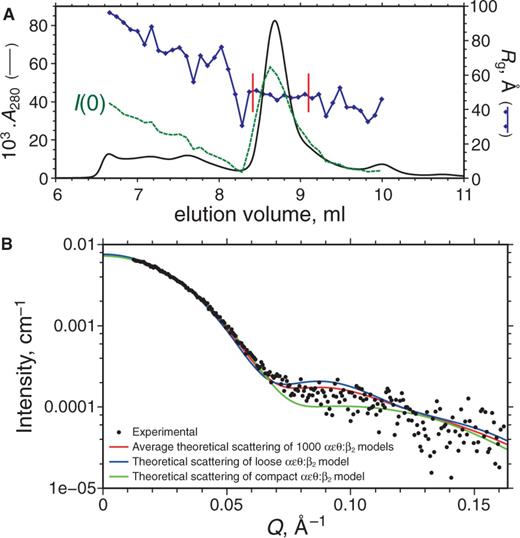
SEC–SAXS measurement of αLεLθ:β2 complex. (A) Elution profile showing the A280 (black continuous line) and SAXS data including the forward scattering intensity I(0) (green-dashed line) and Rg (in blue) calculated by Guinier analysis of five-exposure bins; values of I(0) vary linearly between 0.0007 and 0.0105 cm–1. To obtain the experimental SAXS pattern, exposures were averaged in the region of Rg stability (bounded by vertical red bars) before data reduction and buffer subtraction. (B) Experimental SAXS data for the αLεLθ:β2 complex (scatter plot), for which the pair distance distribution (not shown) indicates Rg = 48.8 ± 0.2 Å and maximum dimension of 152.5 Å. For comparison, the averaged theoretical scattering of 1000 αεθ:β2 models generated by free backbone rotation in segment ε180–182 (Figure 6B) is shown in red; these models have mean Rg = 47.6 Å and mean envelope diameter of 154.2 Å. The theoretical scattering of a typical αεθ:β2 model with a more open (loose) orientation of εθ is shown in blue; Rg = 48.3 Å, envelope diameter = 152.9 Å, and that of an αεθ:β2 model with a compact orientation of εθ is shown in green; Rg = 46.8 Å, envelope diameter = 151.5 Å.
Analysis of the data showed good agreement with the overall dimensions of an initial docking model with a closed α conformation, but indicated too compact packing of the εθ subunits to the α chain (Figure 7B). An ensemble of 1000 alternate structures was generated by allowing free rotation around the backbone dihedral angles of Gly180–Gln182 in ε while disallowing steric clashes with α:β2 (Figure 6B, Supplementary Movie S2 and Supplementary Pymol Session File S2). Averaging over the ensemble resulted in a markedly improved fit of the SAXS data at Q-values in the range of 0.07–0.12 Å−1 (Figure 7), which suggests that εθ is not restrained in a single conformation in the stabilized αεθ:β2 complex, at least in the absence of primer-template DNA.
DISCUSSION
The crystal structure of the α270:εCTS complex solved in the present work allows, for the first time, the building of informed models of the αεθ:β2 replicase complex with primer-template DNA in the polymerization mode (Figure 6A). The high-affinity binding site of the εCTS on the PHP domain of α turned out to be surprisingly remote from the active site of the polymerase. The long Q-linker of ε was found to be highly mobile even in the context of the αεθ:β2 complex, readily accommodating a conformation that allows the CBM located in ε near the C-terminus of the globular exonuclease domain (25) to bind to the well-established protein-binding site of β. It is intriguing to speculate that the exonuclease could swing a long distance from the DNA when its CBM is released from the β2 clamp. Release of the ε CBM from β2 would be a requirement for entry of other β-binding proteins, including repair and translesion polymerases (56) into the replicase complex, and might also occur in the transition from polymerization to proofreading modes (25). The distance between the polymerase and exonuclease active sites in all model structures in Figure 6 is >70 Å (average of 92.3 Å for the ensemble in Figure 6B); that this distance is so large suggests it is very likely that the ε–β contact is broken during transition to the proofreading mode, to allow α to assume a more open structure and access of ε to the mismatched primer terminus. To compensate for the loss of binding affinity of the CBM, the conformational change in α could expose a cryptic-binding site for the ε186 domain (or θ), such that the exonuclease site is appropriately positioned for proofreading. In this scenario, binding of ε186:θ to either the β2 clamp or to α would present a switch between the two modes that is fundamentally different from that observed in simpler polymerases with an integrated proofreading domain, where the transition between polymerization and proofreading modes requires protein-mediated transfer of the 3′ end of the primer over a distance of 20–30 Å between the polymerase and exonuclease active sites (29,30). In contrast, repositioning of the exonuclease domain of Pol III over a sufficiently large distance is perfectly conceivable, as the ε186 domain does not interact to any appreciable degree with the εCTS, α or, as shown here, to β (9). Proofreading would still require disengagement of the mismatched primer-template from the polymerase active site and sliding back of the ds DNA portion through the β2 clamp to allow access of the 3′ primer terminus to the exonuclease active site.
On a technical note, cell-free protein synthesis proved to be a decisive tool in this project, as α270 folded into a defined conformation when expressed by cell-free synthesis but not when it was produced in vivo. Furthermore, overexpression of full-length ε in vivo leads to insoluble protein, which in the past could only be solubilized by a denaturation and refolding protocol (57). Cell-free synthesis of ε in the presence of its natural-binding partners θ and α, however, circumvented this problem, readily yielding the stable ternary αεθ complex (9). Similarly, εCTS59 when expressed by itself was insoluble, but soluble complexes with α270 and mutants thereof were readily obtained by cell-free synthesis. Furthermore, this approach allowed efficient 15N-labeling of individual proteins in selective (58,59) and combinatorial (38,39) labeling schemes, providing a route to NMR resonance assignments of samples of limited solubility and stability. Finally, the cell-free approach is uniquely suited for the incorporation of unnatural amino acids (31), in the present work affording the facile incorporation of the unnatural amino acid Bpa for photo-crosslinking. This method may present a useful tool to probe structures of larger replisomal complexes in the future.
CONCLUSION
The extraordinarily long flexible tether by which the globular domain of the proofreading exonuclease is attached to the polymerase subunit raises the expectation of large conformational changes involved in the transition from the polymerization to the proofreading mode. Future studies may attempt to probe this by single-molecule fluorescence resonance energy transfer (FRET) experiments.
ACCESSION NUMBERS
Protein Data Bank: The coordinates and structure factors of α270:εCTS209–243 and α270:εCTS200–243 fusion proteins have been deposited with accession numbers 4GX8 and 4GX9, respectively.
FUNDING
Australian Research Council [DP0984797 to N.E.D., T.H., K.O. and M.T., DP0877658 to N.E.D. and A.J.O., FT0990287 to A.J.O., FT0991709 to T.H., DP120100561 to T.H. and G.O.]. Funding for open access charge: University of Wollongong.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Prof. P. Schultz for the gene of the Bpa-tRNA-synthetase, Drs Nigel Kirby, Haydyn Mertens and Adrian Hawley for help with SAXS experiments (SAXS/WAXS beamline) and Dr David Jacques for help with X-ray data collection (beamlines MX1 and MX2) at the Australian Synchrotron, Victoria, Australia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments