





Abstract
In a bis-(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP)/transcription factor binding screen, we identified Mycobacterium smegmatis Ms6479 as the first c-di-GMP-responsive transcriptional factor in mycobacteria. Ms6479 could specifically bind with c-di-GMP and recognize the promoters of 37 lipid transport and metabolism genes. c-di-GMP could enhance the ability of Ms6479 to bind to its target DNA. Furthermore, our results establish Ms6479 as a global activator that positively regulates the expression of diverse target genes. Overexpression of Ms6479 in M. smegmatis significantly reduced the permeability of the cell wall to crystal violet and increased mycobacterial resistance to anti-tuberculosis antibiotics. Interestingly, Ms6479 lacks the previously reported c-di-GMP binding motifs. Our findings introduce Ms6479 (here designated LtmA for lipid transport and metabolism activator) as a new c-di-GMP-responsive regulator.
INTRODUCTION
The nucleotide signaling molecule, bis-(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP), represents a widely conserved second messenger in bacteria (1,2). It is synthesized from two molecules of GTP by diguanylate cyclases (DGCs) through their GGDEF domains and is degraded by the EAL and HD-GYP domains of phosphodiesterases (PDEs) (3–7). c-di-GMP participates in the regulation of a wide range of cellular functions and processes, including the biosynthesis of exopolysaccharide matrix substance in biofilms (8–10), bacterial motility (11,12), cell cycle progression (13) and bacterial virulence (14–18).
As a second messenger, c-di-GMP needs to bind to a receptor/effector to exert its regulatory function. Therefore, identifying proteins that directly bind to and sense the levels of c-di-GMP is critical for further understanding the signaling pathway it regulates. In recent years, several types of c-di-GMP receptors/effectors have been successfully identified in different bacterial species. PilZ-containing proteins were the first cellular c-di-GMP receptors/effectors to be identified in Gluconacetobacter xylinus (19) and many proteins of the PilZ family have since been discovered (20–23). c-di-GMP tightly binds to the I-site in the GGDEF domains and to the substrate site in the EAL domains (13,24). Additionally, c-di-GMP has also been shown to bind to riboswitches found in the 5′-untranslated regions of various mRNAs (25).
Several transcription factors involved in the regulation of bacterial biofilm-associated phenotypes have been characterized recently as c-di-GMP receptors/effectors (26–30). FleQ was the first transcriptional regulator to be identified as a c-di-GMP receptor/effector. Upon c-di-GMP binding, FleQ derepresses the expression of several previously characterized target genes such as pel and other EPS genes that are necessary for biofilm formation in Pseudomonas aeruginosa (26). The regulator VpsT can sense c-di-GMP levels to inversely control extracellular matrix production and motility in Vibrio cholerae (27). Clp regulates expression of virulence genes and is itself regulated by c-di-GMP concentrations in Xanthomonas campestris pv. campestris (28,30,31). Other examples include Bcam1349 and MrkH, both of which are c-di-GMP-dependent activators that regulate biofilm formation in Burkholderia cenocepacia (29) and Klebsiella pneumoniae (23), respectively.
Mycobacteria are a large group of slow-growing bacteria that include both pathogens and saprophytes (32). For example, Mycobacterium tuberculosis is the causative agent of tuberculosis (TB) and causes nearly 1.4 million human deaths worldwide every year (33). Mycobacterium smegmatis, a relatively fast-growing and non-pathogenic mycobacterium species, has been used as a model organism to study the gene regulatory mechanisms of other virulent and extremely slow-growing species (34). Recent studies have shown that both M. tuberculosis and M. smegmatis contain enzymes that metabolize c-di-GMP and cause turnovers in c-di-GMP levels (35,36). Deletion of the bifunctional M. smegmatis protein Ms2196, which contains the GGDEF and EAL domains, has been shown to affect long-term survival of the bacteria under conditions of nutritional starvation (35). However, no c-di-GMP receptors/effectors other than proteins containing the GGDEF-EAL domains have yet been found in mycobacteria. Additionally, there is no report on the effect of DGCs or PDEs on colony morphology or antibiotics resistance profile of mycobacterial species.
In this study, we set out to screen for potential c-di-GMP receptors/effectors in a M. smegmatis transcription factor library. We identified a c-di-GMP receptor/effector and the signaling pathway it is associated with. Additionally, a hypothetical transcription factor LtmA was found to act as a c-di-GMP-responsive global regulator that directly controls the expression of a large group of genes. The function of LtmA was further linked to cell wall permeability and antibiotic resistance in M. smegmatis. Based on our results, LtmA represents a new class of c-di-GMP receptors/effectors in bacteria.
MATERIALS AND METHODS
Cloning of the regulatory sequences of Ms6479 target genes and bacterial one-hybrid assays
Ms6479 was cloned into the pTRG vector (Stratagene). Promoters of the M. smegmatis genes were amplified using appropriate primers (Supplementary Table S1) and cloned into the pBXcmT vector (37). Escherichia coli XL1-Blue strain (Stratagene) was used for the routine propagation of all pBXcmT and pTRG recombinant plasmids. Bacterial one-hybrid assays were carried out as described previously (37). Positive growth co-transformants were selected on a selective screening medium plate containing 20 mM 3-AT (3-amino-1,2,4-triazole), 16 µg/ml streptomycin, 15 µg/ml tetracycline, 34 µg/ml chloramphenicol and 30 µg/ml kanamycin. Co-transformants containing the pBX-Rv2031/pTRG-Rv3133 plasmids (37) served as positive controls (CK+) and co-transformants containing the empty vectors pBX and pTRG served as negative controls (CK−).
Expression and purification of recombinant proteins
Mycobacterium smegmatis mc2155 genes were amplified by PCR from mycobacterial genomic DNA using appropriate primers (Supplementary Table S1). Recombinant plasmids, such as pMV261-Ms6479 and pMV261-b1535, and the corresponding recombinant strains were constructed (Supplementary Table S2). The cells were collected and proteins were purified on Ni2+ affinity columns as described previously (38). The elution was dialyzed overnight and stored at −80°C.
Construction of the Ms6479 deletion mutant of M. smegmatis and Southern blot analysis
A pMind-derived (39) suicide plasmid carrying a hygromycin resistance gene was constructed and a sacB-lacZ gene was inserted to confer sensitivity to sucrose as a negative selection marker. Knockout of the Ms6479 gene in M. smegmatis mc2155 was performed as described previously (40). Deletion of the Ms6479 gene was confirmed by Southern blot analysis as described previously (38). The probe consisted of a 300-bp fragment of the upstream region of the Ms6479 gene amplified using appropriate primers. (Supplementary Table S1).
c-di-GMP and c-di-AMP binding analysis for transcription factor proteins
c-di-GMP was enzymatically produced from GTP by the E. coli b1535 DGC protein and then purified as described previously (41,42). c-di-AMP was produced by the Bacillus subtilis DisA protein using ATP and purified according to previously published procedures (43). 32P-labeled c-di-GMP and 32P-labeled c-di-AMP were synthesized using [α-32P]GTP and [α-32P]ATP (3000 Ci/mmol), respectively. Purified transcription factor proteins were co-incubated with 0.125 µM c-di-GMP or c-di-AMP on ice for 10 min. Samples were then irradiated by UV and analyzed by SDS–PAGE as previously described (41,42). Surface plasmon resonance (SPR) analysis for the binding of c-di-GMP with proteins was conducted on a Biacore 3000 instrument (GE Healthcare) according to previously published procedures (44).
SPR analysis for the binding of c-di-GMP with Ms6479
SPR analysis for the binding of c-di-GMP with proteins was conducted on a Biacore 3000 instrument with nitrilotriacetic acid (NTA) sensor chips according to previously published procedures (44). Briefly, to detect the physical interaction between Ms6479 and c-di-GMP, his-tagged Ms6479 proteins were immobilized onto the NTA chips. Purified c-di-GMP or c-di-AMP was used as the ligand and passed over the chips. c-di-GMP or c-di-AMP was diluted in the reaction buffer [50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2] to concentrations of 10, 20 or 50 μM, and injected at 10 ml/min for 5 min at 25°C. Each analysis was performed in triplicate. An overlay plot was produced using the BIA evaluation 3.1 software to depict the interaction between Ms6479 and c-di-GMP. c-di-AMP, heat-denatured Ms6479 protein and an unrelated Ms6022 protein were used as negative controls.
DNA substrate preparation and assays for the interactions between regulator and target DNA fragments
DNA fragments for the DNA-binding activity assays were amplified by PCR from M. smegmatis genomic DNA or synthesized by Invitrogen (Supplementary Table S1). The amplified products were purified and subsequently labeled with [γ-32P] ATP. DNA substrates were co-incubated at 25°C for 30 min or 1 h with various amounts of proteins in a total volume of 20 ml electrophoretic mobility shift assay (EMSA) buffer consisting of 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT and 50 mM NaCl. The reaction mixtures were then subjected to 5% native PAGE. Images were acquired using a Typhoon Scanner (GE Healthcare). Bacterial one-hybrid assays were carried out as described previously (37).
DNase I footprinting assays
The 180-bp promoter regions of Ms6479p3 (both coding and non-coding strands) (Supplementary Table S1) were amplified by PCR using appropriate primers and labeled with fluorescein isothiocyanate (Supplementary Table S1). The products were purified and subjected to the same binding reaction as in EMSA. DNaseI footprinting was performed as described previously (40,45). The ladders were produced by the Sanger dideoxy method using the Ms6479p3f and Ms6479p3r primers (Supplementary Table S1).
Quantitative chromatin immunoprecipitation and real-time PCR analysis
The in vivo interactions between Ms6479 and the target promoters in M. smegmatis were investigated by ChIP assays. Mycobacterium smegmatis cells were fixed with 1% formaldehyde and cross-linked cells were resuspended in 1 ml of TBSTT (TBS 0.2% Triton X-100 0.05% Tween-20). The sample was sonicated on ice, and the complexes were immunoprecipitated with 20 μl 50% proteinA-agarose for 1 h. The immunocomplex was recovered by centrifugation and cross-linking was reversed for 6 h at 65°C. The DNA samples of the input and ChIP were purified and analyzed by PCR (Supplementary Table S3) as described previously (38). For real-time PCR analysis, each PCR (20 μl) contained 10 μl of 2× SYBR Green Master Mix Reagent (Applied Biosystems), 1.0 μl of cDNA samples and 200 nM gene-specific primers (Supplementary Table S4). The PCRs were performed as described previously (38). Amplification specificity was assessed using melting curve analysis. Expression levels of all genes were normalized to the levels of sigma A gene transcripts (46). The degrees of change in expression level were calculated using the 2ΔΔCt method (47).
Determination of mycobacterial growth curves and the effect of antibiotics
Growth patterns of the wild-type (Msm) mycobacterial strain, the ltmA-deleted mutant (Msm ltmA::hyg), the ltmA-overexpressing strain (Msm/pMV261-ltmA) and ltmA-complemented strains (Msm ltmA::hyg/pMV361-ltmA) were examined according to the procedures described previously (48) with some modifications. The ltmA-complemented M. smegmatis strains were constructed by integrating ltmA gene into the chromosome of the ltmA-deleted mutant strains. The ltmA gene was first cloned into pMV361 (49) vector, and the recombinant plasmid, pMV361-ltmA, was transformed into M. smegmatis ltmA::hyg strains. The complementation strain could be selected on the 7H10 medium (complemented with 0.2% glycerol) containing 30 μg/ml kanamycin. To determine mycobacterial growth curves and the effect of antibiotics, M. smegmatis was grown overnight in Middlebrook 7H9 media (complemented with 0.05% Tween-80 and 0.2% glycerol) containing 50 μg/ml Hyg and 30 μg/ml Kan. When cells entered a stationary growth phase (OD600 between 1.5 and 2.0), each culture was diluted (4:100) in 100 ml of fresh 7H9 broth containing the indicated concentration of each antibiotic. The cultures were then allowed to grow further at 37°C with shaking at 160 rpm. Aliquots were taken at the indicated times, and the cultures were plated on 7H10 medium (complemented with 0.2% glycerol) for the determination of colony forming units.
Isolation and detection of c-di-GMP in M. smegmatis
Isolation of c-di-GMP in M. smegmatis was performed according to the procedures previously (50) with modifications. Mycobacterium smegmatis was grown overnight at 37°C in 1L 7H9 media containing 30 μg/ml Kan and the cells were harvested and washed. About 5.5 ml, bacterial cell pellets were obtained and are resuspended in 15 ml ddH2O. To extract intracellular c-di-GMP, the samples were incubated for 20 min on ice with 0.6 M HClO4 and centrifuged at 7000 rpm for 10 min. The supernatant was neutralized to pH 6.0 with 5 M K2CO3 and was recentrifuged to remove the precipitated KClO4. The resultant supernatant was condensed to 0.5 ml. The lyophilized samples (15 μl) were resuspended in HPLC buffer A (100 mM KH2PO4 and 4 mM tetrabutyl ammonium hydrogen sulfate, pH 5.9), filtered (0.22 μm) and injected into C-18 columns for HPLC analysis and separated by reverse-phase HPLC (Waters e2695). Buffer A (100 mM KH2PO4 and 4 mM tetrabutyl ammonium hydrogen sulfate, pH 5.9) and buffer B [75% (v/v) buffer A and 25% (v/v) methanol] were used at a 2:1 gradient at a flow rate of 0.7 ml/min. Nucleotides were detected at 254 nm wavelength.
The relationship of standard curve between c-di-GMP concentration and peak area was acquired through a series of concentration gradient standard c-di-GMP. When 15 μl of a series of 0.05–1 mM standard c-di-GMP was injected into C-18 columns, respectively, for HPLC analysis, a standard curve equation Y = 9 256 866X + 2441 was obtained, in which Y represents peak area (μV × s) and X represents c-di-GMP concentration (mM). The sample peak area, derived from Msm/pMV261-b1535 or Msm/pMV261-b1535 (mut), was put into the standard curve equation and the c-di-GMP concentration of sample could be calculated. Each sample peak area was determined independently for three times. The calculated concentration of c-di-GMP from standard curve was further divided into the folds of sample condensation (11 folds, from 5.5 to 0.5 ml), and the final intracellular concentration of c-di-GMP was obtained.
Microscopic and morphological observation
Mycobacterium smegmatis cells were grown in 7H9 media for 24 h in preparation for crystal violet staining. Cells were harvested by centrifugation and washed twice in double-distilled water. Mycobacterial samples were resuspended and spread on glass slides. After fixation, the samples were stained with 2% crystal violet for 2 min and washed with water to get rid of residual crystal violet. The samples were then dried and observed under a light microscope. Scanning electron microscopy (SEM) was performed according to procedures published previously (38). Colony morphologies of the M. smegmatis strains were investigated by spot-inoculating 3 μl of overnight cultures on 7H10 medium and subsequent incubation at 37°C for 2–3 days.
RESULTS
Mycobacterium smegmatis LtmA is a candidate protein for sensing c-di-GMP
To identify regulatory proteins that specifically sense c-di-GMP in M. smegmatis, about 500 predicted regulatory genes (Genbank accession number CP000480) were cloned and their recombinant plasmids were divided into nine groups. Then, about 50–60 recombinant plasmids in one group were co-expressed and their proteins were co-purified for c-di-GMP binding screen (Supplementary Figure S1A). As shown in Supplementary Figure S1B, we discovered a positive c-di-GMP-binding signal only in a group (lane 3). Every regulator of the group was further purified and used to conduct a [32P]-labeled c-di-GMP/protein binding screen. Finally, LtmA was found to be a candidate regulator (Supplementary Figure S1C, lane 2) that binds to c-di-GMP, as evidenced by a specific and strong autoradiograph signal observed on the gel (Supplementary Figure S1D, lane 2). As expected, Rv1354, a reported M. tuberculosis c-di-GMP-binding protein (36) used as a positive control, was also observed to bind to the second messenger (Supplementary Figure S1D, lane 1). LtmA was purified in soluble form to homogeneity (about 25 kDa) (Supplementary Figure S2) for further functional evaluation. After the protein was cross-linked using UV light, it was analyzed on a PAGE. An obvious 50-kDa band corresponding to LtmA was observed (Figure 1A), suggesting that LtmA is capable of forming dimers in vitro.
![Characterization of the interaction between Ms6479 and c-di-GMP. (A) UV cross-linking assays for the formation of Ms6479 dimer. (B) Cross-linking assay for the specific binding of c-di-GMP with Ms6479. [32P]-labeled GTP (lane 5), [32P]-labeled c-di-AMP (lane 6) and Ms3425 protein (lane 7) were used as negative controls. After irradiating by UV for 20 min, reaction samples were assayed on a 12% (w/v) SDS–PAGE. Representative autoradiograph is shown (also see Supplementary Figure S1). (C) SPR assays for the specific interaction between Ms6479 and c-di-GMP. 6× His-Ms6479 (2.5 μM) was immobilized on the NTA chip and different concentrations of c-di-GMP were passed over the chip. GTP, c-di-AMP and Ms6022 were used as negative controls.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/40/22/10.1093_nar_gks923/3/m_gks923f1p.jpeg?Expires=1750452651&Signature=kluh1DFEuT9IP~vsxQyzEDSfblvzy~EsVkASd~OWoYBEKnOuubOCtNT347bbTxofFVs9YGPoZfLt-q9y-BXV0KH34K5ksdmp6lSfEc-9pWg4vNMMTwRHguzxhXPfSsYEOCL7nuCTV9VplQ~CLoCKEg6V5mKyB7U20AKC2Vf~rBOpJVbF0zLwytn35X5hoLJMky5CDufTc~kBAdEQlCZfVKsbW45c10G35ck70A53eQj~Cfzwfra6IfKBQE8nNlzzzk6TEkOwKGEQHnaCEItEZ0X-fVSnp~FM494A21CZwlMYFzhFrkAbf8p1O7xF0IMwKAUo5vJV-Gx4ahs41bJgMQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of the interaction between Ms6479 and c-di-GMP. (A) UV cross-linking assays for the formation of Ms6479 dimer. (B) Cross-linking assay for the specific binding of c-di-GMP with Ms6479. [32P]-labeled GTP (lane 5), [32P]-labeled c-di-AMP (lane 6) and Ms3425 protein (lane 7) were used as negative controls. After irradiating by UV for 20 min, reaction samples were assayed on a 12% (w/v) SDS–PAGE. Representative autoradiograph is shown (also see Supplementary Figure S1). (C) SPR assays for the specific interaction between Ms6479 and c-di-GMP. 6× His-Ms6479 (2.5 μM) was immobilized on the NTA chip and different concentrations of c-di-GMP were passed over the chip. GTP, c-di-AMP and Ms6022 were used as negative controls.
LtmA specifically binds with c-di-GMP
Next, we conducted cross-linking assays to measure the binding specificity of c-di-GMP with the LtmA protein. We found that LtmA can bind to c-di-GMP, as evidenced by a strong autoradiograph signal corresponding to LtmA bound to radioactively labeled c-di-GMP on a PAGE (Figure 1B, lane 1). As shown in Figure 1B (lanes 2–4), addition of unlabelled c-di-GMP at 10-, 100- and 2000-fold excess to the reaction mixtures competitively inhibited the binding of LtmA to [32P]-c-di-GMP, indicating specificity of LtmA binding to c-di-GMP. Importantly, addition of a different nucleotide (c-di-AMP) at a100-fold higher concentration (12.5 μM) did not alter the binding of LtmA to c-di-GMP (data not shown). In addition, LtmA did not bind with GTP or c-di-AMP molecules under the same conditions, as no band was observed in the autoradiograph (Figure 1B, lanes 5 and 6). Similarly, an unrelated protein, Ms3425, did not bind with c-di-GMP, as no corresponding signal was observed on the gel (Figure 1B, lane 7). These results indicate that LtmA can bind specifically with c-di-GMP.
Furthermore, SPR assays also confirmed the binding specificity of c-di-GMP with LtmA. As shown in Figure 1C, when an increasing amount of c-di-GMP was passed over the 6× His-LtmA immobilized NTA chip, a corresponding increase in response was observed. In particular, when 50 μM c-di-GMP was passed over the chip, a response of ∼400 RU was observed (Figure 1C, left panel). Furthermore, Kd for the specific interaction between LtmA and c-di-GMP was measured to be 0.83 μM, indicating a strong binding affinity between c-di-GMP and LtmA. No response was obtained when either heat-denatured LtmA protein (Figure 1C, left panel) or an unrelated protein, Ms6022, (Figure 1C, right panel) was immobilized on the chip. Consistent with the results of cross-linking assays described earlier, no response was observed when the same amount of GTP or c-di-AMP was passed over the chip (Figure 1C, right panel).
Taken together, these results strongly indicate that LtmA can specifically bind with the second messenger c-di-GMP.
LtmA interacts with the upstream regions of two local genes
The target promoter of the LtmA transcription factor has not yet been identified. We examined the potential binding of LtmA to the upstream regions of several neighboring gene within the LtmA local gene cluster (Figure 2A). As indicated by arrows in Figure 2A, we chose five longer intergenic sequences than 50 bp within the LtmA local gene cluster—Ms6480p, Ms6482p, Ms6484p, Ms6490p and Ms6492p—to test their interactions with LtmA. Using bacterial one-hybrid assays, these upstream DNA fragments were cloned into the reporter vector pBXcmT (37) and co-transformed with pTRG-LtmA into reporter strains. Only the LtmA+Ms6484p and LtmA+Ms6482p co-transformant strains grew well on the screening medium (Figure 2B). No growth was observed for LtmA+negative control promoter Ms6021p co-transformant strains (Supplementary Figure S3) and for the strains containing either LtmA alone or the intergenic sequence alone. These results established Ms6482p and Ms6484p as the potential target sequences of LtmA.
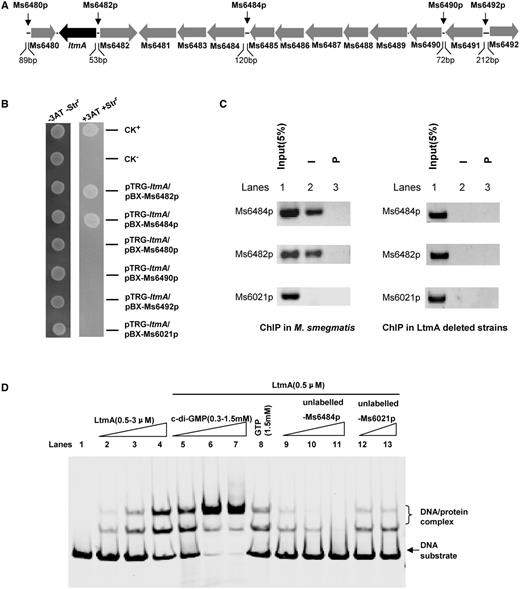
Assays for the interactions between Ms6479 and its neighboring gene upstream regions and the regulation of c-di-GMP. (A) The Ms6479 gene cluster. Several neighboring non-coding regions, which lengths are longer than 50 bp, are indicated by black arrows. (B) Bacterial one-hybrid assays. A pair of pBXcmT/pTRG plasmids was co-transformed into the reporter strain and its growth was monitored together with that of the self-activation controls on selective medium as described in ‘Materials and Methods’ section. Co-transformants containing the pBX-Rv2031/pTRG-Rv3133 plasmids (37) served as positive controls (CK+) and co-transformants containing the empty vectors pBX and pTRG served as negative controls (CK−). (C) ChIP assays. ChIP using preimmune (P) or immune sera (I) raised against Ms6479. The mycobacterial promoter Ms6021p, which does not contain motif sequence, was used as a negative control. (D) EMSA assays for the specific DNA binding activity of Ms6479 on Ms6484 promoter DNA. 32P-labeled Ms6484 promoter DNA substrate was co-incubated with Ms6479 protein in the absence (lanes 1–4) or presence of c-di-GMP (0.3–1.5 mM) (lanes 5–7) or GTP (1.5 mM) (lane 8). The ability of the unlabeled Ms6484 promoter (lanes 9–12) to compete with binding of the labeled Ms6484 promoter with the Ms6479 protein was examined.
Ms6482 is a hypothetical protein-coding gene and Ms6484 encodes a glycosyl transferase (http://www.ncbi.nlm.nih.gov/protein/). We carried out ChIP assays to investigate the specific binding of LtmA to the promoters of Ms6482 and Ms6484 in vivo in M. smegmatis. The two target promoters could be specifically recovered by ChIP using an LtmA antibody (Figure 2C). Importantly, a negative control promoter (Ms6021p) could not be recovered by LtmA antisera. The specificity of LtmA antibodies was confirmed using LtmA deletion strains (Figure 2C, right panel). These results indicate that both Ms6482p and Ms6484p are indeed target promoters of LtmA in M. smegmatis.
c-di-GMP stimulates the binding of LtmA to its target promoter in vitro
We then studied the regulatory role of c-di-GMP on the DNA-binding activity of LtmA. As shown in Figure 2D, LtmA could bind the Ms6484p DNA substrate well in an EMSA because a stepwise increase in the amount of shifted DNA was observed with increasing amounts of the LtmA protein in the reactions (lanes 1–4). Addition of unlabelled Ms6484p at 10- and 100-fold excesses to reaction mixtures competitively inhibited the binding of [32P]-Ms6484p to LtmA (Figure 2D, lanes 9–11). In contrast, an unlabeled and unrelated Ms6021 promoter DNA did not compete for binding of LtmA to labeled Ms6484p (Figure 2D, lanes 12 and 13). We then measured the effect of c-di-GMP on the DNA-binding activity of LtmA. Addition of increasing amounts of c-di-GMP (0.3–1.5 mM) led to corresponding increase in the amounts of shifted DNA substrates (Figure 2D, lanes 5–7). This indicated that c-di-GMP stimulates the DNA-binding activity of LtmA in a concentration-dependent manner (Figure 2D, lanes 5–7). In contrast, GTP had a modest effect on the DNA-binding activity of LtmA (lane 8). These results indicate that c-di-GMP specifically stimulates the ability of LtmA to bind to its target DNA.
LtmA recognizes a palindrome sequence motif
Three truncated DNA substrates covering the promoter region of Ms6484, designated as Ms6484p1, Ms6484p2 and Ms6484p3, were produced to characterize the DNA-binding motif recognized by LtmA. As shown in Supplementary Figure S4A, LtmA was found to bind Ms6484p3, but not Ms6484p2 and Ms6484p1, in EMSA assays.
In order to further map the DNA-binding motif for LtmA, DNase I footprinting assays were conducted. When increasing amounts of the LtmA protein (0–3 µM) were co-incubated with DNaseI and DNA substrates, the region around CTATAGTCTGGACACATGTCCAG AGGTTTC on the coding strand (Figure 3A, lanes 2–4) and the region around GAAACCTCTGGACATGTGTCCAGACTATAG on the non-coding strand (Figure 3B, lanes 2–4) were found to be protected, indicating that these DNA regions contained a binding site for LtmA. Strikingly, c-di-GMP was found to enhance the protective effect of LtmA in these regions in a concentration-dependent manner (Figure 3C, lanes 1–4). This observation is consistent with the results of our EMSA assays in which a stimulatory effect of c-di-GMP on the DNA-binding activity of LtmA was found (Figure 2D).
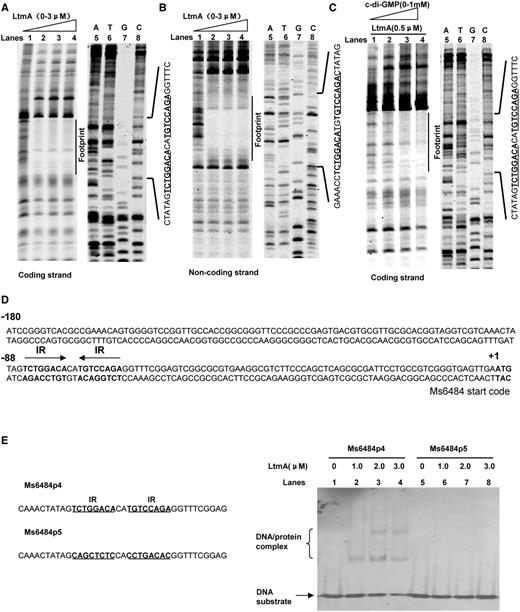
DNA-binding motif assays for Ms6479. DNaseI footprinting assays for the coding (A) and non-coding strands (B) and the effect of c-di-GMP (0–1 mM) on the ability of Ms6479 to protect the target DNA (C). Protection of the promoter DNA by Ms6479 against DNaseI digestion was tested by increasing the amount of Ms6479 (0–3 μM). The ladders are shown and the corresponding nucleotide sequence is listed (lanes 5–8). The protected regions on the coding and non-coding strands are indicated by black bars. (D) Sequence and structural characteristics of the promoter DNA region protected by Ms6479. The regions protected by Ms6479 are underlined. The 18-bp sequences containing the inverted repeats (IR) separated by 2 bp are indicated by a pair of arrows. The translation start codon of Ms6484 is indicated in bold. (E) EMSA assays for the DNA-binding activity of Ms6479 on DNA substrates with wild-type IR sequence (right panel, lanes 1–4) and mutated IR sequences (right panel, lanes 5–8). The DNA substrate was co-incubated with 1.0–3.0 μM of Ms6479 protein. Both the wild-type (Ms6484p4) and mutated (Ms6484p5) DNA substrate sequences are shown in the left panel.
The protected DNA region extended from positions −92 to −63 in the coding strand (Figure 3D). A palindromic motif formed by two inverted repeats (IR, 5′-TCTGGACA-3′) and separated from each other by 2 nt (Figure 3D) was found in the region. EMSAs on short synthesized oligonucleotides confirmed that the motif is required for specific recognition by LtmA. As shown in Figure 3E, LtmA could not bind with the Ms6484p5 fragments in which the two inverted repeats were mutated. In contrast, the protein was able to bind to an Ms6484p4 substrate in which the inverted repeats were intact.
Similarly, two regions in the coding and non-coding strands of the Ms6482 promoter DNA were also found to be protected by LtmA (Supplementary Figure S5A). EMSAs confirmed that a 14-bp motif (CGGACACGTGTCCG) in these regions was required for recognition by LtmA (Supplementary Figures S5B and Supplementary Data). Strikingly, the binding sites for LtmA found in both Ms6484 and Ms6482 promoter DNA fragments contained a common core sequence motif (GGACANNTGTCC).
Taken together, our findings indicate that the putative binding sites for LtmA contain a conserved 12-bp palindromic sequence motifs (GGACANNTGTCC).
LtmA binds with the promoters of diverse genes including 37 lipid transport and metabolism genes
We used the 12-bp sequence motif (GGACANNTGTCC) to search for LtmA target genes in the M. smegmatis genome and only two mismatches in the underlined palindromic sequences at the most are allowed. To perform the searching, the fuzznuc program was employed in local machine with the parameter ‘–pattern GGACANNTGTCC–mismatch 0-2’ against the 500-bp upstream of each coding sequences (51), which were extracted from the M. smegmatis genome using Perl script. We identified a total of 229 potential target promoters covering 336 genes (Supplementary Table S5 and Supplementary Figure S6). We further classified these target genes in terms of clusters of orthologous groups categories. We found that 37 of them are lipid transport and metabolism genes, 21 are transcriptional regulators, 13 are cell wall/membrane biogenesis genes and many are transport- and metabolism-related genes (Supplementary Figure S7). Majority of the target genes are involved in mycolic acid metabolism in mycobacteria (Supplementary Table S6) (52). We further used the WebLogo tool (53) to characterize a more general conserved motif sequence for LtmA binding (Figure 4A and B). Furthermore, all 37 target promoters of lipid transport and metabolism genes could be specifically recovered in a ChIP assay using an LtmA antibody (Figure 4C). Importantly, a negative control promoter (Ms6021p) could not be recovered using the LtmA antibody under the same conditions. These results indicate that LtmA can recognize the promoters of diverse target genes including at least 37 lipid transport and metabolism genes.
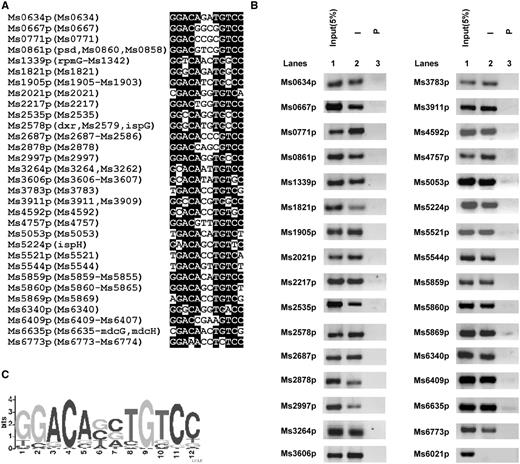
Assays for in vivo DNA-binding activity of Ms6479. (A) The IR-containing motif GGACACATGTCC was used to search the intergenic regions between M. smegmatis Open Reading Frames. The conserved sequence is highlighted by black background. The included genes in the same operon were presented in the brackets, however, the entire gene names were provided in Supplementary Table S5 limited by figure space. (B) ChIP assays for the in vivo association of Ms6479 with the promoter sequences of lipid transport and metabolism target genes using preimmune (P) or immune sera (I) raised against Ms6479. DNA recovered from the immunoprecipitates was amplified. Ms6021p was used as a negative control. (C) Logo assays for the protected region. The logos were generated using the MEME software suite (50).
Overexpression of LtmA affects the mycobacterial sensitivity to crystal violet staining and colony morphology
Most of the potential target genes identified above have been implicated in the synthesis of mycolic acids (Supplementary Table S6), a major constituent of mycobacterial cell wall (52). This suggested that LtmA could be involved in the regulation of cell wall metabolism. To test this hypothesis, we first examined the sensitivity of mycobacteria to crystal violet staining. Examination of the mycobacterial cell walls under a light microscope revealed that the wild-type M. smegmatis strain was stained easily by crystal violet (Figure 5A, left panel). In contrast, very weak crystal violet staining was observed for the LtmA-overexpressed strain (Figure 5A, right panel), suggesting that LtmA is involved in the regulation of mycobacterial cell wall permeability or cell wall composition. This interpretation is consistent with previous conclusions (52) that mycolic acid metabolism affects the permeability of mycobacterial cell wall and sensitivity to crystal violet. However, no obvious change in cell morphology was observed in the LtmA-overexpressed strain when assayed under SEM (Figure 5B) or under the light microscope (Figure 5A). Interestingly, a significant difference was observed in the spot-colony morphology of the cells on 7H10 agar solid medium between the wild-type and LtmA-overexpressed strains (Figure 5C). The wild-type strain presented a typical rough-edged colony and rugose surface (Figure 5C, left panel). In contrast, the overexpression strain had a relatively smooth-edged colony and lacked rugose surface (Figure 5C, right panel). Taken together, these results strongly suggest that LtmA may regulate the sensitivity to crystal violet staining and colony morphology in mycobacteria.
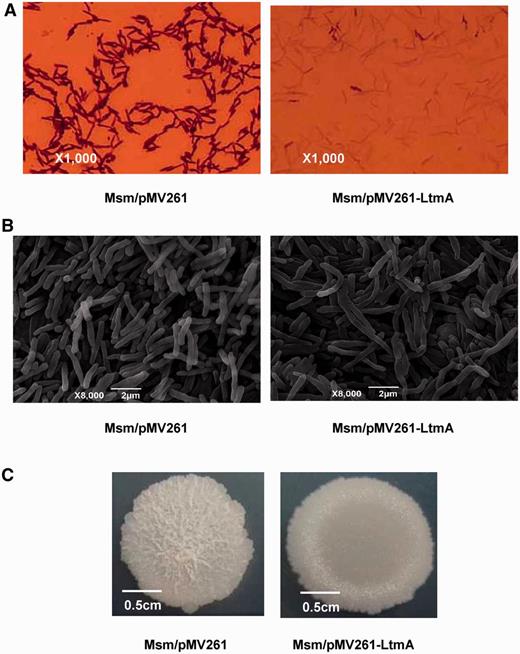
Effects of Ms6479 on M. smegmatis cell and colony morphology. (A) Cells were observed with a light microscope after staining with 2% crystal violet. The Ms6479 overexpression strain Msm/pMV261-Ms6479 (right panel) stained much weaker than the wild-type Msm/pMV261 strain (left panel). (B) Scanning electron microscopy assays of cell morphology. The images were taken at 8000× magnification. Bars, 2 μm. (C) Representative colony morphology of the M. smegmatis spot on 7H10 medium.
LtmA positively regulates the expression of lipid transport and metabolism genes
In order to characterize the function of LtmA, an LtmA-deleted mutant strain of M. smegmatis was generated by a gene replacement strategy (40) (Supplementary Figure S8). We then carried out qRT–PCR analysis to compare the expression levels of lipid transport and metabolism genes in both wild-type and LtmA-deleted mutant M. smegmatis strains to assess the regulatory role of the c-di-GMP-responsive regulator. As shown in Figure 6A and Supplementary Figure S9, expression of most of the tested genes, indicated by an asterisk (*) on top of the column in the figure, was found to be significantly down-regulated (P < 0.05) in the mutant strain compared to the wild-type. In contrast, expression level of the negative control gene Ms6021 was not significantly different between the wild-type and the mutant strains. These results indicate that LtmA functions as a positive regulator in M. smegmatis and are consistent with the results of our LtmA overexpression assays. As shown in Figure 6B and Supplementary Figure S9, compared with the wild-type strain, the expressions of many target genes, indicated by an asterisk (*) on top of the column in the figure, were significantly up-regulated (P < 0.05) when LtmA was overexpressed using a pMV261-derived recombinant plasmid. Importantly, expression level of Ms6021, an unrelated gene, did not change upon overexpression of LtmA. Based on these results, we conclude that LtmA functions as a positive regulator of lipid transport and metabolism genes in M. smegmatis.
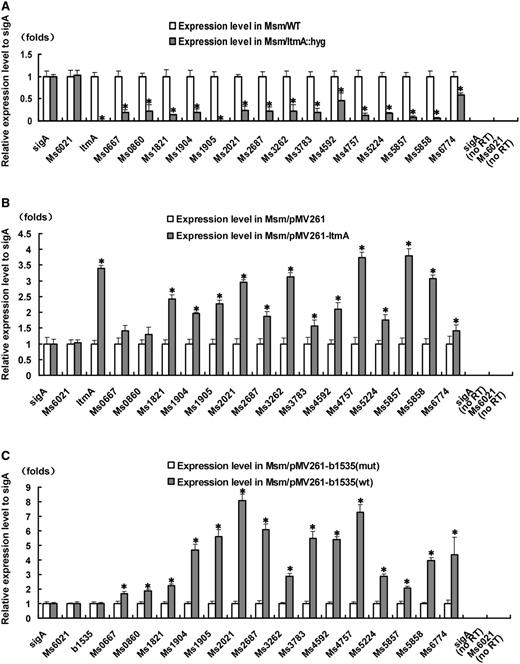
Effects of Ms6479 and c-di-GMP synthesis on the expression of lipid transport and metabolism related genes in M. smegmatis. qRT-PCR assays for determining the relative expression levels of lipid transport and metabolism genes were performed as described in ‘Materials and Methods’ section. Expression levels of genes were normalized using the sigA gene as an invariant transcript and an unrelated Ms6021 gene was used as a negative control. Error bars represent the variant range of the data derived from three biological replicates. The P-values of the relative expression data were calculated by unpaired two-tailed Student’s t-test using GraphPad Prism 5. The P-values of the results (<0.05) are indicated by an asterisk (*) on top of the column in the figure. Relative expression levels of target genes in Msm and Msm/ΔMs6479 (A), in Msm/pMV261 and Msm/pMV261-Ms6479 (B), and in Msm/pMV261-b1535 mut and Msm/pMV261-b1535 wt (C) strains were assayed. b1535wt encodes a diguanylate cyclase that can elevate c-di-GMP levels in M. smegmatis (42). b1535 mut contained mutations that abolished the diguanylate cyclase activity of the wild-type protein. As a positive control, total DNA of each strain was used as template for PCR amplification. The cDNA of the mutant strains and the recombinant strain containing an empty pMV261 vector was used as template in the negative controls. no RT represents genomic DNA contamination control. Data were analyzed using the 2ΔΔCt method (38,47).
LtmA target genes respond to the level of c-di-GMP in M. smegmatis
We further assessed the ability of LtmA target genes to respond to the level of c-di-GMP in vivo in M. smegmatis. Using a previously reported strategy (41), we overexpressed the wild-type b1535wt gene from E. coli (42) and its mutant variant b1535mut to provide different levels of c-di-GMP in M. smegmatis. The b1535wt protein exhibited strong DGC activity, while the b1535mut protein lacked DGC activity (data not shown) in M. smegmatis.
As shown in Figure 6C, the expressions of the tested target genes, indicated by an asterisk (*) on top of the column in the figure, were significantly up-regulated (2- to 8-fold higher) (P < 0.05) in the b1535wt-overexpressing mycobacterial strain compared with those in the strain overexpressing b1535mut. This result is consistent with our observation that LtmA is a positive regulator of its target genes (Figure 6A and B) and that c-di-GMP stimulates the DNA-binding activity of LtmA (Figure 2D). Importantly, no obvious difference was observed in the expression levels of wild-type and mutant DGC genes, indicating that their effects on the targets of LtmA were related to DGC activity, not to their expression level. In addition, no response was observed for the negative control gene Ms6021.
In order to ascertain that c-di-GMP levels were indeed changed in the b1535wt- and b1535mut-overexpressing strains, we quantified cellular c-di-GMP levels in these strains. As shown in Figure 7, when the c-di-GMP control and mycobacterial cell extracts were subjected to HPLC, samples of both b1535wt- and b1535mut-overexpressing mycobacterial strains showed characteristic peaks at about 8.5 min, as was seen for the c-di-GMP control. However, the peak for b1535mut-overexpressing strain was smaller (4.5 μM) compared to that of the b1535wt-overexpressing strain (28 μM), indicating that overexpression of b1535 could indeed enhance cellular c-di-GMP levels in M. smegmatis.
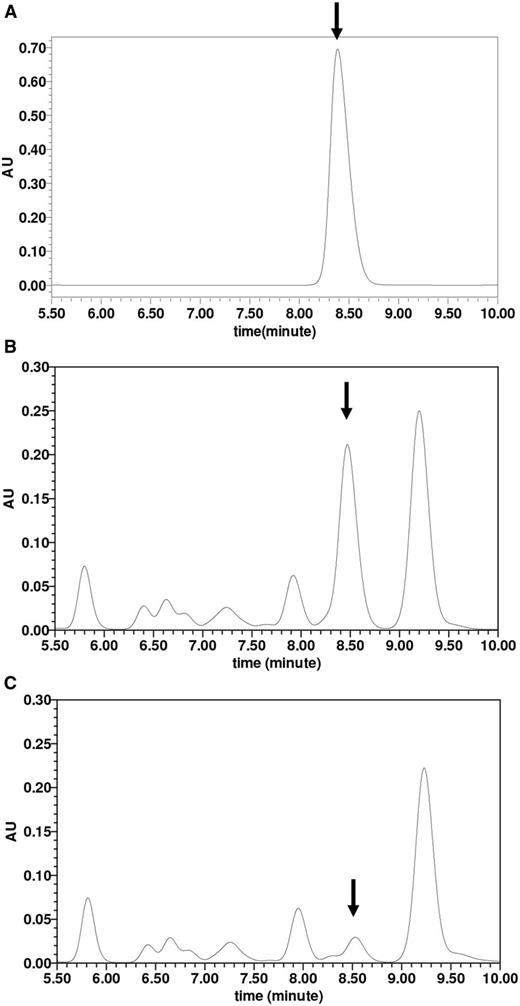
Assays for determining the intracellular c-di-GMP concentration in M. smegmatis. Extraction of mycobacterial nucleotides and detection of c-di-GMP in M. smegmatis were performed as described in ‘Materials and Methods’ section. Together with the purified c-di-GMP positive control (A), the lyophilized samples (15 μl) derived from the Msm/pMV261-b1535 (B) and Msm/pMV261-b1535(mut) (C) were assayed in HPLC buffer A on C-18 columns for HPLC analysis and separated by reverse-phase HPLC as described in ‘Materials and Methods’ section. The peak for c-di-GMP is indicated by arrows. The peak areas from three independent experimental samples for Msm/pMV261-b1535 and for Msm/pMV261-b1535(mut) were put into the standard curve equation Y = 9 256 866X + 2441. The final intracellular concentration of c-di-GMP was obtained for Msm/pMV261-b1535 (28 μM) and for Msm/pMV261-b1535(mut) (4.5 μM).
Taken together, these results indicate that the target genes of LtmA can respond to c-di-GMP levels in M. smegmatis.
LtmA regulates antibiotic resistance in M. smegmatis
We further assayed the regulatory effect of LtmA on the growth of M. smegmatis in response to two anti-TB antibiotics by determining mycobacterial growth curves. No obvious difference was observed in the growth of wild-type, LtmA-deleted and LtmA-overexpressed strains in 7H9 medium in the absence of antibiotics (Figure 8A). However, compared with the wild-type strain, the LtmA-deleted M. smegmatis strain grew more slowly in 7H9 medium containing 8 µg/ml of RFP (Figure 8B) or 5 µg/ml of INH (Figure 8C) and only a modest increase in the number of mycobacterial cells was observed in both cases. We calculated the P-values of the number of LtmA-deleted M. smegmatis cells at two time points, 44 and 48 h (Figure 8B and C), and found that they were <0.05 [indicated by a star ( ) above the error bars in the figure]. In contrast, the LtmA-overexpressed strain grew faster than the wild-type strain under the same conditions (Figure 8B and C). The P-values of the number of cells at these two time points, 44 and 48 h (Figure 8B and C), were calculated and were <0.05 [indicated by an asterisk (*) above the error bars in the figure]. Thus, the difference was statistically significant. Strikingly, when expressing ltmA gene in the LtmA-deleted M. smegmatis strain, the recombinant strain re-obtained a similar growth compared with the wild-type strain with the empty pMV261 plasmid (Figure 8B and C). These results suggest that LtmA positively regulates antibiotic resistance of M. smegmatis and are consistent with our other findings that LtmA stimulates the expression of many mycolic acid metabolism genes and reduces the sensitivity of M. smegmatis to crystal violet staining.
) above the error bars in the figure]. In contrast, the LtmA-overexpressed strain grew faster than the wild-type strain under the same conditions (Figure 8B and C). The P-values of the number of cells at these two time points, 44 and 48 h (Figure 8B and C), were calculated and were <0.05 [indicated by an asterisk (*) above the error bars in the figure]. Thus, the difference was statistically significant. Strikingly, when expressing ltmA gene in the LtmA-deleted M. smegmatis strain, the recombinant strain re-obtained a similar growth compared with the wild-type strain with the empty pMV261 plasmid (Figure 8B and C). These results suggest that LtmA positively regulates antibiotic resistance of M. smegmatis and are consistent with our other findings that LtmA stimulates the expression of many mycolic acid metabolism genes and reduces the sensitivity of M. smegmatis to crystal violet staining.
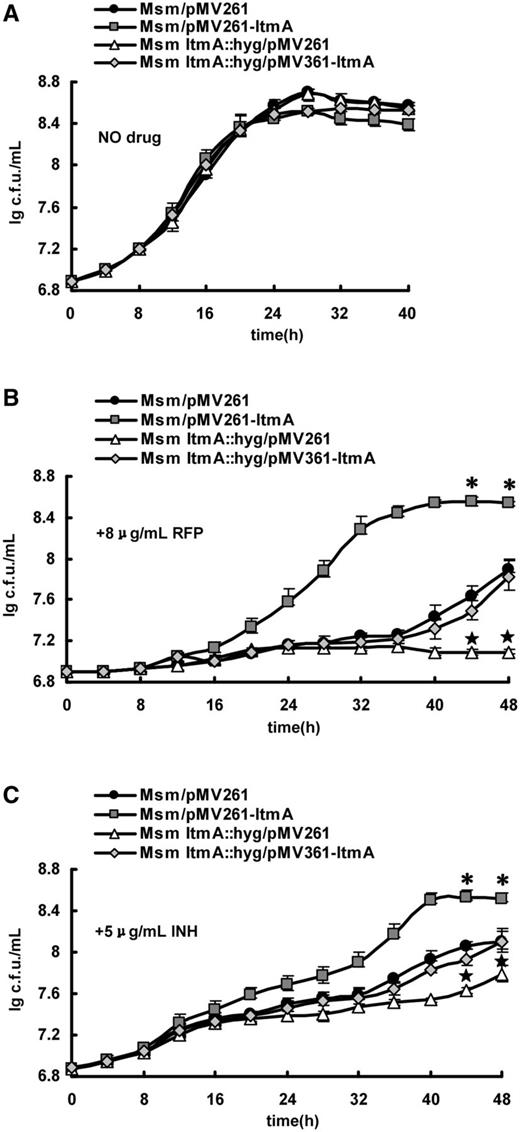
Assays for the effects of LtmA on the antibiotic resistance of M. smegmatis. Growth curves of the wild-type, ltmA-overexpressed, ltmA-deleted and ltmA-complemented strains were determined as described in ‘Materials and Methods’ section. These mycobacterial strains were grown in 7H9 media in the absence of anti-TB antibiotic (A) and in the presence of 8 μg/ml RFP (B) or 5 μg/ml INH (C). Representative growth curves are shown. Error bars represent the variant range of the data derived from three biological replicates. The P-values of the relative growth data were calculated by unpaired two-tailed Student’s t-test using GraphPad Prism 5. The P-values of the results at two time points, 44 and 48 h, were <0.05, which were indicated by an asterisk (*) for LtmA-overexpressed strain or by a star ( ) for LtmA-deleted strain above the error bars in the figure.
) for LtmA-deleted strain above the error bars in the figure.
DISCUSSION
Currently, several transcription factors are known to be c-di-GMP receptor/effectors in bacteria. However, these regulators were linked to c-di-GMP mostly through phenotype or mutant screens (23,29). In the present study, we conducted a c-di-GMP/protein binding assay with radioactively labeled c-di-GMP (54) to identify novel c-di-GMP receptors/effectors. Our strategy had the advantage of not requiring detailed prior information about potential target proteins. We successfully identified a new c-di-GMP receptor/effector designated as LtmA in M. smegmatis. Interestingly, LtmA encodes a hypothetical regulator that contains a TetR-type HTH domain signature and its amino acid sequence is conserved in several mycobacterial species (Supplementary Figure S10). Our findings validate the effectiveness of our strategy, which should be applicable for exploring additional c-di-GMP receptors/effectors with little to no information about their function.
Several c-di-GMP-responsive transcription factors have been reported in other bacterial species. Strikingly, most of these receptors/effectors are involved in the regulation of a limited number of target genes, and their direct target genes remain largely unclear. In the current study, we used DNaseI footprinting experiment in combination with EMSA assays to characterize an 12-bp palindromic motif as the specific DNA sequence recognized by LtmA. Furthermore, LtmA was shown to significantly activate expressions of 37 lipid transport and metabolism related genes in our overexpression experiments. Consistently, expression of these target genes was strikingly down-regulated in LtmA-deleted mutant strains. Taken together, our findings indicate that LtmA functions as an activator of these target genes in M. smegmatis.
Several c-di-GMP binding motifs have been reported in previous studies (55)—the PilZ-like domain in the P. aeruginosa PilZ protein (21), the degenerate GGDEF/EAL domain in the P. aeruginosa PelD protein (8), GEMM (RNA effectors) domain in the V. cholerae Vc1 protein (25) and the AAAδ54 interaction domain in the P. aeruginosa FleQ protein (26). However, we failed to find any of these known c-di-GMP binding motifs within the LtmA protein. Therefore, our results indicate that LtmA represents a novel class of receptors/effectors in M. smegmatis. Interestingly, an ortholog of LtmA, Rv0767c, was also found in the pathogen M. tuberculosis H37Rv and c-di-GMP could stimulate its DNA-binding activity (Supplementary Figure S10).
Mycolic acids are the major components of mycobacterial cell wall, which are largely responsible for the impermeability and lack of effectiveness of many chemotherapeutic agents (56–59). In the present study, a number of cell envelope associated genes were found to be the direct targets of LtmA, including 37 lipid transport and metabolism genes, 13 cell wall/membrane biogenesis genes and more than 50 transport and metabolism genes (Supplementary Figure S7). Many of them are directly involved in metabolism of mycolic acids in mycobacteria (Supplementary Table S6) (60). Based on qRT–PCR assays in recombinant strains overexpressing LtmA, we found that LtmA functions as an activator of these target genes and that these genes can respond to c-di-GMP levels in M. smegmatis. These data strongly suggest that LtmA is involved in the regulation of mycobacterial cell wall metabolism. Consistent with this model, we observed a striking difference in crystal violet staining between wild-type and LtmA-overexpressed strains of M. smegmatis (Figure 5). Compared with the wild-type strain, the recombinant strain showed weak crystal violet staining, which can be explained by inhibition of crystal violet entry through the cell wall in the overexpression strain.
In summary, this study represents the first description of a transcription factor which directly senses c-di-GMP in mycobacteria. LtmA is a broad activator and c-di-GMP can enhance its ability to bind to its target DNA. This finding expands the range of c-di-GMP-responsive regulators identified in bacteria and further studies on it can help improve our understanding of c-di-GMP signaling pathways in mycobacteria.
ACKNOWLEDGEMENTS
The authors thank Tao Cui and Chunhui Gao for their bioinformatic support.
FUNDING
National Natural Science Foundation of China [31025002 and 30930003]; Fundamental Research Funds for the Central Universities [2011PY140]; Creative Research Groups of Hubei and Hubei Chutian Scholar Program [to Z.G.H.]. Funding for open access charge: National Natural Science Foundation of China [31025002].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments