





Abstract
Human purine de novo synthesis pathway contains several multi-functional enzymes, one of which, tri-functional GART, contains three enzymatic activities in a single polypeptide chain. We have solved structures of two domains bearing separate catalytic functions: glycinamide ribonucleotide synthetase and aminoimidazole ribonucleotide synthetase. Structures are compared with those of homologous enzymes from prokaryotes and analyzed in terms of the catalytic mechanism. We also report small angle X-ray scattering models for the full-length protein. These models are consistent with the enzyme forming a dimer through the middle domain. The protein has an approximate seesaw geometry where terminal enzyme units display high mobility owing to flexible linker segments. This resilient seesaw shape may facilitate internal substrate/product transfer or forwarding to other enzymes in the pathway.
INTRODUCTION
The biosynthesis of nucleotides is a crucial process for the production of building blocks for RNA and DNA, as well as to provide nucleotides for additional cellular processes where they, for example, serve as energy carriers or signaling molecules. In the cell, de novo synthesis pathways where nucleotides are synthesized from small metabolites, work in coordination with salvage pathways, where nucleotides are reversibly regenerated from nucleosides and nucleobases, to control the intracellular nucleotide pools. Due to the central role of nucleotides in the human cell, enzymes of the nucleotide metabolism constitute potential anti-proliferative drug targets for the treatment of e.g. cancer or autoimmune diseases (1). Furthermore, enzymes of the nucleotide metabolism in pathogens also provide interesting targets for the development of antibiotics (2). Genetic variations in the human nucleotide metabolism are also known to cause several disorders including gout and Lech–Nyhan syndrome (3).
In the de novo pathways for purine synthesis, phosphoribosyl pyrophosphate is converted to inosine monophosphate in 10 steps (4). In humans, these steps are catalyzed by six different enzymes; three monofunctional, two bifunctional and one tri-functional enzyme (5). Although many of the enzymes of the nucleotide metabolism are well characterized from prokaryotic sources the eukaryotic counterparts are less well characterized. In our current effort to systematically characterize the structure and mechanism of the enzymes of the human nucleotide metabolism (see http://sgc.ki.se/structures.html) we have determined structures of components of the tri-functional human GART (HsGART) (108 kDa, 1010 amino acids). HsGART is composed of three enzyme units: glycinamide ribonucleotide synthetase (GARS, PurD, E.C. 6.3.4.13), aminoimidazole ribonucleotide synthetase (AIRS, PurM, E.C. 6.3.3.1) and glycinamide ribonucleotide transformylase (GARTfase, PurN, E.C. 2.1.2.2.). HsGART thus catalyzes steps 2, 3 and 5 of the de novo purine synthesis pathway (Figure 1).
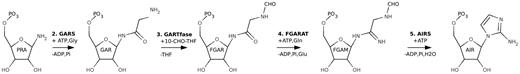
Steps 2–5 of the purine de novo synthesis pathway. Enzymes and intermediates are named as defined in the text.
The N-terminal enzyme unit GARS catalyses the second step of the purine pathway and uses phosphoribosylamine (PRA), the product of PRPP amidotransferase (PPAT, PurF, E.C. 2.4.2.14), as well as ATP and glycine to make glycinamide ribonucleotide (GAR), ADP and Pi (Figure 1). The C-terminal GARTfase unit carries out the third-reaction step in the synthesis by forming N-formylglycinamide ribonucleotide (FGAR) from GAR using 10-formyltetrahydrofolate as a cofactor (Figure 1). Finally the middle domain of HsGART, AIRS, uses formylglycinamidine ribonucleotide (FGAM) and ATP to form aminoimidazole ribonucleotide (AIR), ADP and Pi (Figure 1). The central fourth step of the purine pathway is performed by phosphoribosyl formylglycineamide amidotransferase (FGARAT, purL, E.C.6.3.5.3), which is encoded by a separate gene (purL) (1).
Notably, PRA, the substrate of GARS, is very unstable and hydrolyzes spontaneously to ribose 5-phosphate and has a half-life of only 5 s at 37°C (6). PRA has therefore been suggested to be transferred directly from PPAT to GARS (7). However, interactions between these protein components are believed to be transient as no direct interactions have been observed (6).
Being an enzyme in the core nucleotide metabolism, HsGART is a potential target for anti-proliferative drugs. The C-terminal GARTfase domain uses folate as a cofactor, and inhibitors of folate-dependent enzymes have been demonstrated to have anti-tumor activity in vivo (8). HsGART is localized to chromosome 21 (9) and trisomy of this chromosome is observed in Down syndrome. Individuals with Down syndrome have also been shown to have increased levels of serum purines that may be caused by the overexpression of HsGART (10).
Here, we present the structures of two functional domains of HsGART: GARS and AIRS. Together with the previous structures of the GARTfase domain of HsGART (11–12), this completes the structural characterization of the individual functional units of HsGART, thereby providing a structural framework for understanding substrate specificity and catalytic mechanism, as well as for structure-based drug design. Furthermore, we present the low-resolution structure of the complete tri-functional enzyme obtained by combining small angle X-ray scattering (SAXS) data with the high-resolution crystal structures, revealing the overall architecture of the protein. The latter analysis might also have implications for HsGART’s role as a potential core unit of the recently discovered multi-enzyme complex, ‘the purinosome’, which is assembled in response to purine levels in the cell (13).
METHODS
Cloning
The clone for the GART gene was obtained from National Institute of Health Mammalian Gene Collection (MGC; accession no. BC107712). The sequences encoding residues 1–430 (GARS) and 1–1003 (GARS–AIRS–GARTfase) were amplified by PCR and inserted into pNIC28-Bsa4 vector by ligation independent cloning. Constructs include an N-terminal tag containing a 6-His sequence (MHHHHHHSSGVDLGTENLYFQSM). For expression, the pNIC28-Bsa4 vector containing the insert was transformed into Escherichea coli BL21(DE3) gold pRARE2 strain.
Protein expression
GARS
One hundred and fifty milliliter of LB supplemented with 8 g/l glycerol, 50 µg/ml kanamycin and 34 µg/ml and chloramphenicol was inoculated and grown at 37°C overnight. Of the overnight cultures, 120 ml were used to inoculate 3 l (4 × 750 ml) of Terrific Broth (TB) supplemented with 8 g/l glycerol and antibiotics. Cells were grown in TunAir flasks at 37°C until OD600 reached 1.5. Cultivations were cooled down to 18°C and protein expression was induced by addition of 0.5 mM IPTG at OD600 of 2.5 and the growth was allowed to continue overnight. Cells were harvested (OD600 15.2) by centrifugation (5500g, 20 min, 4°C). The resulting pellet (93.8 g wet cell weight) was frozen at −80°C.
GARS–AIRS–GARTfase
One hundred and fifty milliliter LB media supplemented with glycerol and antibiotics was inoculated and grown overnight at 37°C. TB media (3 l in 4 × 750 ml) supplemented with glycerol, antibiotics and 200 µl BREOX FMT 30 anti-foam solution (Cognis Performance Chemicals UK Ltd) was inoculated with 80 ml of the overnight cultures. Cells were grown at 37°C and when OD600 reached the value of 2 the temperature was cooled down to 18°C and the culture induced with 0.5 mM IPTG. Protein expression was continued overnight. The cells (106 g) were harvested by centrifugation (4400g, 10 min, 4°C) and frozen.
Protein purification
GARS
Cells were thawed and resuspended in lysis buffer (0.5 ml/g of cells) containing 100 mM HEPES pH 8.0, 500 mM NaCl, 10% glycerol, 10 mM imidazole and 0.5 mM TCEP. Protease inhibitor cocktail (one tablet of complete, EDTA-free, Roche applied Science), ∼0.25 mg of lysozyme (Sigma) and 1000 U of benzonase (Merc) was added and the sample was sonicated using Sonics VibraCell at 80% amplitude for 3 min (pulsed 4 s on, 4 s off). The sample was centrifuged for 60 min at 34000g. Soluble fraction was decanted and filtered through a 0.45 µm filter. The purification was conducted on an ÄKTA Prime (GE Healthcare). A HisTrap HP 5 ml (IMAC) (GE Healthcare) column was equilibrated with binding buffer (20 mM HEPES pH 7.5, 10 mM imidazole, 500 mM NaCl, 10% glycerol, 0.5 mM TCEP). The sample was loaded and the column was supplemented with buffer containing 25 mM imidazole. The protein was eluted with buffer supplemented with 500 mM imidazole. The second purification step, gel-filtration, was conducted on a Superdex 200 High Load 16/60 Prep Grade (GF), (GE Healthcare) using a gel filtration buffer containing 20 mM HEPES, pH 7.5, 300 mM NaCl, 10% glycerol, 0.5 mM TCEP. The protein eluted in a single peak from the GF column. Fractions containing monomeric protein were pooled and the protein was concentrated with Vivaspin (Sartorius Stedim Biotech) with a MWCO of 10 kDa to 21.5 mg/ml in a volume of 0.72 ml. Small aliquots were flash frozen in liquid nitrogen and stored at −80°C.
GARS–AIRS–GARTfase
Lysis buffer (1 ml/g cell pellet) supplemented with protease inhibitor cocktail, ∼0.25 mg of lysozyme and 1000 U of benzonase was added to the thawed pellet prior to sonication. The sample was centrifuged; the soluble fraction was decanted and filtered. Protein purification was carried out on an ÄKTAxpress system (GE Healthcare) operated by UNICORN. Prior to purification, columns were equilibrated with binding buffer (1 ml HiTrap Chelating HP, GE Healthcare) and gel filtration buffer (Superdex 200). The protein sample was loaded on the HiTrap Chelating column and washed with binding buffer followed by a wash with buffer supplemented with 25 mM imidazole. Bound protein was eluted from the IMAC columns with buffer supplemented with 500 mM imidazole and loaded onto a gel filtration column. Fractions containing GARS–AIRS–GART were pooled and fresh TCEP was added to the final concentration of 2 mM. Concentration was performed using a Vivaspin to a concentration of 12.4 mg/ml in a volume of 0.5 ml and flash frozen.
Crystallization, data collection and structure determination
GARS
Crystals were obtained by hanging drop vapor diffusion. Protein solution (15.7 mg/ml, 1 μl) containing also 4 mM ATP and 4 mM glycine was mixed with precipitant solution (1 μl) containing 16% PEG 3350, 100 mM Bis–Tris pH 5.2 and 300 mM lithium sulfate. The drops were equilibrated at 20°C over 500 μl of well solution. A crystal was dipped into a cryo solution (100 mM Bis–Tris pH 5.2, 300 mM lithium sulfate, 17% PEG-3350, 200 mM NaCl and 20% glycerol) and flash-frozen in liquid nitrogen. Data were collected at the ESRF beamline ID14-2 and processed with XDS (14) (Table 1).
Data and refinement statistics
| GARS | AIRS | |
|---|---|---|
| Beamline | ID14-2 (ESRF) | BL 14.1 (BESSY) |
| Wavelength (Å) | 0.93300 | 1.00 |
| Space group | P21 | P21212 |
| Resolution (Å) | 20–2.45 (2.50–2.45) | 15–2.10 (2.15–2.10) |
| Cell dimensions | ||
| a, b, c (Å) | 70.3, 79.8, 113.9 | 80.7, 81.0, 98.3 |
| β (°) | 104.1 | |
| Resolution (Å) | 2.45 | 2.1 |
| Rmergea | 0.049 (0.341) | 0.08 (0.39) |
| I/σ(I) | 22.15 (3.94) | 28.70 (8.06) |
| Completeness (%) | 99.2 (91.7) | 99.6 (100) |
| Redundancy | 5.6 (3.4) | 14.8 (14.9) |
| Refinement | ||
| Number of reflections | 42 544 | 38 171 |
| Rworkb/Rfreec | 0.174/0.221 | 0.194/0.240 |
| No. of atoms | ||
| Protein | 6262 | 4695 |
| Water | 111 | 170 |
| Other | 98 | 15 |
| B-factors (Å2) | ||
| Protein | 60.3 | 32.9 |
| Water | 51.6 | 34.4 |
| Other | 73.9 | 33.2 |
| RMSD | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.546 | 1.477 |
| Ramachandran plot (%)d | ||
| Favored regions | 95.79 | 98.86 |
| Additionally allowed regions | 4.09 | 1.14 |
| GARS | AIRS | |
|---|---|---|
| Beamline | ID14-2 (ESRF) | BL 14.1 (BESSY) |
| Wavelength (Å) | 0.93300 | 1.00 |
| Space group | P21 | P21212 |
| Resolution (Å) | 20–2.45 (2.50–2.45) | 15–2.10 (2.15–2.10) |
| Cell dimensions | ||
| a, b, c (Å) | 70.3, 79.8, 113.9 | 80.7, 81.0, 98.3 |
| β (°) | 104.1 | |
| Resolution (Å) | 2.45 | 2.1 |
| Rmergea | 0.049 (0.341) | 0.08 (0.39) |
| I/σ(I) | 22.15 (3.94) | 28.70 (8.06) |
| Completeness (%) | 99.2 (91.7) | 99.6 (100) |
| Redundancy | 5.6 (3.4) | 14.8 (14.9) |
| Refinement | ||
| Number of reflections | 42 544 | 38 171 |
| Rworkb/Rfreec | 0.174/0.221 | 0.194/0.240 |
| No. of atoms | ||
| Protein | 6262 | 4695 |
| Water | 111 | 170 |
| Other | 98 | 15 |
| B-factors (Å2) | ||
| Protein | 60.3 | 32.9 |
| Water | 51.6 | 34.4 |
| Other | 73.9 | 33.2 |
| RMSD | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.546 | 1.477 |
| Ramachandran plot (%)d | ||
| Favored regions | 95.79 | 98.86 |
| Additionally allowed regions | 4.09 | 1.14 |
Values for the highest resolution shell are shown in parentheses.
aRmerge = Σi |Ii − 〈I〉| / Σ 〈I〉, where I is an individual intensity measurement and 〈I〉 is the average intensity for this reflection with summation over all data.
bRwork is defined as Σ||Fobs| − |Fcalc|| / Σ |Fobs|, where Fobs and Fcalc are observed and calculated structure-factor amplitudes, respectively.
cRfree is the R-factor for the test set (5% of the data).
dAccording to molprobity (44).
Data and refinement statistics
| GARS | AIRS | |
|---|---|---|
| Beamline | ID14-2 (ESRF) | BL 14.1 (BESSY) |
| Wavelength (Å) | 0.93300 | 1.00 |
| Space group | P21 | P21212 |
| Resolution (Å) | 20–2.45 (2.50–2.45) | 15–2.10 (2.15–2.10) |
| Cell dimensions | ||
| a, b, c (Å) | 70.3, 79.8, 113.9 | 80.7, 81.0, 98.3 |
| β (°) | 104.1 | |
| Resolution (Å) | 2.45 | 2.1 |
| Rmergea | 0.049 (0.341) | 0.08 (0.39) |
| I/σ(I) | 22.15 (3.94) | 28.70 (8.06) |
| Completeness (%) | 99.2 (91.7) | 99.6 (100) |
| Redundancy | 5.6 (3.4) | 14.8 (14.9) |
| Refinement | ||
| Number of reflections | 42 544 | 38 171 |
| Rworkb/Rfreec | 0.174/0.221 | 0.194/0.240 |
| No. of atoms | ||
| Protein | 6262 | 4695 |
| Water | 111 | 170 |
| Other | 98 | 15 |
| B-factors (Å2) | ||
| Protein | 60.3 | 32.9 |
| Water | 51.6 | 34.4 |
| Other | 73.9 | 33.2 |
| RMSD | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.546 | 1.477 |
| Ramachandran plot (%)d | ||
| Favored regions | 95.79 | 98.86 |
| Additionally allowed regions | 4.09 | 1.14 |
| GARS | AIRS | |
|---|---|---|
| Beamline | ID14-2 (ESRF) | BL 14.1 (BESSY) |
| Wavelength (Å) | 0.93300 | 1.00 |
| Space group | P21 | P21212 |
| Resolution (Å) | 20–2.45 (2.50–2.45) | 15–2.10 (2.15–2.10) |
| Cell dimensions | ||
| a, b, c (Å) | 70.3, 79.8, 113.9 | 80.7, 81.0, 98.3 |
| β (°) | 104.1 | |
| Resolution (Å) | 2.45 | 2.1 |
| Rmergea | 0.049 (0.341) | 0.08 (0.39) |
| I/σ(I) | 22.15 (3.94) | 28.70 (8.06) |
| Completeness (%) | 99.2 (91.7) | 99.6 (100) |
| Redundancy | 5.6 (3.4) | 14.8 (14.9) |
| Refinement | ||
| Number of reflections | 42 544 | 38 171 |
| Rworkb/Rfreec | 0.174/0.221 | 0.194/0.240 |
| No. of atoms | ||
| Protein | 6262 | 4695 |
| Water | 111 | 170 |
| Other | 98 | 15 |
| B-factors (Å2) | ||
| Protein | 60.3 | 32.9 |
| Water | 51.6 | 34.4 |
| Other | 73.9 | 33.2 |
| RMSD | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.546 | 1.477 |
| Ramachandran plot (%)d | ||
| Favored regions | 95.79 | 98.86 |
| Additionally allowed regions | 4.09 | 1.14 |
Values for the highest resolution shell are shown in parentheses.
aRmerge = Σi |Ii − 〈I〉| / Σ 〈I〉, where I is an individual intensity measurement and 〈I〉 is the average intensity for this reflection with summation over all data.
bRwork is defined as Σ||Fobs| − |Fcalc|| / Σ |Fobs|, where Fobs and Fcalc are observed and calculated structure-factor amplitudes, respectively.
cRfree is the R-factor for the test set (5% of the data).
dAccording to molprobity (44).
For molecular replacement, protein data bank (PDB) entry 1GSO was trimmed according to the sequence alignment using MrBUMP (15) and CHAINSAW (16). The edited model was used as an input for MOLREP (17), which located two molecules in the asymmetric unit. Model building and refinement were done with COOT (18) and REFMAC5 (19), respectively. TLS refinement with three groups per monomer was carried out with the groups selected according to the suggestion of TLS motion determination server (20–21) (Table 1). The coordinates and structure factors are available from the PDB under accession ID 2QK4.
AIRS
Crystals were obtained using the hanging drop vapor diffusion method. Notably, 0.2 mg/ml of chymotrypsin was added to the protein solution prior to crystallization. One microliter of protein solution (12.4 mg/ml) was mixed with 1 µl of well solution containing 0.1 M Bis–Tris pH 5.2, 27% PEG 3350 and 0.2 M ammonium sulfate. Plate was incubated at 20°C and crystals appeared in a couple of days. A crystal was dipped into a cryo solution containing 0.1 M Bis–Tris pH 5.2, 25% PEG 3350 and 200 mM ammonium sulfate, 200 mM NaCl and 20% glycerol before flash frozen in liquid nitrogen. Data collection was carried out at BESSY BL14-1 and processed with XDS (14) (Table 1).
The structure was solved with molecular replacement using PHASER (22) with the PDB entry 1CLI as a search model. Model building and refinement were done with COOT (18) and REFMAC5 (19). One TLS group per protein molecule was used in the refinement (Table 1). The structure revealed that N- and C-terminal domains were removed by chymotrypsin and only the AIRS domain was crystallized. According to mass spectrometry from the chymotrypsin treated protein the size of this domain was 34 968 Da and the most probable sequence for the segment consisted of residues 467–794. The coordinates and structure factors are published in the PDB under accession id 2V9Y.
SAXS and rigid-body modeling
Aliquotes of the full-length HsGART were prepared at concentrations between 0.5 and 7 mg/ml in the gel filtration buffer. Synchrotron SAXS experiments were performed at station 2.1 of the SRS Daresbury, UK according to standard procedures (23) using sample-to-detector distances of 1 m and 4.5 m covering a momentum transfer range of 0.008Å−1 < q < 0.75Å−1 with q = 4 π sin(Θ)/λ (where 2Θ is the scattering angle and λ the X-ray wavelength of 1.54Å). The background signal was deducted by means of the scattering from the buffer alone. Data processing was carried out along with previously published methods (24) including the calculation of the radius of gyration and maximum molecular dimension. Low-resolution ab initio shapes were restored with the program GASBOR (25) using a 2-fold symmetry constraint. Due to the expected conformational plasticity of the dimeric full-length construct, ab initio shapes were used to scrutinize possible domain arrangements as starting condition for rigid-body modeling that was subsequently carried out with the program BUNCH (26) to fit the experimental scattering data. In view of the known high-resolution structures for all three enzyme domains rigid-body modeling offers an obvious advantage over shape reconstruction so as to adequately explore the conformational space of domain positions as well as of the extended and mobile linker segments. Specifically, the following assumptions were made during rigid-body modeling: (i) AIRS forms a dimer similar to the crystallographic one in the full-length enzyme. (ii) The N-terminal helix of AIRS, 441–463, was modeled according to the EcAIRS structure and the flexible G1 loop was not modeled. (iii) Residues 787–792 were modeled rather than using their conformation observed in the AIRS crystal structure as these result from crystallographic contacts, which most likely do not represent a biologically relevant conformation.
Structure analysis and figures
Superpositions used in the text and figures were made using the SSM superposition algorithm in COOT (18,27). Structural representations were made using Pymol (28). Electrostatic potential was calculated with APBS (29) using solvent and protein dielectric constants 80 and 2, respectively. Docking was done using GOLD 3.2 through the Hermes interface and the docking wizard (30). Goldscore was used to rank the docking results.
RESULTS AND DISCUSSION
Structure of GARS
Overall structure
The structure of GARS belongs to the superfamily of ATP-grasp proteins (31). The N-terminal GARS unit of HsGART has an α/β-fold consisting of four structural sub-domains similar as described by Wang et al. (7) (Figures 2A and 3A) for the bacterial homolog, E. coli GARS (EcGARS). The domains are named N (N-terminal), B, A and C (C-terminal). Domains N, A and C form the core of the enzyme with B being located towards the periphery. HsGARS was co-crystallized with ATP, which is bound between the A and B domains in the structure. The co-substrate glycine was also included in the crystallization, but was not found in the structure, most likely due to crystallization at low pH. The asymmetric unit of the crystal contains two monomers, where residues 0–429 and 0–430 were modeled in the electron density for monomers A and B, respectively. Both monomer models are missing two short loops in the B-domain (A: 153–155 and 178–184; B: 154–156 and 179–182).
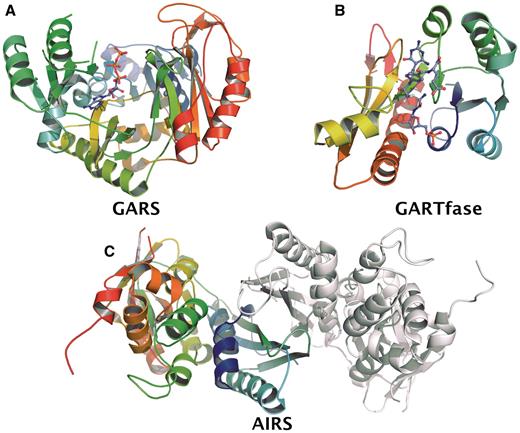
Schematic presentation of crystal structures of the HsGART domains. (A) GARS in complex with ATP. (B) Ternary complex of GARTfase with 10-(trifluoroacetyl) 5,10-dideazaacyclic-5,6,7,8-tetrahydrofolic acid and substrate glycinamide ribonucleotide (PDB ID. 1RBY). (C) Dimeric structure of AIRS.
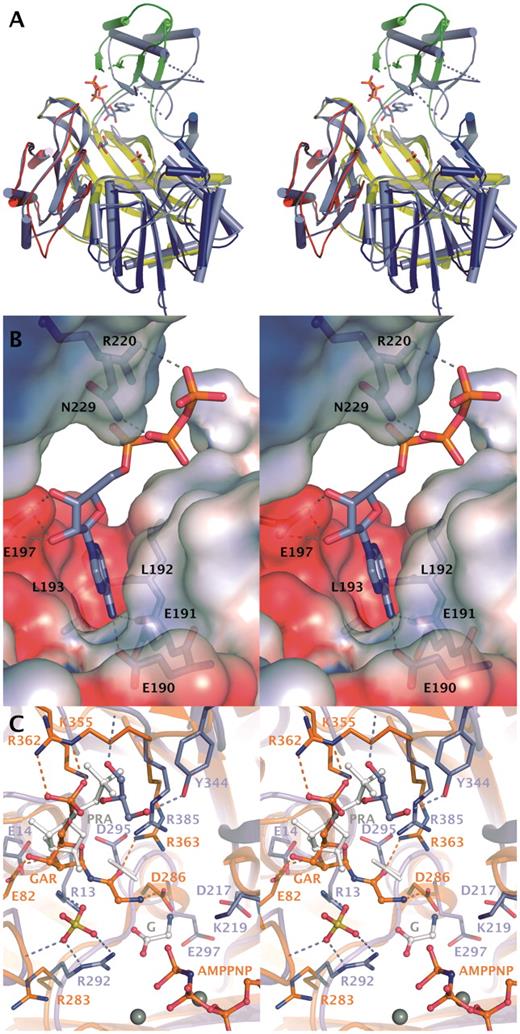
Substrate binding to GARS. (A) Conformational change in GARS induced by binding of ATP. Cartoon model of GARS is shown in slate blue together with the ATP molecule, glycerol and sulfate ion. Comparison with EcGARS crystallized in apo form reveals the movement of Domain B. Domains of EcGARS are colored blue (N), green (B), yellow (A) and red (C). (B) ATP-binding cleft in GARS. Hydrogen bonds between ATP and residues are shown. Electrostatic potential is mapped from solvent accessible surface and is shown from −4 kT (red) to +4 kT (blue). (C) Comparison of GARS with EcGART (orange) crystallized with AMPNP, Mg2+ and GAR (35). Modeled position of PRA is shown in white. Active site of GARS contains a sulfate ion and a glycerol molecule in addition to ATP. Glycine observed in GkGARS structures (2YS6) is shown with white carbon atoms. Ligands in the active sites are shown as ball-and-stick models and Mg2+-ions as grey spheres. Rotamer change in Asp295 is indicated as white alternative conformation.
ATP binding
The closest structural homolog of GARS is EcGARS (7), which shares 51% sequence identity and superimposes with a RMSD value of 1.7Å for 381 Cα atoms. The B-domains of GARS and EcGARS are however in a significantly different conformation relative to the core structure (Figure 3A). When the B-domain (residues 123–191) is omitted in the structural alignment, the fit is better with a RMSD value of 1.2Å for 347 Cα’s (Figure 3A). Also the B-domains by themselves superimpose well (RMSD 1.6 Å). The largest movements of the B-domain are as extensive as 16Å or an angle of 27° between EcGARS residues Gly178, Phe189 and Gly178. All GARS enzyme structures reveal B-domains in a different orientation with respect to the core enzyme most likely due to crystal packing effects. In the structures of GARS from Geobacillus kaustophilus (GkGARS; 2YRW, 2YRX, 2YS6 and 2YS7) and Aquifex aeolicus (AaGARS; 2YW2, 2YYA), where ATP is bound, nucleotide binding does not induce large changes in the orientation of the B domain. In another enzyme of the ATP-grasp family, PurK or AIRC of E. coli, ADP binding was seen to induce a small 2.5Å movement of the B-domain (32). This demonstrates the mobility of the B-domain in GARS enzymes. ATP binding alone does not appear to stabilize the catalytic conformation as non-competent conformations have been observed in AaGARS (2YW2) and reported here in GARS.
In GARS, hydrophobic side chains align the adenosine-binding site and adenosine also makes conserved hydrogen bonds to the backbone carbonyl of Glu190, backbone amide of Leu193 and to carboxyl group of Glu191 (Figure 3B). Glu197 is coordinating the ribose by hydrogen bonding to the hydroxyl groups. The phosphates of ATP, interacting with Arg220 and Asn229 of GARS (Figure 3B), are less well coordinated as the B-factors of the ATP vary from 40–65 Å2 for the adenine moiety to 90–110 Å2 for the phosphate atoms. We also crystallized GARS in the presence of Mg2+ excess, but this did not result in a more ordered structure of the ATP molecule (data not shown). In the light of the reported ordered, sequential mechanism of GARS, with PRA binding first, then ATP and glycine (33), the flexibility of the ATP phosphates suggest that the ATP binding seen in the GARS crystal structure represents ATP in a ‘waiting position’.
Binding of PRA
We have not been able to determine the structure of GARS with either its substrate PRA or glycine, but based on homologous structures we propose binding sites for these substrates in the active site of GARS (Figure 3C and discussions below). The closest structural homolog of GARS, after the prokaryotic GARS enzymes, was found through a DALI search (34). This is the E. coli PurT-encoded glycinamide ribonucleotide transformylase (EcGART) (PDB ID. 1EZ1; (35). EcGART catalyses an alternative formylation reaction utilizing ATP and formate and proceeds through a formyl phosphate intermediate (35). EcGART was solved in complex with Mg2+, AMPPNP and GAR—the product of GARS (35). The ATP conformation and the catalytic core were shown to be similar to the one in d-alanine–d-alanine ligase, although the acceptor substrates are quite different. Structural comparison of GARS with EcGART, which use the structurally most similar substrate, might indicate how a ternary substrate complex is interacting in GARS and thereby shed light on the binding mode of PRA and glycine (Figure 3C).
Superposition of the structures reveals that in GARS, the phosphates of ATP are in a different position compared to the ternary-like complex of EcGART, and have swung out of the active site as discussed above. Due to similarities in the catalytic mechanisms, it is however plausible that ATP in the ternary complex of GARS, will be in a similar conformation as ATP in a ternary-like complex of EcGART, despite all residues coordinating Mg2+ ions being non-conserved residues (Glu267 of EcGART replaced by Gly276). This also suggests that GAR, the product of GARS and substrate of EcGART, will bind in the same pocket in both enzymes, providing a similar geometric relationship to ATP in a ternary complex. Although the pockets appear to have similar size and topology in the two enzymes, it is likely, that GAR (and PRA) will bind to GARS in a somewhat different manner than in EcGART. Between GARS and EcGART only Arg385/363, which binds to the glycine moiety of GAR in EcGART, is conserved (Figure 3C). The distribution of positively charged residues in the binding pocket appears reversed. At the site where the phosphate of GAR is binding in EcGART there are three positively charged residues Lys355, Arg362 and Arg363, whereas in GARS there is only one (Arg385). On the opposite side of the pocket, EcGART has one positively charged residue, Arg283, while in GARS Arg13, Arg292 and a positive dipole of the short helix 75–80 are found. A sulfate ion is bound in the GARS structure between Arg13, Arg292 and capping the short helix. Sulfate or phosphate ions are found at this site also in other crystal structures of GARS enzymes. A bound glycerol molecule is also observed near the PRA-binding site of GARS, close to the phosphate group of GAR in EcGART (Figure 3A and C). Notably, in some crystal structures of homologous proteins (Thermus thermophilus GARS-2IP4 and GkGARS-2YRX) sulfate or phosphate ions are present at this site and thus despite of the presence of glycerol in the GARS structure, it is tempting to suggest that this could form the binding site for the PRA phosphate. Only a 2 Å shift in the phosphate position seen in GAR of EcGART would be required to enable hydrogen bond formations with the backbone amides and Arg363 (Figure 3C). The key residue for coordinating GAR in EcGART, Arg362, is not conserved in GARS enzymes. The second sulfate-binding site found in the human GARS structure may therefore represent the binding site of the product phosphate. Glycine binding Arg292 may furthermore stabilize the acyl phosphate intermediate as has been suggested previously (35).
This substrate binding mode would require only small changes in GARS, as compared with EcGART (Figure 3C). The potential candidate residue orienting the ribosyl of PRA, similarly to Glu82 in EcGART, would be Glu14. These residues reside in different secondary-structure elements showing a complementary change from Glu to Gly and vice versa. If the glycerol-binding site reflects the PRA phosphate site then some of the interactions with the ribose moiety may be mediated by water molecules.
Glycine binding
Studies of EcGARS have indicated that the enzyme is very specific for glycine (33). Because of the sequential binding of substrate molecules, with glycine being last in the sequence, it appears that ATP as well as PRA might contribute to the glycine-binding site. It has been proposed that glycine would bind directly in-between ATP and PRA (36). Glycine was indeed found between ATP and PRA sites in one of the GkGARS structures (2YS6) making hydrogen bonds with the Asp295 carboxylate group (GARS numbering), Asp217, Glu297 and Arg292 (Figure 3C), which are conserved in GARS. A binding site for glycine between ATP and PRA would be in agreement with the mechanism proceeding via an acyl phosphate intermediate, which could then react with PRA on the other side of glycine (33). In the structure of GARS, there is some residual density at the glycine-binding site, albeit this density could not be unambiguously interpreted as a glycine. In d-alanine–d-alanine ligase the binding site for alanine overlaps with the proposed glycine-binding site in GARS (37).
Three binding sites—implications of structures for catalysis
Residues and structures implicated in the catalytic site of GARS are shown in Figure 3C. The initial step of the reaction is the phosphoryl transfer from ATP to glycine. Glycine is coordinated by two conserved acidic residues Asp217 and Glu297 as well as backbone carboxyl of Arg292 (2YS6). This places glycine appropriately to perform an in-line nucleophilic attack on the γ-phosphate of ATP (Figure 3C). We proposed that the ATP seen in GARS is in a waiting position while in e.g. EcGART structure (35), ATP binds in a catalytically more relevant orientation. It should be noted that the B-loop interacting with the γ-phosphate of ATP (2YW2, 2YYA) is missing in most of the structures and that the B-loop sequence, GLAAGK, is conserved in all the GARS enzymes discussed here. The negatively charged transition state of this first reaction step is likely to be stabilized by a Mg2+ ion, backbone amides of the B-loop and possibly Lys219.
In the second part of the reaction, the reactive amine of PRA should reside closer to the intermediate than in the model presented in Figure 3C. A conformational change is thus required prior or during the reaction. The acylphosphate intermediate could be stabilized by Arg292 and Arg13 that form a sulfate-binding site in the GARS structure. Asp295 also needs a rotamer change upon substrate binding. This would place Asp295 appropriately to bind to the amine of PRA and to interact with Arg13. This change would enable it to act as a general base similarily to the Asp286–Arg363 pair as suggested for EcGARS (35).
Crystal structure of AIRS
Overall structure
Several attempts of making a soluble construct for the AIRS domain had failed. Instead a longer construct (1–1003) containing all three enzyme components was crystallized in the presence of chymotrypsin and during crystallization the N- and C-terminal domains were separated and only the AIRS domain crystallized (38). The monomer A in the asymmetric unit has better electron density and the model consists of residues 475–792, while in monomer B residues 475–785 were modeled (Figure 2B). A flexible loop corresponding to residues 770–773 does not have electron density and was not modeled in either subunit. The monomer of AIRS can be divided into two domains, Domains A (475–590) and B (603–792), as described for the E. coli enzyme (EcAIRS) (39). A dimer interface is formed by a 4-stranded β-sheet from Domain A of each subunit (Figure 2). According to DALI (34) the most similar structure to the human AIRS is EcAIRS giving a RMSD of 1.3Å for 294 Cα superimposed. The sequence identity between human AIRS domain and E. coli AIRS is 47%. In the crystal structure of AIRS, we observe similar dimerization through the β-sheets as seen in EcAIRS (39).
A glycine rich loop (GGLGGFGA; G1) in the EcAIRS is believed to be involved in the binding of ATP (39). This loop is not visible in the AIRS structure electron density (Figure 4A). A second-glycine-rich region (GVGTK; G2) is located in a deep cleft near the sulfate-binding site and has been suggested to be involved in binding of the FGAM (39). The AIRS structures belong to the PurM superfamily, as do the structure of formylglycineamide ribonucleotide amidotransferase from Thermotoga maritime (TmFGARAT) (40). The members of the PurM superfamily are believed to share similar catalytic mechanism via iminophosphate intermediates (39–40). The structure of TmFGARAT has been solved in several complexes, e.g. in presence of an ATP analog (AMPPCP; 2HRY) and formylglycinamide ribonucleotide (FGAR and AMPPCP; 2HS4) (40). AIRS and TmFGARAT have a sequence identity of 22% spanning residues 520–795. When superposed the structures align well yielding a RMSD value of only 2.1 Å per 200 Cα atoms and 2.9 Å (433 Cα atoms) in the case of dimeric AIRS. Although enzymes catalyze different reactions the comparison of the structures can provide some clues about ATP and FGAM binding in AIRS.
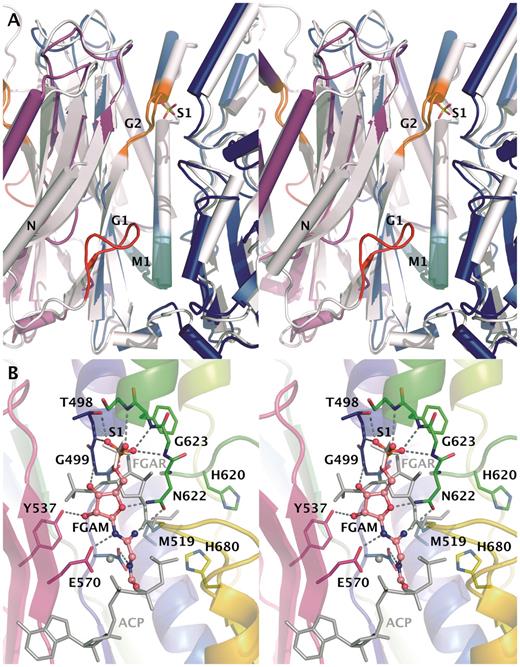
Active-site cavity at AIRS dimer interface. (A) Comparison of AIRS and EcAIRS structures. Monomers of AIRS are shown in blue and magenta and EcAIRS is white. Glycine rich motifs G1 and G2 are colored red and orange, respectively. Conserved M1 motif thought to form ATP-binding site is in cyan. The S1 sulfate located close to the G2 motif is shown. The N-terminal helix of EcAIRS used in the rigid body fitting of SAXS data is labeled with ‘N’. (B) Potential binding mode of substrate to AIRS. Superpositioned FGAR and AMPPcP-Mg of TmFGARAT are shown in grey on top of the AIRS structure with monomer A colored from blue to read, monomer B in magenta and sulfate in white. Docking pose of FGAR to the AIRS structure is shown as a ball-and-stick model.
Binding of FGAM
Three sulfate ions are bound to the two AIRS monomers in the asymmetric unit. S1 sulfate ions are situated between the two domains near the G2 motif (Figure 4A). S2 sulfate is bound in proximity to positively charged residues Arg604, His674 and Lys676 on the protein surface only in subunit B and it is participating in the crystal contacts. Sulfate S1 is found in both the AIRS and EcAIRS structures (39) with a sulfur–sulfur distance of only 0.6 Å (Figure 4A). As both FGAM and ATP contain phosphate, sulfate sites could correspond to either of their binding sites. It is however more likely that sulfate S1 binds at the FGAM-binding site, because of its proximity to the FGAR-binding site in TmFGARAT (Figure 4B). In the superimposed model, sulfate ion S1 is situated 5 Å away from the phosphate of FGAR in comparison to 10 Å for the γ-phosphate of the ATP analog (Figure 4B). The G2 motif predicted to form the FGAM-binding site is lining the FGAR-binding site when AIRS and TmFGARAT structures are compared (Figure 4B). The long distance between the FGAR phosphate and the sulfate ion indicates a different binding mode of FGAM in AIRS (Figure 4B). Upon direct superposition of TmFGARAT, there are clashes between FGAR and AIRS enzyme that make the same binding mode unlikely in AIRS. In TmFGARAT there are two conserved histidines shown to be important for catalysis (40). Potential residues that could be involved in the stabilization of reaction intermediates are the conserved His620 and His680, different from those of TmFGARAT. They are lining the nearby pocket that is covered by Met519 in the AIRS crystal structure (Figure 4B). It could be possible that after the phosphoryl transfer reaction these two histidines could have a role in the following cyclization as has been suggested (40). This would require a conformational change making them available to the intermediate. It is possible that the conformation of the active site is affected by low pH of 5.5 used in the crystallization and the conformation seen in the crystal is not catalytically competent. This could be caused by protonation of His680. This pH induced change would be similar to the one seen in GARTfase (12).
ATP-binding mode and implications for the mechanism
The PurM superfamily contains a conserved motif M1 (Dx(4,)GAxP) which forms a helix-turn-strand structure (Figure 4A) (39). This motif is believed to participate in ATP and magnesium binding (39–40). Comparison of the AMPPcP TmFGARAT complex and HsAIRS and EcAIRS structures gives a picture of the ATP-binding mode in AIRS enzymes. Residues involved in magnesium binding and residues making up the hydrophobic surrounding of the adenine moiety are strongly conserved (40). The different complexes of TmFGARAT and nucleotides show similar positions of α- and β-phosphates while the γ-phosphates are taking different conformations. Most interactions with the phosphate moiety of AMPPcP are from magnesium ions and water, thus making it harder to predict the binding. Therefore, while the adenosine-binding motif is likely the same in AIRS, EcAIRS and TmFGARAT, the interactions with the phosphate groups may be somewhat different. The N-terminus of EcAIRS contains a glycine rich G1 loop, which is believed to be involved in the ATP binding (41). This loop is not visible in the AIRS structure but when AIRS, EcAIRS and TmFGARAT are superimposed, it is evident that the G1 loop is positioned close to the ATP-binding site of TmFGARAT and thus this loop may be important in binding of ATP also in AIRS—it may become ordered upon ATP binding (Figure 4A).
Quaternary structure
The crystal structures reported here together with those previously published (11–12) complete the structural scaffold of human tri-functional GART: high-resolution structures of all individual globular domains from HsGART are now available. In addition, we also produced a close to full-length construct of HsGART, which we did not succeed in crystallizing in an intact form. However, we performed SAXS experiments on this full-length enzyme in solution in order to determine its overall conformation and to visualize relative arrangements of domains.
The conformational space taken up by full-length HsGART as derived from SAXS is shown in Figure 5. The experimental profile (see Supplementary Figure S1) characterizes a dimeric enzyme with a maximum molecular dimension of 205 ± 10 Å and a radius of gyration of 64.1 ± 0.5 Å. Considering that a compact macromolecule of similar mass (220 kDa) would be expected to yield a radius of gyration of ∼40Å (42), the dimer of full-length HsGART can only be described by an extended conformation. This feature is strongly linked with the mobile and/or unstructured nature of polypeptide segments connecting the globular enzyme domains. The conformational flexibility has been presented by depicting multiple structural arrangements of GARTfase and GARS domains with respect to the central AIRS dimer (Figure 5). These models are consistent with the experimental SAXS data and demonstrate the putative seesaw-like domain arrangements with GARTfase and AIRS domains of the dimer organized in a linear fashion and GARS domains arranged around AIRS dimer. Yet, due to the globular shapes of GARTfase and GARS domains, the SAXS data is not sensitive enough to differentiate among specific domain orientations (Supplementary Figure S2). This structural scenario implies that the linker regions enable the sampling of a large conformational space where domain crossing or swapping in the dimeric molecule is also possible. Taking into account that the molecular symmetry of the full-length enzyme is restricted to the dimer forming, central AIRS domain, rigid-body modeling was performed without constraints yet preserving the symmetry of the AIRS dimer. This procedure produced models, which reliably fit the experimental data (with an average goodness-of-fit value of χ = 1.8; Supplementary Figure S1). However, it is reasonable to assume that no unique structure in solution would alone account for the observed scattering behavior. A more realistic scenario will therefore encompass the presence of many conformations in solution (including conformations that may be more or less compact compared to those shown in Figure 5) highlighting the dynamic nature of this multi-domain macromolecule,
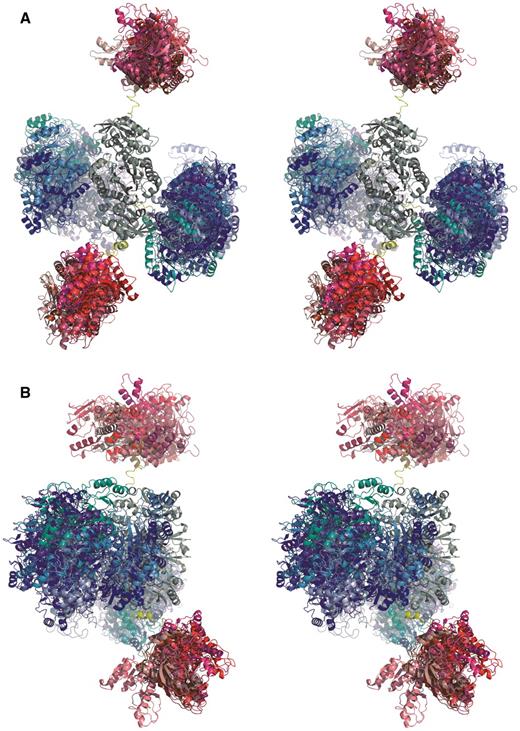
Overall conformations of full-length HsGART deduced from solution X-ray scattering (SAXS). Ten rigid-body models superimposed with respect to the AIRS dimer thus highlighting the conformational space taken up by GARS (shades of blue) and GARTfase (shades of red) domains in both monomers. The non-symmetry constrained rigid-body models are shown only. One of the models also includes the linker segments connecting globular domains (yellow). (A) View along the AIRS (grey) dimer axis. (B) Orientation rotated 90° along y-axis.
Li et al. (39) proposed a dimeric model of this tri-functional enzyme, which is similar, yet not identical to the experimental models presented here. Based on the crystal structures it was estimated that the distance between GARS and GARTfase domains is between 20 and 30 Å, whereas in our SAXS derived models it appears to be in the range of 30–50 Å. It should be kept in mind that there are many potential domain dispositions, in particular the C-terminal GARTfase domain appears to sample a large conformational space. This behavior was also seen in SAXS experiments with a protein construct consisting of only AIRS and GARTfase domain (data not shown). This is not surprising since the two domains are connected via a long linker that is predicted to be disordered. The flexibility of GARS and GARTfase domains is not unexpected considering that the tri-functional enzyme catalyzes three non-consecutive reactions of the pathway and the need for transferring unstable substrate to GARS. Taken together this indicates that this protein may function as a central gathering component of the ‘purinosome’ (13). PRA substrate is tunneled from PPAT to GARS catalyzing the second step of the reaction. This interaction between PPAT and GARS proposed earlier (7) is supported by the full-length model derived from SAXS experiments as the PRA binding crevice is directed away from the central AIRS dimer. The product, GAR, is then transferred to the C-terminal GARTfase. GARTfase is then able to pass the product further to FGAMS, which is again able to interact with both GART and the central AIRS dimer catalyzing the following step of the reaction. One way to speed up biosynthetic pathways is through multi-functional enzymes or enzyme complexes that would help accumulating intermediates at appropriate locations concomitant with an increase of localized substrate concentration.
A multi-domain enzyme assembly with a conformational heterogeneity and flexibility as seen in HsGART may have several biological implications and advantages. Cycling through a complex pathway such as the de novo purine biosynthesis, which involves the interaction of several enzymes with diverse functions, requires a variable as well as an adaptable structural scaffold. The conformational flexibility appears not only to allow for molecular associations but also dissociations taking place efficiently. Benkovic and coworkers (13) coined the expression of ‘purinosome’ in view of a dynamic multi-enzyme complex formed during de novo purine biosynthesis that is regulated depending on cellular purine levels. The conformation of full-length HsGART presented here would portray a convenient structural framework for a larger enzyme complex to be formed during catalysis. Interestingly, a recent study (43) seems to suggest that metabolite-dependent organization into large-scale protein assemblies in response to cellular nutrient stress may be a more general phenomenon in cell biology.
CONCLUSIONS
We have described the crystal structures of two enzymatic domains of HsGART thus furnishing structures of all domains of this tri-functional enzyme. Comparisons with structures of homologous enzymes allowed us to clarify potential substrate binding modes of the enzymes. Our efforts to combine crystal structures with shape models derived from SAXS successfully reveal the overall conformation of full-length HsGART in solution, but at the same time demonstrate its dynamics and significant structural flexibility. This characteristic plasticity is likely to have implications in view of the recently emerged ‘purinosome’ complex. The challenge will be to form and preserve complexes of HsGART with various proteins that are suitable for structural studies investigating conformations of large complexes and interactions between enzymatic reactions.
ACCESSION NUMBERS
2QK4, 2V9Y.
FUNDING
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. Work was also supported by Swedish Cancer Society and Swedish Research Council (to P.N.) and Academy of Finland grant (no. 128322 to L.L.). Funding for open access charge: Structural Genomics Consortium.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We acknowledge BESSY and ESRF for synchrotron radiation facilities and we would like to thank beam line personnel for assistance in using beam lines BL 14.1 and ID 14-2, respectively. The Science and Technologies Facilities Council, UK is acknowledged for provision of X-ray scattering time (beam line 2.1) at the SRS at Daresbury Laboratory.
REFERENCES
Author notes
Present address: Pål Stenmark, Center for Biomembrane Research, Department of Biochemistry and Biophysics, Stockholm University, SE-10691 Stockholm, Sweden.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments