





Abstract
DNA loop organization by nuclear scaffold/matrix attachment is a key regulator of gene expression that may provide a means to modulate phenotype. We have previously shown that attachment of genes to the NaCl-isolated nuclear matrix correlates with their silencing in HeLa cells. In contrast, expressed genes were associated with the lithium 3,5-diiodosalicylate (LIS)-isolated nuclear scaffold. To define their role in determining phenotype matrix attached regions (MARs) on human chromosomes 14–18 were identified as a function of expression in a primary cell line. The locations of MARs in aortic adventitial fibroblast (AoAF) cells were very stable (r = 0.909) and 96% of genes attached at MARs are silent (P < 0.001). Approximately one-third of the genes uniquely expressed in AoAF cells were associated with the HeLa cell nuclear matrix and silenced. Comparatively, 81% were associated with the AoAF cell nuclear scaffold (P < 0.001) and expressed. This suggests that nuclear scaffold/matrix association mediates a portion of cell type-specific gene expression thereby modulating phenotype. Interestingly, nuclear matrix attachment and thus silencing of specific genes that regulate proliferation and maintain the integrity of the HeLa cell genome suggests that transformation may at least in part be achieved through aberrant nuclear matrix attachment.
INTRODUCTION
The continuum of gene expression that enables specialized cell function is one of the hallmarks of cellular differentiation. This is initiated through changes in nuclear architecture that occur throughout development (1) and differentiation (2) beginning with lineage specification from embryonic stem cells (3). The dynamic restructuring of the nucleus appears to be both a cause and consequence of alterations in gene expression (4,5) and it has been suggested that these changes are facilitated by attachment to the nuclear matrix (6–8).
The same processes that are essential to normal cellular development and differentiation, when gone awry, can lead to transformation. For example, the introduction of DNA double-strand breaks during lymphocyte differentiation by either endogenous nucleases or genotoxic agents may, respectively, be an integral part of the differentiative pathway or serve to disrupt normal cellular function (9). It has become clear that disruption is often preceded by changes in nuclear matrix proteins or sites of attachment (10). Such changes in nuclear matrix attachment have been correlated with deleterious alterations in gene expression (11) that are likely achieved in a cell type-specific manner (8,12,13).
The nuclear scaffold/matrix organizes the genome into approximately 60 000 looped domains that are bound by specific interactions of the genome to the proteinaceous network at scaffold/matrix attachment regions (S/MARs). In many cases, these interactions facilitate the formation of gene containing potentiated looped domains that are poised for transcription (14,15). Recruitment to transcription factories may be mediated by nuclear scaffold/matrix attachment (16) such that actively transcribed genes co-localize within the same transcription factories (17). Furthermore, MARs can indirectly influence transcription by insulating nearby genes and are often used in transgenic constructs for this purpose (18). Though matrix attachment in intergenic regions can protect genes from silencing, the presence of MARs within genes correlates with silencing in humans, with exceptions (19).
Regions of the genome that attach to the nuclear scaffold or matrix can be physically distinguished and have been operationally defined. MARs are defined by their preferential isolation with 2 M NaCl that disrupts the non-nuclear matrix DNA–protein interactions. This permits the unbound DNA to loop out from the MARs enabling the resolution of each fraction following nuclease digestion. In comparison, scaffold-attached DNA, SARs, are isolated in a similar manner using 25 mM LIS (lithium 3,5-diiodosalicylate). It is clear from both small scale (20) and genomic (19) studies that these differential isolation methods can identify distinct sites of what has been interchangeably termed sites of scaffold and matrix attachment. Visual evidence does suggests that there is a single network within the nucleus (21,22), but whether these isolation protocols reveal different parts of the same structure remains to be unequivocally established.
While multiple methods have been utilized as a means to isolate a cohesive nuclear non-histone protein body (23–25), 2 M NaCl and 25 mM LIS are the most commonly used biochemical fractionation methods to prepare this structure. Utilizing these methods, we have recently shown that the HeLa S3 genome is organized by a unique complement of S/MARs. MAR presence within genes is correlated with silencing, whereas SARs within a 10-kb region upstream of a gene is correlated with the expression of that gene (19). This suggested that nuclear matrix attachment is responsible for genomic repression while nuclear scaffold attachment positively influences gene expression. The identification of sites with complementary function suggested that the two extraction methods were able to isolate interactions with different groups of proteins that are perhaps components of the same network.
The functional contribution of the nuclear scaffold/matrix to gene regulation is likely to be driven by both cell type specific, or facultative, as well as constitutive attachment. The locations of facultative attachment sites are expected to differ in various cell types according to specific requirements for each cell during the cell cycle and as a function of the differentiative state. However, the correlation of matrix attachment with gene silencing and scaffold attachment with gene expression that has been observed should be maintained. Accordingly, nuclear matrix attachment-correlated genomic silencing should be observed in all cell types with variability in specific sites of attachment reflective of the gene expression profile.
To begin to define the role of differential attachment in cell type-specific gene expression, we have identified S/MARs in the aortic adventitial fibroblast (AoAF) primary cell line. This cell line was not transformed to increase proliferation capacity. Interestingly, a significant portion of genes uniquely expressed in AoAF cells were associated with the nuclear matrix in HeLa cells and silenced. Consistent with a cell type-specific scaffold/matrix attachment mechanism that controls both expression and silencing, the majority of these expressed genes were associated with the nuclear scaffold in the AoAF cells. The results of these studies are reported in the following.
MATERIALS AND METHODS
S/MAR preparation and identification
S/MARs and loop regions were prepared by either NaCl or LIS extraction from AoAF cells (Cambrex Bio Science Walkersville, Inc.) as described (19). Nuclear halos were prepared in solution from isolated AoAF nuclei with either timed exposure to 2 M NaCl or dounce homogenization in the presence of 25 mM LIS using approximately 1 × 107 cells. After extraction, the halos were pelleted then washed gently with REact® 3 restriction buffer (Invitrogen, Carlsbad, CA, USA). The halos were then resuspended in restriction buffer and the loops were separated from the nuclear matrix/scaffold-associated DNA by digestion with 400 U of EcoRI (Invitrogen) at 37°C for 3 h. Subsequent to restriction digestion, the matrix/scaffold fractions were pelleted and loop containing supernatants were removed and placed in separate tubes. Both loop and matrix/scaffold restriction fragments were then freed from any nuclear proteins by overnight digestion at 55°C with 50 µg/ml of proteinase K buffered with 50 mM Tris–HCl buffer, pH 8.0, containing 50 mM NaCl, 25 mM EDTA (ethylenediaminetetraacetic acid) and 0.5% SDS (sodium dodecyl sulfate). DNA was recovered and purified using a Quantum-prep matrix (BioRad, Hercules, CA, USA) then resuspended in deionized water. Purified loop and scaffold/matrix DNA was then analyzed by aCGH. This analysis utilized the array containing human chromosomes 14–18 (Array 7 of 8 array set) from the Nimblegen Systems CGAR0150-WHG8 CGH isothermal oligonucleotide array system (Nimblegen Systems, Inc., Madison, WI, USA) which provides a median probe spacing of 713 bp.
Array data were q-spline normalized using the NimbleGen data analysis suite then rank comparisons were undertaken using Sigma Stat (http://www.systat.com) to assess concordance between biological replicates. Regions of significance were identified initially as probes with a Log2 signal ratio in the top 2.5% of the ranked signal. This set of regions was refined by the inclusion of only those top probes that had an additional two neighboring probes with similar signal located within a distance of 3 kb. The restriction fragments that contained probes meeting these criteria were then analyzed for consistent signal across the entire fragment. Regions displaying consistent signal for each replicate were then compared between the two independent biological replicates. All S/MAR locations identified are available in Supplementary Table 1.
Several regions on chromosome 16 were randomly selected for real-time PCR verification of aCGH signal distribution. All PCRs were performed in triplicate. The concentration of the initial template was calculated by the KLab PCR algorithm and ratios of scaffold/matrix enrichment are calculated in reference to loop DNA as described (26). This was compared to the analogous ratio calculated from the array signals, and significant concordance was observed between array identification and PCR validation. All primer sequences and ratios are available in Supplementary Table 2.
Expression analysis
The expression profile of the AoAF cells used for aCGH analysis was determined. Total RNA was isolated using RNeasy (Qiagen Inc., Valencia, CA, USA) then amplified using the Illumina RNA amplification system (Ambion, Austin, TX, USA). A total of 750 ng was used for hybridization to Illumina Sentrix Human-8 v2 Expression BeadChip arrays; then data were analyzed using the Illumina Bead Studio software suite. For each reporter, the average signal was q-spline normalized and expressed genes were identified by signal values higher than internal spike-in controls for expression (Smax>3000). Ontological analysis was carried out using either Panther (27) or David (http://david.abcc.ncifcrf.gov/). All other comparisons and statistical calculations were carried out using Sigma Stat (http://www.systat.com).
Permutation analysis
MAR locations were permuted to statistically assess the correlation of gene distance with expression. Genes were segregated into three categories based on the transcript signal level: ‘expressed’, genes with signal levels above the value for low expression based on experimental spike in controls (i.e. Smax > 3000); ‘silent’ genes (Smax < 300); and ‘intermediate’ genes in which confidence in a silent or expressed state could not be assigned (300 < Smax > 3000). Three different methods of permutation, i.e. unstructured randomization, semi-structured and structured permutation were utilized to determine the significance of the location of MARs relative to the three categories of expression (Supplementary Table 3). Unstructured randomization of MAR locations maintained the length of each individual MAR on each chromosome. This method of randomization was reiterated 50 times. The average enrichment of randomized MARs in each gene category was compared with the observed enrichment to determine significance by chi-squared analysis using 1 df and Yate's correction for continuity. This inferred the significance of the absolute MAR location relative to the gene. In comparison, the semi-structured permutation maintained the length of each MAR and their spacing relative to one another, but inverted their location on each chromosome arm. In this manner MAR clusters are retained but their locations are changed. Their distribution relative to the three categories of genes should not change significantly if their spacing is not essential to their correlation with expression. The enrichment of permuted MARs in each gene category from this method of analysis was compared with the observed enrichment to determine significance by chi-squared analysis using 1 df and Yate's correction for continuity. Lastly, a structured permutation was computed using megabase blocks of sequence and inverting the MAR locations end to end within the blocks. These megabase blocks are generally much larger than the average gene but smaller than G or other cytological bands. This method of permutation maintains MAR locations relative to chromosomal banding patterns but changes their locations relative to genes. The relative MAR spacing is disrupted at the boundaries of the 1 Mb blocks. This test was repeated 50 times by changing the block start locations by 200 kb each time. Significance was determined by chi-squared analysis as above.
RESULTS AND DISCUSSION
Gene-poor regions are enriched at the nuclear matrix
MARs were isolated from AoAF cells by 2 M NaCl to remove the majority of histones while leaving the nuclear matrix/DNA interactions unperturbed. The use of 2 M NaCl is far in excess of the 0.6 M NaCl that can remove all histones from any region of the genome regardless of condensation state (28). This permitted the unconstrained loop DNA to diffuse away from the nuclear matrix forming a peripheral halo, as shown in Figure 1A. The loop DNA halo was separated from matrix-associated DNA by restriction digestion, then the loop and matrix fractions were labeled with Cy-3 and Cy-5, respectively. These fractions were then competitively hybridized to NimbleGen Systems human whole genome CGH CGAR0150-WHG8CGH array 7 containing human chromosomes 14–18 to yield chromosome-wide profiles of nuclear matrix association. The normalized signal ratios of the loop to matrix signal from independent biological replicates were calculated and displayed a high level of concordance (r = 0.909). To assess matrix enrichment at the global level, the signal ratios were averaged between replicates and plotted as a function of their position along each chromosome as shown in Figure 1B–F. Negative signal ratios represent regions of nuclear matrix enrichment (as displayed by blue bars); whereas, positive signal ratios mark loop enrichment (green bars).
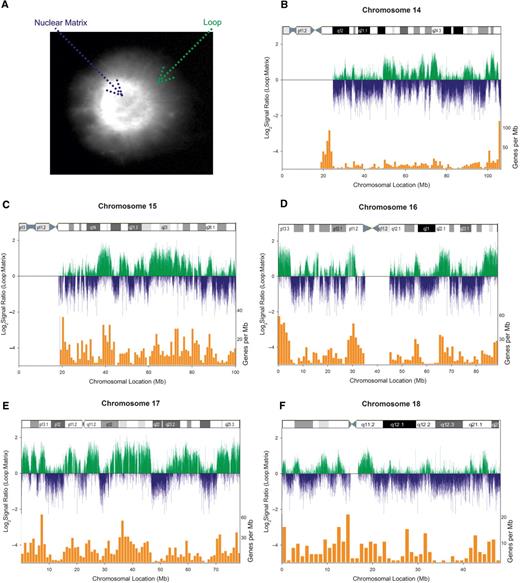
Nuclear matrix attachment correlates with gene poor regions. (A) Disruption of non-nuclear matrix proteins with 2 M NaCl allowed loop DNA to radiate away from MARs of the genome, forming a peripheral halo which was visualized with Dapi. (B–F) Fractionated loop (green) and matrix (blue) pools of DNA were differentially labeled with Cy 3 and Cy 5, respectively, then competitively hybridized to a CGH array containing human chromosomes 14–18 to generate the genomic profile of nuclear matrix enrichment. Comparison with gene density (orange bars) and chromosomal G banding revealed matrix enrichment in gene-poor, AT-rich G bands.
MARs were identified using a three-tier process that we have previously shown to minimize the rate of identification false positives (19). First, only the probes exhibiting a signal ratio (log2[loop/matrix]) in the extreme 2.5% of all signals were considered. Second, to remove potentially spurious signals, additional neighboring probes with a similar signal distribution were required. Finally, since resolution is determined by the length of each individual restriction fragment, the entire restriction fragment containing the region of interest was required to display an average signal consistent with the primary observation. Regions meeting these three levels of scrutiny from each independent biological replicate were then compared to identify MARs. This analysis identified 4224 MARs of which 1011 were identified on chromosome 14 933 on chromosome 15 920 on chromosome 16 716 on chromosome 17 and 644 on chromosome 18 (Supplementary Table 1).
As illustrated in Figure 1, comparison of G banding with gene density (orange bars, overlaid) revealed a positive correlation of loop enrichment with gene density on all chromosomes analyzed while the gene poor G bands were nuclear matrix enriched. Figure 2 highlights a darkly staining G band within 14q12. A 1 Mb chromatin loop extending from 30 028 735 bp to 31 053 836 bp is bounded by two NaCl-isolated MARs. The organization of this gene-rich heterochromatic region reflects poising by matrix attachment regions that likely act as boundary elements. Complementary attachment throughout this NaCl nuclear matrix-bounded loop region to the LIS-isolated nuclear scaffold may serve to structurally poise intervening genes for expression (e.g. SCFD1) or even recruit factors necessary for transcription. In accord with previous observations in other cell types (19), this establishes that the global profiles of nuclear matrix enrichment are similar across cell types.
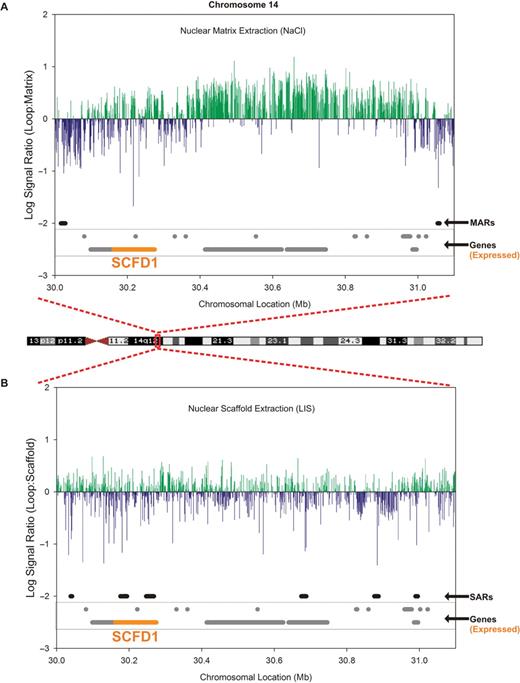
Nuclear scaffold/matrix-mediated organization is a function of gene density rather than chromosomal condensation. (A) Analysis of nuclear matrix attachment within the G band at chromosome 14q12 reveals the presence of a large 1 025 101 bp loop bounded by MARs as identified using the three-tiered process for identification. This particular gene-rich region contains from left to right: SYF2P, KIAA1333, SCFD1, UBE2CP1, RPL12P5, RPL27P1, COCH, STRN3, MIRN624, AP4S1, HECTD1, NARSP, ATP5GP4, LOC728852, C14orf126, LOC644223, GPR33 and NUBPL (genes for which expression was measured are in bold and offset from others in graph). (B) In contrast, many SARs are present within this G banded region.
The bimodal distribution of genomic loops created by MARs
Loop size was then estimated as a function of the bimodally distributed MARs (Supplementary Figure 1). The first group of MARs was spaced from 87 bp to 2217 bp apart representing ∼22% of the loops with an average size of 640 bp (±46 bp). The second group of MARs was spaced from 3.3 kb to 970 kb apart and contained ∼70% of the loops with an average size of 58 kb (±9.3 kb). A subset of S/MARs is expected to serve an ORI origins of replication initiation function (29) and it has been proposed that loop size may be reflective of the varying speed of replication with smaller loops corresponding to faster replicated segments and larger loops to more slowly replicated DNA (30). MARs that function as ORIs are likely contained within the various sized loops observed.
Nuclear matrix-bound genes are transcriptionally silent
Although many MARs are located in gene-poor regions, a subset of MARs overlap genes on each chromosome. In HeLa cells, MARs that overlapped genes were correlated with silenced genes (19). To determine whether this was a general property, nuclear matrix attachment was assessed as a function of the distribution of transcripts in AoAF cells. Transcript profiles were established using the Illumina WG8 v2.0 RefSeq bead array system and expressed genes are identified as a function of the spike-in control (Smin > 3000). The results are summarized in Table 1. Approximately 10% of the genes queried were nuclear matrix bound (i.e. 300 of 2945 genes). These were essentially separate from the 415 genes that were expressed. Strikingly, ∼96% of the genes containing MARs were silent (P < 0.001) and attached to the nuclear matrix by an average of four MARs per gene (Supplementary Table 4). These silencing MARs displayed no discernable preference for introns or exons, tethering each gene to the nuclear matrix along its entire length.
AoAF cell nuclear matrix attachment correlates with silencing
| Chromosomes | All genes | Expressed genes | Genes overlapped by MARs | Average no. of MARs per gene | Expressed genes overlapped by MARs |
|---|---|---|---|---|---|
| 14 | 433 | 64 | 76 | 4 | 2 |
| 15 | 534 | 72 | 60 | 4 | 1 |
| 16 | 758 | 112 | 53 | 5 | 4 |
| 17 | 1058 | 147 | 67 | 3 | 1 |
| 18 | 162 | 20 | 44 | 3 | 4 |
| Chromosomes | All genes | Expressed genes | Genes overlapped by MARs | Average no. of MARs per gene | Expressed genes overlapped by MARs |
|---|---|---|---|---|---|
| 14 | 433 | 64 | 76 | 4 | 2 |
| 15 | 534 | 72 | 60 | 4 | 1 |
| 16 | 758 | 112 | 53 | 5 | 4 |
| 17 | 1058 | 147 | 67 | 3 | 1 |
| 18 | 162 | 20 | 44 | 3 | 4 |
For each gene that was represented by probes on both the expression (Illumina) and aCGH (NimbleGen) microarrays, the correlation between expression and matrix attachment was measured. Of the subset of genes that contain MARs, they contain an average of four MARs per gene. The observed correlation between matrix attachment and gene silencing is statistically significant for all chromosomes (P < 0.001).
AoAF cell nuclear matrix attachment correlates with silencing
| Chromosomes | All genes | Expressed genes | Genes overlapped by MARs | Average no. of MARs per gene | Expressed genes overlapped by MARs |
|---|---|---|---|---|---|
| 14 | 433 | 64 | 76 | 4 | 2 |
| 15 | 534 | 72 | 60 | 4 | 1 |
| 16 | 758 | 112 | 53 | 5 | 4 |
| 17 | 1058 | 147 | 67 | 3 | 1 |
| 18 | 162 | 20 | 44 | 3 | 4 |
| Chromosomes | All genes | Expressed genes | Genes overlapped by MARs | Average no. of MARs per gene | Expressed genes overlapped by MARs |
|---|---|---|---|---|---|
| 14 | 433 | 64 | 76 | 4 | 2 |
| 15 | 534 | 72 | 60 | 4 | 1 |
| 16 | 758 | 112 | 53 | 5 | 4 |
| 17 | 1058 | 147 | 67 | 3 | 1 |
| 18 | 162 | 20 | 44 | 3 | 4 |
For each gene that was represented by probes on both the expression (Illumina) and aCGH (NimbleGen) microarrays, the correlation between expression and matrix attachment was measured. Of the subset of genes that contain MARs, they contain an average of four MARs per gene. The observed correlation between matrix attachment and gene silencing is statistically significant for all chromosomes (P < 0.001).
To determine the significance of the distribution of MARs on gene expression relative to a background distribution, MAR locations were permuted. The enrichment of permuted MARs within expressed and non-expressed genes were compared to that experimentally observed. The results of this analysis are presented in Supplementary Table 3. Interestingly, complete randomization of all MAR locations revealed that when a gene is bound to the nuclear matrix it is likely silent. There was a striking absence of MARs within expressed genes as compared to a random model (P < 0.001). The nuclear matrix is likely one player that mediates gene silencing.
This raised the issue of whether these observations were due in part to the observed clustering of MARs, i.e. structural spacing. MAR locations were then permuted such that MAR spacing was retained but MARs were located in different positions along the chromosome. Comparison of the permuted locations to the observed locations revealed that the absence of MARs within expressed genes was significantly different from random (P < 0.05). This suggested that MAR–MAR spacing, i.e. co-localization is an integral component of gene silencing.
The global analysis of matrix enrichment (Figure 1) revealed that many MARs cluster in gene-poor G bands along each chromosome. To determine whether the absence of MARs within expressed genes is due to this global trend rather than specific locations, MARs were permuted locally to maintain their relationship with the chromosomal banding pattern. This analysis revealed a significant difference between the absence of experimentally identified MARs from expressed genes and that of the background model (P < 0.01). From a genomic perspective, the data support the view that the correlation of matrix attachment within a gene and silencing is due to the position of the MAR with respect to silenced genes rather than a result of the distribution of MARs within and between G Bands. In this manner, MARs within genes may confer a general silent chromatin state (no precise location needed) which is required to be absent from expressed genes (precise location of MARs needed).
Cell type-specific nuclear matrix attachment
Cell type-specific gene expression reflects identity through facultative expression within a background of constitutively expressed genes. Accordingly, it was expected that comparison of the expression profile of AoAF cells with that of HeLa cells would reveal differences consistent with cell type-specific function and that these would be reiterated through nuclear matrix association. Adventitial fibroblast cells provide support for the aorta as part of the vasculature and are involved in cardiovascular disease response (31,32). For example, after endoluminal vascular injury, they migrate from the adventitia into the neointima to participate in vascular remodeling and plaque formation (33). Thus, it is expected that AoAF gene expression will be strictly controlled in this specialized cell type and that genes involved in cellular proliferation and migration, intercellular signaling and inflammatory response would be expressed. Comparatively, HeLa cells are epithelial cells derived from a cervical adenocarcinoma that have been propagated in culture for >50 years. Their expression program is expected to be perturbed relative to normal cervical epithelium with genes associated with a proliferative cancer phenotype expressed (34).
It was expected that genes differentially expressed in AoAF cells would have divergent nuclear matrix association profiles when compared to the immortalized HeLa cell line. Accordingly, genes expressed in AoAF cells but silent in HeLa cells should be preferentially bound to the HeLa cell nuclear matrix. To test this tenet, nuclear matrix attachment status of genes expressed in AoAF cells on chromosomes 14–18 but not expressed in HeLa cells was assessed. The results are summarized in Table 2. Initial analysis of AoAF MARs revealed that nuclear matrix attachment within genes robustly correlated with silencing. As expected, contrasting profiles of specific sites of nuclear matrix attachment were observed when cell types were compared. A significant portion of genes uniquely expressed in AoAF cells were associated with the nuclear matrix in the HeLa cells and silenced. Of the 415 genes expressed in AoAF cells on the chromosomes queried, 160 genes were silent in HeLa cells. Approximately one-third of these differentially expressed genes (53 of 160; P < 0.001) were also bound to the nuclear matrix in HeLa cells. This is consistent with the view that nuclear matrix attachment is at least part of a cell type-specific silencing mechanism. It is of note that a recent study by Wang et al. (35) concluded that overrepresented histone modification modules can only account for 25% of gene expression. This reiterates that gene expression, or silencing, is a symphony of multiple mechanisms working in concert. What is apparent is that nearly all of the differentially expressed genes bound to the nuclear matrix in HeLa cells are attached to the nuclear scaffold in AoAF cells. Thus, the state of expression appears in part to be controlled by complementary attachments to the non-histone protein body that can be distinguished from one another by both the methods of isolation and function.
Differential expression correlates with changes in nuclear matrix attachment
| Chromosomes | Genes expressed in AoAF cells | AoAF expressed genes silenced in HeLa | Subset containing AoAF MARs | Subset containing HeLa MARs |
|---|---|---|---|---|
| 14 | 64 | 28 | 0 | 15 |
| 15 | 72 | 34 | 0 | 12 |
| 16 | 112 | 41 | 1 | 11 |
| 17 | 147 | 47 | 1 | 8 |
| 18 | 20 | 10 | 3 | 7 |
| Chromosomes | Genes expressed in AoAF cells | AoAF expressed genes silenced in HeLa | Subset containing AoAF MARs | Subset containing HeLa MARs |
|---|---|---|---|---|
| 14 | 64 | 28 | 0 | 15 |
| 15 | 72 | 34 | 0 | 12 |
| 16 | 112 | 41 | 1 | 11 |
| 17 | 147 | 47 | 1 | 8 |
| 18 | 20 | 10 | 3 | 7 |
Genes represented on both the expression and aCGH arrays were analyzed for differences between cell types. Many genes that are expressed in AoAF cells are bound to the nuclear matrix and silent in HeLa cells (i.e. subset containing HeLa MARs), while relatively few are nuclear matrix bound in the AoAF cells (i.e. subset containing AoAF MARs).
Differential expression correlates with changes in nuclear matrix attachment
| Chromosomes | Genes expressed in AoAF cells | AoAF expressed genes silenced in HeLa | Subset containing AoAF MARs | Subset containing HeLa MARs |
|---|---|---|---|---|
| 14 | 64 | 28 | 0 | 15 |
| 15 | 72 | 34 | 0 | 12 |
| 16 | 112 | 41 | 1 | 11 |
| 17 | 147 | 47 | 1 | 8 |
| 18 | 20 | 10 | 3 | 7 |
| Chromosomes | Genes expressed in AoAF cells | AoAF expressed genes silenced in HeLa | Subset containing AoAF MARs | Subset containing HeLa MARs |
|---|---|---|---|---|
| 14 | 64 | 28 | 0 | 15 |
| 15 | 72 | 34 | 0 | 12 |
| 16 | 112 | 41 | 1 | 11 |
| 17 | 147 | 47 | 1 | 8 |
| 18 | 20 | 10 | 3 | 7 |
Genes represented on both the expression and aCGH arrays were analyzed for differences between cell types. Many genes that are expressed in AoAF cells are bound to the nuclear matrix and silent in HeLa cells (i.e. subset containing HeLa MARs), while relatively few are nuclear matrix bound in the AoAF cells (i.e. subset containing AoAF MARs).
The group of genes that are differentially expressed in AoAF cells but nuclear matrix bound and silenced in HeLa cells is provided in Table 3. As expected, several genes involved in cellular proliferation (e.g. c14ORF11, EPB41L3), stress response (e.g., PRKCA, MAP4K5) inflammatory response (e.g. CMTM4) and cellular adhesion/receptor signaling (e.g. FLRT2, CDH11 and CDH2) were expressed in AoAF cells yet silenced in HeLa cells. The presence of genes such as FLRT2 that encode extracellular matrix like proteins, in combination with similarly regulated genes encoding proteins that interact with the extracellular matrix, e.g. ITGA11 and involved in migration, i.e. CDH11 and CDH2, likely aid in the maintenance of vascular elasticity in response to environmental cues.
Differentially expressed genes are nuclear scaffold associated
| Chromosomes | Gene name | Biological process/function | SAR within 10-kb upstream | SAR within gene |
|---|---|---|---|---|
| 14 | SCFD1 | Exocytosis | − | + |
| 14 | C14orf11 | Stimulates proliferation | + | + |
| 14 | KIAA0423 | Mitosis | + | + |
| 14 | MAP4K5 | Protein phosphorylation | − | + |
| 14 | PYGL | Glycogen metabolism | − | + |
| 14 | KIAA0831 | Chromosome partitioning | − | + |
| 14 | DACT1 | Regulates WNT signaling | − | + |
| 14 | FUT8 | Transferase activity | − | + |
| 14 | PCNX | Neurogenesis | + | + |
| 14 | SIPA1L1 | Cell adhesion | − | + |
| 14 | RBM25 | Pre-mRNA processing | − | − |
| 14 | JDP2 | mRNA transcription regulation/tumor suppressor | − | + |
| 14 | C14orf179 | Cytoskeleton | − | − |
| 14 | FLRT2 | Cell surface receptor mediated signal transduction | − | + |
| 14 | BTBD7 | Homo/heteromeric protein dimerization | − | + |
| 15 | AQR | Viral RNA-dependent RNA polymerase | − | + |
| 15 | EIF2AK4 | Protein phosphorylation/translational regulation | + | + |
| 15 | FLJ43339 | Unclassified | + | − |
| 15 | CCDC32 | Unclassified | − | + |
| 15 | AP4E1 | Receptor-mediated endocytosis | − | + |
| 15 | LEO1 | mRNA transcription elongation | − | − |
| 15 | TPM1 | Cell motility | − | − |
| 15 | OAZ2 | Apoptosis/oncogenesis | − | + |
| 15 | ITGA11 | Cell adhesion | + | + |
| 15 | SIN3A | mRNA transcription regulation | + | + |
| 15 | UBE2Q2 | Protein modification | − | + |
| 15 | CHSY1 | Protein glycosylation | − | + |
| 16 | CREBBP | mRNA transcription regulation | − | + |
| 16 | KIAA0350 | Autoimmunity | − | + |
| 16 | ARHGAP17 | Signal transduction/cell structure | − | + |
| 16 | NSMCE1 | Genome stability | + | + |
| 16 | XPO6 | Nuclear export to cytoplasm | − | + |
| 16 | CHD9 | mRNA transcription regulation | − | + |
| 16 | AMFR | Proteolysis | + | + |
| 16 | GTL3 | Unclassified | − | − |
| 16 | CDH11 | Cell adhesion | + | + |
| 16 | CMTM4 | Inflammatory response/migration | − | + |
| 16 | AP1G1 | Receptor-mediated endocytosis | − | + |
| 17 | C17orf25 | Carbon metabolism | + | + |
| 17 | TTC19 | Unclassified | − | − |
| 17 | ZNF207 | Nucleic acid metabolism | − | − |
| 17 | CCDC47 | Unclassified | − | − |
| 17 | ERN1 | Protein phosphorylation | − | + |
| 17 | TEX2 | Unclassified | − | − |
| 17 | PRKCA | Protein phosphorylation/cell proliferation and differentiation | − | + |
| 17 | WIPI1 | Protein complex assembly | + | − |
| 18 | EPB41L3 | Tumor progression | − | + |
| 18 | RNMT | mRNA capping | + | + |
| 18 | CABLES1 | Proliferation/differentiation | − | − |
| 18 | CDH2 | Cell adhesion | + | + |
| 18 | HDHD2 | Phosphate metabolism | − | + |
| 18 | KIAA0427 | Protein biosynthesis | − | + |
| 18 | DYM | Skeletal development/brain function | − | + |
| Chromosomes | Gene name | Biological process/function | SAR within 10-kb upstream | SAR within gene |
|---|---|---|---|---|
| 14 | SCFD1 | Exocytosis | − | + |
| 14 | C14orf11 | Stimulates proliferation | + | + |
| 14 | KIAA0423 | Mitosis | + | + |
| 14 | MAP4K5 | Protein phosphorylation | − | + |
| 14 | PYGL | Glycogen metabolism | − | + |
| 14 | KIAA0831 | Chromosome partitioning | − | + |
| 14 | DACT1 | Regulates WNT signaling | − | + |
| 14 | FUT8 | Transferase activity | − | + |
| 14 | PCNX | Neurogenesis | + | + |
| 14 | SIPA1L1 | Cell adhesion | − | + |
| 14 | RBM25 | Pre-mRNA processing | − | − |
| 14 | JDP2 | mRNA transcription regulation/tumor suppressor | − | + |
| 14 | C14orf179 | Cytoskeleton | − | − |
| 14 | FLRT2 | Cell surface receptor mediated signal transduction | − | + |
| 14 | BTBD7 | Homo/heteromeric protein dimerization | − | + |
| 15 | AQR | Viral RNA-dependent RNA polymerase | − | + |
| 15 | EIF2AK4 | Protein phosphorylation/translational regulation | + | + |
| 15 | FLJ43339 | Unclassified | + | − |
| 15 | CCDC32 | Unclassified | − | + |
| 15 | AP4E1 | Receptor-mediated endocytosis | − | + |
| 15 | LEO1 | mRNA transcription elongation | − | − |
| 15 | TPM1 | Cell motility | − | − |
| 15 | OAZ2 | Apoptosis/oncogenesis | − | + |
| 15 | ITGA11 | Cell adhesion | + | + |
| 15 | SIN3A | mRNA transcription regulation | + | + |
| 15 | UBE2Q2 | Protein modification | − | + |
| 15 | CHSY1 | Protein glycosylation | − | + |
| 16 | CREBBP | mRNA transcription regulation | − | + |
| 16 | KIAA0350 | Autoimmunity | − | + |
| 16 | ARHGAP17 | Signal transduction/cell structure | − | + |
| 16 | NSMCE1 | Genome stability | + | + |
| 16 | XPO6 | Nuclear export to cytoplasm | − | + |
| 16 | CHD9 | mRNA transcription regulation | − | + |
| 16 | AMFR | Proteolysis | + | + |
| 16 | GTL3 | Unclassified | − | − |
| 16 | CDH11 | Cell adhesion | + | + |
| 16 | CMTM4 | Inflammatory response/migration | − | + |
| 16 | AP1G1 | Receptor-mediated endocytosis | − | + |
| 17 | C17orf25 | Carbon metabolism | + | + |
| 17 | TTC19 | Unclassified | − | − |
| 17 | ZNF207 | Nucleic acid metabolism | − | − |
| 17 | CCDC47 | Unclassified | − | − |
| 17 | ERN1 | Protein phosphorylation | − | + |
| 17 | TEX2 | Unclassified | − | − |
| 17 | PRKCA | Protein phosphorylation/cell proliferation and differentiation | − | + |
| 17 | WIPI1 | Protein complex assembly | + | − |
| 18 | EPB41L3 | Tumor progression | − | + |
| 18 | RNMT | mRNA capping | + | + |
| 18 | CABLES1 | Proliferation/differentiation | − | − |
| 18 | CDH2 | Cell adhesion | + | + |
| 18 | HDHD2 | Phosphate metabolism | − | + |
| 18 | KIAA0427 | Protein biosynthesis | − | + |
| 18 | DYM | Skeletal development/brain function | − | + |
Genes expressed in AoAF cells, but bound to the nuclear matrix and silenced in HeLa cells, were analyzed for nuclear scaffold attachment within and up to 10 kb upstream of each gene. The majority of genes (81%) were found to contain a SAR either within the gene, upstream of the gene or both suggesting a scaffold-mediated mechanism for tissue-specific expression.
Differentially expressed genes are nuclear scaffold associated
| Chromosomes | Gene name | Biological process/function | SAR within 10-kb upstream | SAR within gene |
|---|---|---|---|---|
| 14 | SCFD1 | Exocytosis | − | + |
| 14 | C14orf11 | Stimulates proliferation | + | + |
| 14 | KIAA0423 | Mitosis | + | + |
| 14 | MAP4K5 | Protein phosphorylation | − | + |
| 14 | PYGL | Glycogen metabolism | − | + |
| 14 | KIAA0831 | Chromosome partitioning | − | + |
| 14 | DACT1 | Regulates WNT signaling | − | + |
| 14 | FUT8 | Transferase activity | − | + |
| 14 | PCNX | Neurogenesis | + | + |
| 14 | SIPA1L1 | Cell adhesion | − | + |
| 14 | RBM25 | Pre-mRNA processing | − | − |
| 14 | JDP2 | mRNA transcription regulation/tumor suppressor | − | + |
| 14 | C14orf179 | Cytoskeleton | − | − |
| 14 | FLRT2 | Cell surface receptor mediated signal transduction | − | + |
| 14 | BTBD7 | Homo/heteromeric protein dimerization | − | + |
| 15 | AQR | Viral RNA-dependent RNA polymerase | − | + |
| 15 | EIF2AK4 | Protein phosphorylation/translational regulation | + | + |
| 15 | FLJ43339 | Unclassified | + | − |
| 15 | CCDC32 | Unclassified | − | + |
| 15 | AP4E1 | Receptor-mediated endocytosis | − | + |
| 15 | LEO1 | mRNA transcription elongation | − | − |
| 15 | TPM1 | Cell motility | − | − |
| 15 | OAZ2 | Apoptosis/oncogenesis | − | + |
| 15 | ITGA11 | Cell adhesion | + | + |
| 15 | SIN3A | mRNA transcription regulation | + | + |
| 15 | UBE2Q2 | Protein modification | − | + |
| 15 | CHSY1 | Protein glycosylation | − | + |
| 16 | CREBBP | mRNA transcription regulation | − | + |
| 16 | KIAA0350 | Autoimmunity | − | + |
| 16 | ARHGAP17 | Signal transduction/cell structure | − | + |
| 16 | NSMCE1 | Genome stability | + | + |
| 16 | XPO6 | Nuclear export to cytoplasm | − | + |
| 16 | CHD9 | mRNA transcription regulation | − | + |
| 16 | AMFR | Proteolysis | + | + |
| 16 | GTL3 | Unclassified | − | − |
| 16 | CDH11 | Cell adhesion | + | + |
| 16 | CMTM4 | Inflammatory response/migration | − | + |
| 16 | AP1G1 | Receptor-mediated endocytosis | − | + |
| 17 | C17orf25 | Carbon metabolism | + | + |
| 17 | TTC19 | Unclassified | − | − |
| 17 | ZNF207 | Nucleic acid metabolism | − | − |
| 17 | CCDC47 | Unclassified | − | − |
| 17 | ERN1 | Protein phosphorylation | − | + |
| 17 | TEX2 | Unclassified | − | − |
| 17 | PRKCA | Protein phosphorylation/cell proliferation and differentiation | − | + |
| 17 | WIPI1 | Protein complex assembly | + | − |
| 18 | EPB41L3 | Tumor progression | − | + |
| 18 | RNMT | mRNA capping | + | + |
| 18 | CABLES1 | Proliferation/differentiation | − | − |
| 18 | CDH2 | Cell adhesion | + | + |
| 18 | HDHD2 | Phosphate metabolism | − | + |
| 18 | KIAA0427 | Protein biosynthesis | − | + |
| 18 | DYM | Skeletal development/brain function | − | + |
| Chromosomes | Gene name | Biological process/function | SAR within 10-kb upstream | SAR within gene |
|---|---|---|---|---|
| 14 | SCFD1 | Exocytosis | − | + |
| 14 | C14orf11 | Stimulates proliferation | + | + |
| 14 | KIAA0423 | Mitosis | + | + |
| 14 | MAP4K5 | Protein phosphorylation | − | + |
| 14 | PYGL | Glycogen metabolism | − | + |
| 14 | KIAA0831 | Chromosome partitioning | − | + |
| 14 | DACT1 | Regulates WNT signaling | − | + |
| 14 | FUT8 | Transferase activity | − | + |
| 14 | PCNX | Neurogenesis | + | + |
| 14 | SIPA1L1 | Cell adhesion | − | + |
| 14 | RBM25 | Pre-mRNA processing | − | − |
| 14 | JDP2 | mRNA transcription regulation/tumor suppressor | − | + |
| 14 | C14orf179 | Cytoskeleton | − | − |
| 14 | FLRT2 | Cell surface receptor mediated signal transduction | − | + |
| 14 | BTBD7 | Homo/heteromeric protein dimerization | − | + |
| 15 | AQR | Viral RNA-dependent RNA polymerase | − | + |
| 15 | EIF2AK4 | Protein phosphorylation/translational regulation | + | + |
| 15 | FLJ43339 | Unclassified | + | − |
| 15 | CCDC32 | Unclassified | − | + |
| 15 | AP4E1 | Receptor-mediated endocytosis | − | + |
| 15 | LEO1 | mRNA transcription elongation | − | − |
| 15 | TPM1 | Cell motility | − | − |
| 15 | OAZ2 | Apoptosis/oncogenesis | − | + |
| 15 | ITGA11 | Cell adhesion | + | + |
| 15 | SIN3A | mRNA transcription regulation | + | + |
| 15 | UBE2Q2 | Protein modification | − | + |
| 15 | CHSY1 | Protein glycosylation | − | + |
| 16 | CREBBP | mRNA transcription regulation | − | + |
| 16 | KIAA0350 | Autoimmunity | − | + |
| 16 | ARHGAP17 | Signal transduction/cell structure | − | + |
| 16 | NSMCE1 | Genome stability | + | + |
| 16 | XPO6 | Nuclear export to cytoplasm | − | + |
| 16 | CHD9 | mRNA transcription regulation | − | + |
| 16 | AMFR | Proteolysis | + | + |
| 16 | GTL3 | Unclassified | − | − |
| 16 | CDH11 | Cell adhesion | + | + |
| 16 | CMTM4 | Inflammatory response/migration | − | + |
| 16 | AP1G1 | Receptor-mediated endocytosis | − | + |
| 17 | C17orf25 | Carbon metabolism | + | + |
| 17 | TTC19 | Unclassified | − | − |
| 17 | ZNF207 | Nucleic acid metabolism | − | − |
| 17 | CCDC47 | Unclassified | − | − |
| 17 | ERN1 | Protein phosphorylation | − | + |
| 17 | TEX2 | Unclassified | − | − |
| 17 | PRKCA | Protein phosphorylation/cell proliferation and differentiation | − | + |
| 17 | WIPI1 | Protein complex assembly | + | − |
| 18 | EPB41L3 | Tumor progression | − | + |
| 18 | RNMT | mRNA capping | + | + |
| 18 | CABLES1 | Proliferation/differentiation | − | − |
| 18 | CDH2 | Cell adhesion | + | + |
| 18 | HDHD2 | Phosphate metabolism | − | + |
| 18 | KIAA0427 | Protein biosynthesis | − | + |
| 18 | DYM | Skeletal development/brain function | − | + |
Genes expressed in AoAF cells, but bound to the nuclear matrix and silenced in HeLa cells, were analyzed for nuclear scaffold attachment within and up to 10 kb upstream of each gene. The majority of genes (81%) were found to contain a SAR either within the gene, upstream of the gene or both suggesting a scaffold-mediated mechanism for tissue-specific expression.
Interestingly, several genes that are required to modulate proliferation (e.g. EPB41L3) and maintain genomic stability (e.g. NSMCE1 and KIAA0831) contain MARs and are silenced in HeLa cells. One of these genes, EPB41L3, has been shown to suppress growth and increase cellular attachment in MCF-7 breast cancer cells (36). Matrix-induced silencing of EPB41L3, NSMCE1 and KIAA0831, genes involved in cell division, may resolve the observed excessive proliferation marked by aneuploidy. This is in addition to other genomic aberrations marked by the integration of papillomaviruses (37) within the MAR of the c-MYC region (38–40).
Aberrant silencing of genes involved in the cellular stress response including SAPK/JNK and p38MAPK proteins required for cellular senescence (41–45) may also contribute to a proliferative phenotype. For example, PRKCA, which activates JNK (46,47), is nuclear matrix bound and silenced in HeLa cells. The regulation of other genes-encoding proteins of the stress response pathway like MAP4K5 and CREBBP suggests that targeting these genes to the nuclear matrix serves as a reiterative mechanism to ensure the silence of this pathway. This would also reduce the ability of HeLa cells to respond to stress as exemplified by nuclear matrix attachment and silencing of EIF2AK4. The protein encoded by EIF2AK4 is a member of a family of kinases that play a role in the down regulation of protein synthesis in response to stress. Silencing would likely permit cellular function to continue in a suboptimal environment to extend lifespan. This is in contrast to AoAF cells that in culture are limited to approximately 10 passages (Cambrex Bio Science Walkersville, Inc.).
The genome is expected to contain a combination of both constitutive and facultative MARs. Facultative MARs would be involved in functions such as cell type-specific gene expression. Comparatively, constitutive MARs may resolve as structural organizers of the genome. There are four genes that are bound to the nuclear matrix by five MARs that are not silenced in AoAF cells. These genes represent a subset of the 12 genes in total that escape nuclear matrix-mediated silencing in AoAF cells. They are also bound to the nuclear matrix in HeLa cells, where they are silent. Their constitutive attachment suggests that the MARs may function to maintain genome structure, while as above, tissue-specific regulation is governed by other factors.
Genes bound to the nuclear matrix and silenced in HeLa cells are bound to the nuclear scaffold and expressed in AoAF cells
We have observed a global correlation between expressed genes and attachment to a LIS-isolated nuclear scaffold in HeLa cells (19). Therefore, it was expected that genes expressed in AoAF cells, but matrix associated and silenced in HeLa cells, would be associated with the nuclear scaffold to promote or maintain gene expression in AoAF cells. The profile of scaffold attachment was measured for the 53 genes that are expressed in AoAF cells but bound to the nuclear matrix and silenced in HeLa cells. Interestingly, in 81% of these cases (43 of 53 genes), SARs were present either within the gene itself or within a 10-kb region upstream of the gene, including the extended promoter region (P < 0.001). This emphasizes that cell type-specific expression could be mediated through the attachment of genes to the nuclear scaffold. There is an average of two SARs per gene with no overwhelming preference for a specific location within a gene (Supplementary Table 4). The reduction in the number of attachment sites compared to MARs when the genes are silenced in HeLa cells is consistent with the view that scaffold attachment poises a gene for transcription, while matrix attachment within a gene acts as a tether to sequester and inhibit transcription.
The differential roles of nuclear matrix and nuclear scaffold attachment, as well as the ability to isolate different sites of attachment, suggested that there may be sequence characteristics that can distinguish MARs from SARs. Sequence analysis of the 107 MARs and 106 SARs that organize the 53 differentially expressed genes defined above revealed no distinguishing characteristics. The 58.06% AT content of MARs is not significantly different from that of 60.12% for the SARs. Neither is substantially different from the genomic background. The length of S/MAR attachment is expected to be ∼1000–1500 bp. This is somewhat shorter than the median length of restriction fragments analyzed (3.4 kb for the MARs and 4.6 kb for the SARs). Despite a significant computational effort a clear recognition sequence has yet to emerge. Preliminary in silico analysis suggests that sites of attachment for both the PTBP (polypyrimidine tract-binding protein) and SATB (special AT-rich sequence-binding protein) are perhaps enriched in the AoAF SAR fraction. Higher resolution strategies are currently being explored to test this tenet and identify the specific sequence characteristics that define an S/MAR.
The role of cell type-specific gene expression in cellular identity is an area intense effort. However, the role of nuclear scaffold/matrix organization of differentially expressed genes in defining and maintaining identity is only beginning to be realized. We have shown, at the genomic level, that cell type-specific gene expression is differentially maintained by nuclear scaffold association to promote appropriate expression and nuclear matrix association to inhibit inappropriate expression. While many of the differentially regulated genes presented in this study are specifically involved in cell type-specific functions of AoAF cells, several have been implicated in the aberrant proliferation and genomic instability that are characteristic of cancer cells. Their silencing in HeLa cells suggests that aberrant nuclear matrix association may lead to increased proliferation capacity in cancerous and transformed cell lines. This may provide clues to the role of the nuclear matrix in maintaining a ‘normal’ cell population.
FUNDING
National Institutes of Health (grant HD36512); Wayne State University Research Enhancement Program in Computational Biology (to S.A.K.). Funding for open access charge: Charlotte B. Failing Professorship.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Dr D. Rappolee and A. Platts for their helpful comments and discussion throughout this study. The aCGH data reported in this study will be available at GEO GSE13792.

{kind=link}
{kind=link}
Comments