





Abstract
The multifunctional mammalian apurinic/apyrimidinic (AP) endonuclease (APE) participates in the repair of AP sites in the cellular DNA as well as participating in the redox regulation of the transcription factor function. The function of APE is considered as the rate-limiting step in DNA base excision repair. Paradoxically, an unbalanced increase in APE protein leads to genetic instability. Therefore, we investigated the mechanisms of genetic instability that are induced by APE. Here, we report that the overexpression of APE protein disrupts the repair of DNA mismatches, which results in microsatellite instability (MSI). We found that expression of APE protein led to the suppression of the repair of DNA mismatches in the normal human fibroblast cells. Western blot analysis revealed that hMSH6 protein was markedly reduced in the APE-expressing cells. Moreover, the addition of purified Mutα (MSH2 and MSH6 complex) to the extracts from the APE-expressing cells led to the restoration of mismatch repair (MMR) activity. By performing MMR activity assay and MSI analysis, we found that the co-expression of hMSH6 and APE exhibited the microsatellite stability, whereas the expression of APE alone generated the MSI-high phenotype. The APE-mediated decrease in MMR activity described here demonstrates the presence of a new and highly effective APE-mediated mechanism for MSI.
INTRODUCTION
The major human apurinic and apyrimidinic (AP) endonuclease APE (also known as Hap1, Apex and Ref-1), which is homologous to Escherichia coli exonuclease III, plays a central role for both short-patch and long-patch base excision repair ( 1 – 3 ). APE acts in the base excision repair pathways to hydrolyze the abasic sites that arise from the enzymatic removal of damaged purine and pyrimidine bases, and APE also cleaves the abasic sites arising from the spontaneous hydrolysis of damaged bases. In addition to its AP endonuclease activity, this enzyme also exhibits 3′→5′ exonuclease, phosphodiesterase, 3′-phosphatase and Rnase H activities ( 4 , 5 ). APE has recently been shown to have a 3′-mismatch exonuclease activity ( 6 ), and also a nucleotide incision repair activity ( 7 ), so it might well be considered as a proofreading enzyme. APE is a multifunctional protein that is not only responsible for the repair of AP sites, but it also has been implicated in the redox regulation of the transcription factor DNA binding activity via the reduction of conserved cysteine residues in the DNA binding domains of several transcription factors ( 1 ). These factors include the activator protein-1 (AP-1), c-Fos, c-Jun, nuclear factor (NF)-κB, p53, HIF-1α and Pax protein. Furthermore, APE acts as a negative regulator of its own gene and other genes too, such as those genes coding for the parathyroid hormones ( 8 ).
Paradoxically, the microsatellite instability (MSI)-high group has a significantly high APE activity, and a higher APE activity appears to generate MSI in the dinucleotide repeats ( 9 ). In addition, expression of APE in human erythroleukemia cells generates frameshift mutations in the microsatellite markers ( 9 ), and the overexpression of APE results in an increase of 40% in the frequency of micronuclei and 33% in sister chromatid exchanges of CHO mutant cells ( 10 ). This suggests that an increase in the APE expression contributes to MSI; however, the mechanisms by which APE produce MSI are unknown.
In this study, we explore how APE generates MSI. We show that APE expression in the human fibroblast GM00637 cells inhibits mismatch repair (MMR) activity. We also provide evidence that APE decreases the level of hMSH6 protein, which is a key component in the MMR pathway. In addition, we show that the addition of hMutSα (heterodimers of hMSH2/hMSH6) to the extracts of APE-expressing cells restores the MMR activity. Moreover, we show that APE overexpression in the GM00637 cells generates frameshift mutations in the microsatellite markers having dinucleotide repeats, and also that hMSH6 expression inhibits the APE-mediated generation of MSI. Our results have an implication for how APE induces MSI.
MATERIALS AND METHODS
Cell culture
The human fibroblast GM00637 cells (Coriell Institute for Medical Research) were maintained in Eagle's minimum essential medium (EMEM) that was supplemented with 10% fetal bovine serum (FBS). The HEC59 and HEC59-chr2 cells (a generous gift from Dr Richard Boland, University of California) were cultured in Iscove's modified Dulbecco's medium (IMDM) that was supplemented with 10% FBS.
Preparation of the constructs and clones
Human APE cDNA and human MSH6 (hMSH6) cDNA were amplified by RT–PCR using the APE oligo primer (5′-ATG CCG AAG CGT GGG AAA AAG G-3′, 5′-TCA CAG TGC TAG GTA TAG GGT G-3′) and the hMSH6 oligo primer (5′-ATG TCG CGA CAG AGC ACC CTG-3′, 5′-CTA TAA TTC CTT AAT CAA AGT CAG-3′), respectively, from human fibroblast GM00637 cells. After confirming the DNA sequences, the APE cDNA was cloned into a pcDNA3 mammalian expression vector, and the hMSH6 cDNA was cloned into a pcDNA3/Hygro mammalian expression vector (Invitrogen). The transfections were performed using the LipofectAMINE method (Promega) according to the manufacturer's instructions. After transfection, the cells were incubated in complete medium containing 400 µg/ml G418 or 200 µg/ml hygromycine for 4–5 weeks. The cell clones that were resistant to G418 (for pc DNA3/APE) or hygromycine (for pcDNA3/Hygro-hMSH6) were isolated and analyzed.
Construction of adenoviral vector encoding hAPE cDNA
The APE cDNA was cloned into a pShuttle vector (Invitrogen, Carlsbad, CA) after confirming the DNA sequence. The newly constructed plasmid pShuttle-hAPE was then doubly digested with PI-SceI/I-CeuI, and the purified product was ligated using Adeno-X DNA. The DNA was linearized with PacI and purified before Liopfectamine (Invitrogen) transfection of HEK293 cells. After transduction, HEK293 cells layers were overlaid with agarose and assessed for viral plaque formation at 10 days. For virus collection, the cells were lysed with three consecutive freeze-thaw cycles, and the virus was collected from the supernatant. The virus titer was ∼1 × 10 7 p.f.u./ml, which was determined using an end-point dilution assay. A vector carrying the β-galactosidase gene LacZ (Ad-LacZ) was used to monitor the efficiency of transduction by the viral vectors and a nonspecific transgene expression controls.
Purification of human MutSα
Human MutSα was purified from the nuclear extracts of HeLaS3 cells, as described with some modifications ( 11 ). In brief, the samples used for hMutSα purification were precipitated from HeLa nuclear extracts between 30 and 65% ammonium sulfate saturation. The extract was diluted three times before being loaded onto a single-stranded DNA cellulose column (1 × 1.5 cm; United States Biochemical). The proteins were eluted by a 10 ml of 0.2–1 M NaCl gradient in buffer A; the fractions containing the MSH2–MSH6 complex were diluted to bring the conductivity of the pool to an equivalent to 100 mM NaCl before being loaded onto Mono S (HR5/5). The bound proteins were eluted with a 20 ml of 0.1–1 M NaCl gradient in buffer A. the purified hMutSα was immediately supplemented with 1.0 mg/ml BSA and 10% (w/v) sucrose, frozen in liquid N 2 , and stored at −80°C. Purified hMutSα retained full activity under these storage conditions for at least 3 months. SDS–PAGE indicated that the purified protein contained only two polypeptides, 160 kDa hMSH6 and 105 kDa hMSH2, based on Coomassie Brilliant blue staining ( Figure 6A ).
Western blotting
The cells were washed with PBS and lysed at 0°C for 30 min in a lysis buffer (20 mM HEPES, pH 7.4, 2 mM EGTA, 50 mM glycerol phosphate, 1% Triton X-100, 10% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 1 mM Na 3 VO 4 and 5 mM NaF). The protein content was determined using the Bio-Rad dye-binding microassay (Bio-Rad, Hercules, CA), and 20 µg protein per lane was electrophoresed on 10% SDS polyacrylamide gels. The proteins were blotted onto Hybon ECL membranes (Amersham-Phamacia, Biotech) and immunoblotting was carried out with anti-MSH2, anti-MSH6, anti-hMLH1 and anti-α-tubulin antibodies (Pharmingen), and also with hPMS2 (Calbiochem) The blotted proteins were then detected using an enhanced chemiluminescence detect system (iNtRON, Biotech, Seoul, Korea).
Northern blotting
The total RNA was prepared using Trizol (Gibco-BRL), separated by electrophoresis and transferred to a nitrocellulose filter in 20× SSC, and then baked at 80°C for 2 h. The filters were hybridized using a 32 P-labeled hMSH6 cDNA probe. After hybridization, the same membrane was re-probed with a 32 P-labeled GAPDH cDNA probe. Hybridization was carried out in 50% formamide, 10% dextran sulfate, 1% SDS, 1 M NaCl at 42°C for 16 h, followed by two 10 min washes at room temperature with 2× SSC and one 30 min wash at 65°C in 2× SSC-1% SDS.
In vitro MMR assay
The in vitro MMR activities of the control cells and the APE-expressing cells were measured using the M13mp2 lacZ α-complementation method as described previously ( 12 ). Briefly, the MMR repair activities were investigated using heteroduplex DNA that contained a G–T, G–G or G–A mismatch in the lacZ α-complementation gene in the M13mp2 DNA. The mismatch was at position +88, and a nick was present in the (−) strand at position −264. The repair reactions (in 25 µl vol) contained 30 mM HEPES (pH 7.8), 7 mM MgCl 2 , 4 mM ATP, 200 µM each of CTP, GTP and UTP, 100 µM each of dATP, dGTP, dTTP and dCTP, 40 mM creatine phosphate, 100 mg/ml creatine phosphokinase, 15 mM sodium phosphate, 5 ng of purified heteroduplex DNA and 50 µg of the extracted nuclear protein. The incubation was carried out at 37°C for 30 min. The DNA heteroduplex was then purified and introduced into a mutS strain via electroporation. When introduced into an E.coli strain deficient in methyl-directed heteroduplex repair ( mutS ), an unrepaired mismatch heteroduplex yields a ‘mixed’ plaque phenotype due to expression of both strands of the M13mp2 DNA. However, if repair of the mismatched base situated on the (−) strand takes place during incubation with a human cell extract, the percentage of mixed plaques decreases and the percentage of colorless (+) plaques increases, such that ratio of pure blue to pure colorless plaques is reduced. The DNA MMR activity was calculated from the following formula: 100% × [1 − (percentage of mixed colored plaques developed from the reaction containing the extract/percentage of mixed colored plaques developed from the reaction containing no extract)].
MSI analysis
Genomic DNA was extracted from each of the cell populations (the parental control cells, empty vector transfected cells, APE-expressing cells, APE and pcDNA3 expressing cells, and the APE and hMSH6 expressing cells), using the standard protocol. The markers for MSI were chosen based on the recommendations from the National Cancer Institute Workshop on Microsatellite Instability ( 13 ). The following panel of five loci was examined: BAT 25, BAT 26, D5S346, D2S123 and D17S250. PCR amplifications were performed with 100 ng of purified genomic DNA in an MJ Research Thermocycler (PTC-100; MJ Research, Watertown, MA). Subsequently, PCR products were analyzed by 8% denaturing polyacrylamide gel electrophoresis. MSI was readily determined by the presence of clearly visualized altered allelic shifts in the PCR products when compared with the control DNA derived from GM00637 cells. MSI status was independently evaluated by two blinded observers, and PCRs for microsatellite analysis were performed at least three times for all samples that exhibited MSI. The cells overexpressing APE or both APE and hMSH6 were characterized as having high-frequency MSI (MSI-High) if two or more of the five markers showed instability; they were characterized as having low-frequency MSI (MSI-Low) if only one of the five markers showed instability; or if no MSI was found, then they were classified as having microsatellite stability.
Data analysis
Data in all experiments are represented as mean ± SD. Statistical comparisons were carried out using unpaired t -test. P -values < 0.05 were considered to be statistically significant.
RESULTS
APE inhibits MMR activity
To investigate whether the APE expression influenced the DNA MMR activity, the human APE gene was subcloned into the pcDNA3 vector to form pcDNA3–APE. This construct was then transfected into the GM00637 human fibroblast cells. Several stable transfected cell lines were established after the cell selection was carried out for 5 weeks using G418. Western blot analysis revealed that the APE expression levels in the APE transfected GM00637 clones (clones-1, -3, -6 and -7) were significantly higher than those APE expression levels of the parental cells and the empty vector transfected cells ( Figure 1 ).
The nuclear extracts were then prepared and examined for their MMR ability using three heteroduplexes containing these different DNA mismatches that were derived from bacteriophage M13mp2 DNA. Unrepaired heteroduplex plasmids generated mixed-color colonies when transfected into E.coli . Repaired reaction by cell lysate resulted in decreased numbers of mixed-color colonies. As shown in Figure 2 , parental GM00637 cell extracts and pcDNA3-transfected GM00637 cell extracts efficiently repaired all three mispairs. However, APE expressing GM00637 clones were deficient in MMR function and had limited MMR activities. These results demonstrate that the APE-expressing cells exhibited a decrease in MMR activity.
APE-expressing cells exhibit reduced hMSH6 protein level
To determine whether APE protein affects the level of expression of the proteins involved in the MMR pathway, such as hMSH2, hMSH6, hMHL1 and hPMS2, we used SDS–PAGE to separate the whole cell protein extracts from the parental control cells, the empty vector transfected cells and the APE expressing GM00637 clone-3 and clone-7. The western blotting results using specific polyclonal antibodies against hMSH2, hMSH6, hMHL1 and hPMS2 showed a significant decrease in the level of MSH6 protein in the APE-expressing cells ( Figure 3 ). The hMSH6 signal intensities were calculated as percentages ± SD of the α-tubulin signal. The levels of MSH6 protein expression were reduced by 84 ± 3.3% ∼ 88 ± 3.9% in the APE-expressing cells compared with the parental cells ( Figure 3 , right panel), whereas the expression of the other proteins were similar to those of the control cells. To investigate whether APE can also suppress hMSH6 expression in other human cells, Ad-APE or Ad-LacZ was infected into human pancreas cancer cell lines PC3 and human colon cancer cell line DLD1. Forty-eight hours after infection, cells were harvested and hMSH6 expression levels were determined. Western blot analysis revealed that the hMSH6 expression levels in the three individual Ad-APE infected cells were significantly lower than that of the mock and Ad-LacZ infected cells ( Figure 4A ). The G:G mismatch activity was next examined, and it was found that APE expressing PC3 and DLD1 cells showed reduced G:G MMR activity ( Figure 4B ). These results demonstrate that hMSH6 expression can be suppressed by APE.
To further investigate whether the decreased hMSH6 expression in the APE-expressing cells occurs at the transcriptional level, we carried out northern blotting using a probe for hMSH6. We found that the levels of hMSH6 mRNA were not different between the control cells and the APE-expressing cells ( Figure 5 ).
The addition of MutSα to extracts of the APE clones restored mismatch repair
As MSH6 combines with MSH2 (to form the MutSα) is responsible for the initial recognition of a mismatched nucleotide ( 14 ), it was decided to determine whether the addition of purified human MutSα to the APE clones' extracts could restore the MMR activity. Thus, the extracts from the GM00637 clone-3 and the clone-7 in the presence or absence of purified MutSα ( Figure 6A ) were tested for their ability to correct a heteroduplex that contained a G:G mismatch. We found that the addition of purified recombinant MutSα to the extracts of the APE-expressing clones resulted in the restoration of the G:G MMR ( Figure 6B ). For controls, the MMR activities were measured in an MMR-deficient human cell line and in its complementary cell line: MSH2 -deficient HEC59 and HEC59-chr2; these are derivatives of the HEC59 cells in which the wild-type MSH2 expression has been restored via a chromosome 2 transfer. The HEC59 extracts were deficient for the repair of a G:G mismatch, whereas the addition of recombinant MutSα to the HEC59 extracts restored this MMR ( Figure 6B ). These results suggest that the APE expression results in the lack of a functional hMSH2 and hMSH6 heterodimer (the hMutSα), and this leads to a decrease in MMR activity.
Expression of hMSH6 suppresses the APE-mediated increase in microsatellite instability
To investigate whether the APE-mediated decrease in hMSH6 contributes to the APE-induced genomic instability, the hMSH6 gene was subcloned into the pcDNA3 vector. The APE and the hMSH6 constructs were cotransfected into the GM00637 cells, and stable cell lines were established after selection was done using G418 and hygromycine for 5–6 weeks. Western blot analysis revealed that the hMSH6 expression levels in the hMSH6 transfected GM00637 clone-2, clone-4 and clone-5 were significantly higher than those hMSH6 expression levels of the empty vector transfected cells ( Figure 7A ). We then measured the MMR activity, and found that the MMR activities were restored in the hMSH6 expressing cells from 83 to 98% of the activity present in the parental GM00637 cells ( Figure 7B ).
In order to determine whether down-regulation of the hMSH6 gene was involved in the APE-induced MSI, we analyzed the five microsatellite markers that are used in colorectal cancer ( 13 ). The cells were assessed for MSI at ∼55 population doublings after selection was done using antibiotics. When two or more of the five microsatellite markers showed MSI, based on the MSI-criteria, MSI-High was then scored for the cells. If only one of the five markers showed MSI, then this was scored as MSI-low for the cells, and if none of the five markers showed instability, the cell line was considered to display microsatellite stable. MSI was observed in all four APE-expressing clones, with five meeting criteria for MSI-H (MSI in two or more loci), whereas the expression of APE combined with hMSH6 showed microsatellite stability ( Table 1 ). These data suggest that unrepaired mismatch lesions resulting from the suppression of the hMSH6 gene could be an important cause of the MSI displayed by the APE-expressing cells.
DISCUSSION
We have concluded that APE overexpression interferes with the repair of DNA mismatches. Because the DNA MMR mechanisms are essential for the maintenance of the genomic integrity ( 14 ), impairment of the MMR alone is sufficient to cause genetic instability. Suppression of the hMSH6 gene via APE overexpression is likely to be responsible for the cellular inability to efficiently repair DNA mismatches, and this results in the MSI activity described in this study. Our results may explain the previous observations showing that APE overexpression, ironically enough, results in increased MSI. Our results are consistent with the possibility that the adaptive imbalanced increase in APE protein may contribute to carcinogenesis by interfering with the repair of DNA mismatches.
MMR removes the DNA mismatches that have evaded proofreading during the replication process. This versatile post-replicative repair system efficiently corrects single base mismatches and the loops of 1–3 extrahelical nucleotides that arise during the replication of the repetitive DNA tracts ( 15 – 18 ). Error correction is initiated with the binding by one of two mismatch recognition complexes that have overlapping specificities, and this ensures the efficient repair of all of the common replication errors. The MutSα and MutSβ mismatch recognition factors are heterodimers of hMSH2/hMSH6 and hMSH2/hMSH3, respectively. The MutSα complex is able to bind to and participate in the repair of single base–base mismatches and of those containing insertion/deletion loops, whereas the MutSβ complex recognizes the insertion/deletion loops that are essentially larger than the single base mispairs ( 19 – 23 ). The data presented here show that the MMR activity, as measured by monitoring the repair of A:C, G:G and G:T mispairs, is markedly reduced in the APE-expressing human fibroblast GM00637 cells ( Figure 2 ). In order to address the question as to what kind of MMR protein might be involved in the APE-dependent reduction of MMR activity, we compared the levels of MMR protein in the control cells and in the APE-expressing cells, and we found that the MSH6 protein was significantly decreased in the APE-expressing cells ( Figure 3 ). Because the hMSH6 protein can bind to mismatched DNA when it is complexed with MSH2 to form the MutSα heterodimers, we next investigated if a reduced hMSH6 level contributed to the APE-dependent decline in the MMR activity. Thus, we purified the MutSα complex and performed MMR assays after adding MutSα to the APE-expressing cell extracts. We found that purified MutSα was able to restore MMR activity in the APE-expressing cells ( Figure 6 ). Therefore, these data suggest that the reduced hMSH6 protein contributed to the suppression of MMR activity in the APE-expressing cells.
The mechanisms of MSI have been evaluated, and especially for hereditary nonpolyposis colon cancer (HNPCC). These patients have many mutations in their colon tissue and they frequently develop colon cancer during early adulthood. The etiology of the mutagenic propensity noted in HNPCC comes from the defective DNA MMR enzymes, and these defective enzymes are due to inactivating germline mutations in the MMR genes ( 24 – 27 ). The cells with a defective DNA MMR demonstrate a mutation rate 100-fold greater than that of normal cells, and this causes the accumulation of potentially deleterious mutations throughout the genome. These mutations preferentially affect the repetitive DNA sequences and they are responsible for the occurrence of MSI. APE activity was positively correlated with the degree of MSI instability for the ulcerative colitis colon tissue, and overexpression of APE protein in the human erythroleukemic cells produced the MSI-high phenotype ( 9 ). In addition, the overexpression of APE protein in an xrcc1 −/− background led to an increased number of spontaneous sister chromatid exchanges as well as the formation of micronuclei ( 10 ). Moreover, a significant increase in APE protein expression has been demonstrated for such malignancies as epithelial ovarian cancers, cervical cancer tissues and their cell lines, prostate cell tumors, gliomas, rhabdomyosarcoma and germ cell tumors ( 28 – 33 ). A higher level of APE protein expression was also reported to be associated with tumor progression ( 34 ). This suggests that the imbalanced increase in APE protein may lead to genetic instability and so contribute to carcinogenesis. We have now shown that a decreased hMSH6 expression contributes to the APE-mediated MSI. First, the APE overexpression suppressed the DNA MMR activity, and this defect was reversed by the expression of hMSH6 protein ( Figure 7 ). In addition, the cells with the co-expression of hMSH6 and APE protein exhibited microsatellite stability, whereas the expression of APE protein alone generated the MSI-high phenotype ( Table 1 ). Therefore, the MMR deficiency can arise from an APE overexpression, and this results in the generation of MSI.
Free radicals are especially prevalent in active and chronic inflammation ( 35 ). The production of free radicals induces somatic mutations that can result in genetic instability, and this can ultimately lead to carcinogenesis ( 36 ). MSI has been shown to occur in tissues that are undergoing inflammation ( 37 – 39 ). The prevailing hypothesis for the cause of the MSI seen in chronic inflammatory disease is that the overexpression of free radicals eventually overwhelms the ability of the cell to repair the DNA damage prior to replication ( 40 , 41 ). Free radicals enhance the expression of APE in gastric epithelial cells, and this is similar to the observations for other cell types such as fibroblast, marcrophages, HeLa, B cells and Chinese hamster ovarian cells ( 42 – 46 ). In addition, the gastric epithelial cells isolated from those individuals chronically infected with Helicobacter pylori were noted to express higher levels of APE protein ( 47 ). Interestingly, the level of hMSH6 protein is dramatically reduced in erythroleukemia cells exposed to free radicals ( 48 ). hMSH6 protein was also significantly decreased in the rheumatoid arthritis synovium where abundant MSI was observed ( 49 ). Moreover, an increased cancer risk occurs in the tissues of the body undergoing chronic inflammation ( 50 , 51 ). Therefore, the suppression of the MMR activity due to APE expression induced by the free radicals may be an underlying mechanism for inflammation-mediated oncogenesis.
Although our study established that the APE expression produced MSI by suppressing MMR activity, other repair enzymes such as 3-methyladenine DNA glycosylase ( 9 ), DNA polymerase β ( 52 ) and DNA polymerase κ ( 53 ) can also generate genetic instability when these proteins are overexpressed. However, the expression of 3-methyladenine DNA glycosylase, DNA polymerase β or DNA polymerase κ did not affect hMSH6 expression or the MMR activity in our system (data not shown). Ongoing experiments are being carried out to address this issue, and they involve examining the MSI generating mechanisms in response to the overexpression of 3-methyladenine DNA glycosylase, DNA polymerase β or DNA polymerase κ.
The important question that remains to be answered is the detailed mechanisms of the APE-mediated decreased MSH6 protein level. One hypothesis is that APE may affect the MSH6 stability. Because APE acts as a redox regulator, APE may affect the redox-sensitive residues of MSH6, resulting in the stimulation of MSH6 degradation. Although four MMR proteins have similar percentages of aromatic and sulfur-containing residues, MSH6 is more unstable than MSH2 and MLH1 in response to oxidative stress ( 48 ). Differential susceptibility to oxidative modification has also been reported in plasma and mitochondria proteins ( 54 , 55 ). Furthermore, APE is found to be a component of protein complexes that binds to negative calcium response element (nCaRE) ( 56 ), Ku70(Ku86) ( 57 ) and heterogeneous nuclear ribonucleoprotein L (hnRNP-L) ( 58 ), where it may down-regulate expression of those genes ( 59 , 60 ). Therefore, APE may interact with a MSH6 protein, leading to the triggering the MSH6 degradation. It is also possible that APE may affect the hMSH6 protein synthesis. Accordingly, investigations aimed at determining the detailed mechanism of the APE-associated decrease in the MSH6 protein level are currently under way.
In summary, MMR activity is markedly suppressed in APE-expressing human fibroblast GM00637 cells, and this suppression corresponds to the demonstrated declines in the levels of MSH6 protein, a known key component in the MMR pathway. Furthermore, the APE-mediated decrease in the MSH6 expression results in a reduced cellular MMR capacity as well as the increased generation of MSI. The potential role of APE to generate MSI may have clinically important implications for cancer susceptibility and carcinogenesis.
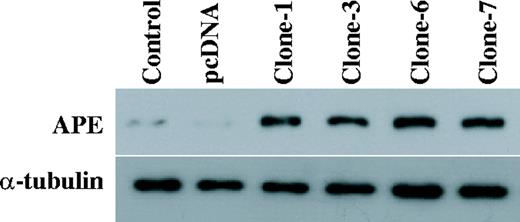
Expression of APE in GM00637 cells. The total protein was extracted from the parental control cells (control), the empty vector transfected cells (pcDNA) and the APE expressing GM00637 cells (clones-1, -3, -6 and -7). An aliquot of 50 µg of the total protein was loaded on SDS polyacrylamide gel for western blot analysis. The antibodies against APE were used. The detection of α-tubulin was used as the loading control.
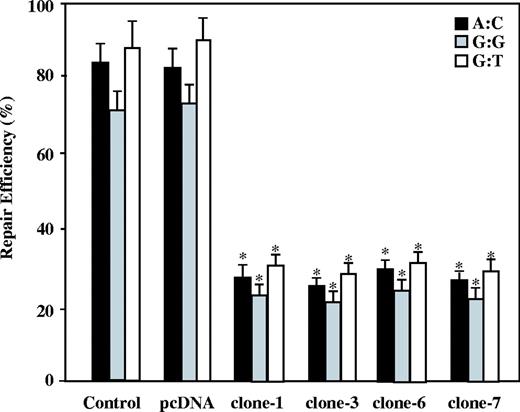
Mismatch repair efficiency in the APE-expressing cells. The nuclear extracts of the parental cells (control), the empty vector transfected cells (pcDNA) and the APE expressing GM00637 cells (clones-1, -3, -6 and -7) were compared in terms of the A:C (in black), G:G (in gray) and G:T (in white) MMR efficiencies. The results are based on counting 500–1000 plaques per variable. The repair efficiency was calculated as the percentage of decrease in the mixed-color colonies. The values are a mean ± SD from three separate experiments. Asterisk denotes P < 0.05.
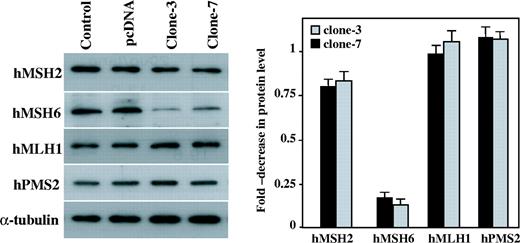
Expression of the MMR protein levels in the APE-expressing cells. In the left panel, the protein extracts of the parental cells (control), the empty vector transfected cells (pcDNA) and the APE expressing GM00637 cells (clone-3 and clone-7) were examined by western blot using Bcl-2, hMSH2, hMSH6, hMLH1 and hPMS2 antibodies. The detection of α-tubulin was used as the loading control. In the right panel, the expression levels were quantified by densitometry and they were expressed as the fold-decrease compared with the control cells. The values are a mean ± SD from three separate experiments.
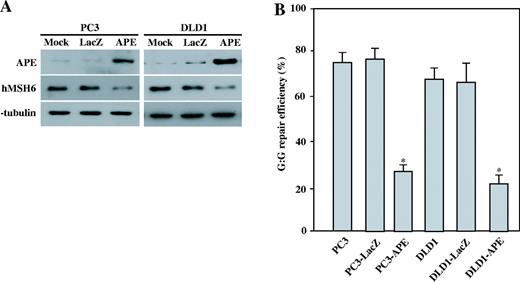
hMSH6 expression after the adenovirus-mediated transfer of APE in human cells. ( A ) PC3 and DLD1 cells were transfected with Ad-LacZ or Ad-APE at an m.o.i. of 50, and the cells were harvested 48 h after the infection. Protein extracts prepared 48 h after the infection with mock, Ad-LacZ or Ad-APE. µg of the total protein was loaded on SDS polyacrylamide gel for western blot analysis. The antibodies against APE and hMSH6 were used. The detection of α-tubulin was used as the loading control. ( B ) The nuclear extracts of the parental PC3 and DLD1 cells, Ad-LacZ infected cells and Ad-APE infected cells were used to determine the G:G MMR efficiencies. The results are based on counting 500–1000 plaques per variable. The repair efficiency was calculated as the percentage of decrease in the mixed-color colonies. The values are a mean ± SD from three separate experiments. Asterisk denotes P < 0.05.
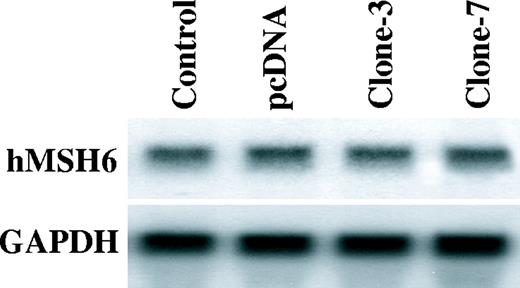
The hMSH6 mRNA levels in the APE-expressing cells. Northern blots against the total RNA for the hMSH6 and GAPDH in the parental cells (control), the empty vector transfected cells (pcDNA) and the APE expressing GM00637 cells (clones-3 and clone-7).

The hMutSα restores the repair of the G:G mismatches to the APE-expressing cells. ( A ) Purified hMutSα (hMSH2-hMSH6 heterodimer) was separated on a SDS–PAGE (8%), which was stained with Coomassie blue. The first lane of gel contains pre stained protein size marker (MBI, Fermentas), and the second lane contains hMSH6 (160 kDa) and hMSH2 (105 kDa). ( B ) The nuclear extracts of the control cells (637) and the APE expressing GM00637 cells (637 clone-3 and clone-7), and the HEC59 cells and its complementary cells were assayed for the repair of a G:G mispair in the presence or absence of 40 ng of the purified recombinant hMutSα. The results are based on counting 500–1000 plaques per variable. The repair efficiency was calculated as the percentage of decrease in the mixed-color colonies. The values are a mean ± SD from three separate experiments. Asterisk denotes P < 0.05.
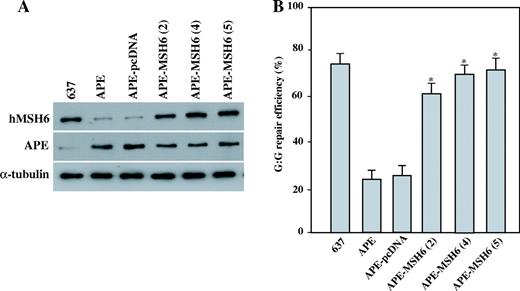
MMR activities in the APE and hMSH6 expressing cells. ( A ) The total proteins were extracted from parental GM00637 cells (637), APE expressing GM00637 clone-2 cells (APE), APE and pcDNA3 expressing GM00637 cells (APE-pcDNA), and APE and hMSH6 expressing GM00637 clones (APE-MSH6-2, -4, -5). An aliquot of 50 µg of the total protein was loaded on SDS polyacrylamide gel for western blot analysis. The antibodies against hMSH6 and APE were used. The detection of α-tubulin was used as the loading control. ( B ) The nuclear extracts of the A parental GM00637 cells (637), the APE expressing GM00637 clone-2 cells (APE), the APE and pcDNA3 expressing GM00637 cells (APE-pcDNA), and the APE and hMSH6 expressing GM00637 clones (APE-MSH6-2, -4, -5) were used to determine the G:G mismatch repair efficiencies. The results are based on counting 500–1000 plaques per variable. The repair efficiency was calculated as the percentage of decrease in the mixed-color colonies. The values are a mean ± SD from three separate experiments. Asterisk denotes P < 0.05.
MSI statue in GM00637 human fibroblast cells overexpressing APE (clones-1, 3, 6, 7) or both APE and hMSH6 (clones-2, 4, 5)
| Cell line | Microsatellite marker | MSI level | ||||
|---|---|---|---|---|---|---|
| BAT25 | BAT26 | D2S123 | D13S170 | D17S250 | ||
| GM00637 | − | − | − | − | − | Microsatellite stable |
| GM00637 + pcDNA | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE (1) | − | − | + | + | − | MSI-High |
| GM00637 + APE (3) | + | − | + | + | − | MSI-High |
| GM00637 + APE (6) | − | + | + | − | − | MSI-High |
| GM00637 + APE (7) | − | + | − | + | − | MSI-High |
| GM00637 + APE + hMSH (2) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (4) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (5) | − | − | − | − | − | Microsatellite stable |
| Cell line | Microsatellite marker | MSI level | ||||
|---|---|---|---|---|---|---|
| BAT25 | BAT26 | D2S123 | D13S170 | D17S250 | ||
| GM00637 | − | − | − | − | − | Microsatellite stable |
| GM00637 + pcDNA | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE (1) | − | − | + | + | − | MSI-High |
| GM00637 + APE (3) | + | − | + | + | − | MSI-High |
| GM00637 + APE (6) | − | + | + | − | − | MSI-High |
| GM00637 + APE (7) | − | + | − | + | − | MSI-High |
| GM00637 + APE + hMSH (2) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (4) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (5) | − | − | − | − | − | Microsatellite stable |
MSI statue in GM00637 human fibroblast cells overexpressing APE (clones-1, 3, 6, 7) or both APE and hMSH6 (clones-2, 4, 5)
| Cell line | Microsatellite marker | MSI level | ||||
|---|---|---|---|---|---|---|
| BAT25 | BAT26 | D2S123 | D13S170 | D17S250 | ||
| GM00637 | − | − | − | − | − | Microsatellite stable |
| GM00637 + pcDNA | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE (1) | − | − | + | + | − | MSI-High |
| GM00637 + APE (3) | + | − | + | + | − | MSI-High |
| GM00637 + APE (6) | − | + | + | − | − | MSI-High |
| GM00637 + APE (7) | − | + | − | + | − | MSI-High |
| GM00637 + APE + hMSH (2) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (4) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (5) | − | − | − | − | − | Microsatellite stable |
| Cell line | Microsatellite marker | MSI level | ||||
|---|---|---|---|---|---|---|
| BAT25 | BAT26 | D2S123 | D13S170 | D17S250 | ||
| GM00637 | − | − | − | − | − | Microsatellite stable |
| GM00637 + pcDNA | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE (1) | − | − | + | + | − | MSI-High |
| GM00637 + APE (3) | + | − | + | + | − | MSI-High |
| GM00637 + APE (6) | − | + | + | − | − | MSI-High |
| GM00637 + APE (7) | − | + | − | + | − | MSI-High |
| GM00637 + APE + hMSH (2) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (4) | − | − | − | − | − | Microsatellite stable |
| GM00637 + APE + hMSH (5) | − | − | − | − | − | Microsatellite stable |
This work was supported by a grant of the National Cancer Control R&D Program 2003, Ministry of Health & Welfare, Republic of Korea, and by a grant of the Molecular and Cellular BioDiscovery Research Program from the Ministry of Science and Technology. Funding to pay the Open Access publication charges for this article was provided by Ministry of Health & Welfare and the Ministry of Science and Technology, Republic of Korea.
Conflict of interest statement . None declared.
REFERENCES
Doetsch, P.W. and Cunningham, R.P.
Izumi, T. and Mitra, S.
Barzilay, G. and Hickson, I.D.
Wilson, D.M., III, Takeshita, M., Grollman, A.P., Demple, B.
Chou, K.M. and Cheng, Y.C.
Gros, L., Ishchenko, A.A., Ide, H., Elder, R.H., Saparbaev, M.K.
Hofseth, L.J., Khan, M.A., Ambrose, M., Nikolayeva, O., Xu-Welliver, M., Kartalou, M., Hussain, S.P., Roth, R.B., Zhou, X., Mechanic, L.E., et al.
Sossou, M., Flohr-Beckhaus, C., Schulz, I., Daboussi, F., Epe, B., Radicella, J.P.
Drummond, J.T., Li, G.M., Longley, M.J., Modrich, P.
Youn, C.K., Cho, H.J., Kim, S.H., Kim, H.B., Kim, M.H., Chang, I.Y., Lee, J.S., Chung, M.H., Hahm, K.S., You, H.J.
Boland, C.R., Thibodeau, S.N., Hamilton, S.R., Sidransky, D., Eshleman, J.R., Burt, R.W., Meltzer, S.J., Rodriguez-Bigas, M.A., Fodde, R., Ranzani, G.N., Srivastava, S.
Kolodner, R.D. and Marsischky, G.T.
Harfe, B.D. and Jinks-Robertson, S.
Jiricny, J. and Nystrom-Lahti, M.
Kolodner, R.D.
Modrich, P. and Lahue, R.
Marsischky, G.T., Filosi, N., Kane, M.F., Kolodner, R.
Gradia, S., Acharya, S., Fishel, R.
Genschel, J., Littman, S.J., Drummond, J.T., Modrich, P.
Marsischky, G.T. and Kolodner, R.D.
Mazurek, A., Berardini, M., Fishel, R.
Fishel, R., Lescoe, M.K., Rao, M.R., Copeland, N.G., Jenkins, N.A., Garber, J., Kane, M., Kolodner, R.D.
Leach, F.S., Nicolaides, N.C., Papadopoulos, N., Liu, B., Jen, J., Parsons, R., Peltomaki, P., Sistonen, P., Aaltonen, L.A., Nystrom-Lahti, M.
Papadopoulos, N., Nicolaides, N.C., Wei, Y.F., Ruben, S.M., Carter, K.C., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D., Fraser, C.M., Adams, M.D.
Nicolaides, N.C., Papadopoulos, N., Liu, B., Wei, Y.F., Carter, K.C., Ruben, S.M., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D., Fraser, C.M.
Moore, D.H., Michael, H., Tritt, R., Parsons, S.H., Kelley, M.R.
Xu, Y., Moore, D.H., Broshears, J., Liu, L., Wilson, T.M., Kelley, M.R.
Kelley, M.R., Cheng, L., Foster, R., Tritt, R., Jiang, J., Broshears, J., Koch, M.
Bobola, M.S., Blank, A., Berger, M.S., Stevens, B.A., Silber, J.R.
Thomson, B., Tritt, R., Davis, M., Kelley, M.R.
Robertson, K.A., Bullock, H.A., Xu, Y., Tritt, R., Zimmerman, E., Ulbright, T.M., Foster, R.S., Einhorn, L.H., Kelley, M.R.
Fritz, G., Grosch, S., Tomicic, M., Kaina, B.
Ward, P.A., Warren, J.S., Johnson, K.J.
Wiseman, H. and Halliwell, B.
Brentnall, T.A., Chen, R., Lee, J.G., Kimmey, M.B., Bronner, M.P., Haggitt, R.C., Kowdley, K.V., Hecker, L.M., Byrd, D.R.
Brentnall, T.A., Crispin, D.A., Bronner, M.P., Cherian, S.P., Hueffed, M., Rabinovitch, P.S., Rubin, C.E., Haggitt, R.C., Boland, C.R.
Suzuki, H., Harpaz, N., Tarmin, L., Yin, J., Jiang, H.Y., Bell, J.D., Hontanosas, M., Groisman, G.M., Abraham, J.M., Meltzer, S.J.
Loeb, K.R. and Loeb, L.A.
Brentnall, T.A., Crispin, D.A., Bronner, M.P., Cherian, S.P., Hueffed, M., Rabinovitch, P.S., Rubin, C.E., Haggitt, R.C., Boland, C.R.
Ramana, C.V., Boldogh, I., Izumi, T., Mitra, S.
Grosch, S., Fritz, G., Kaina, B.
Flaherty, D.M., Monick, M.M., Carter, A.B., Peterson, M.W., Hunninghake, G.W.
Frossi, B., Tell, G., Spessotto, P., Colombatti, A., Vitale, G., Pucillo, C.
Hsieh, M.M., Hegde, V., Kelley, M.R., Deutsch, W.A.
Ding, S.Z., O'Hara, A.M., Denning, T.L., Dirden-Kramer, B., Mifflin, R.C., Reyes, V.E., Ryan, K.A., Elliott, S.N., Izumi, T., Boldogh, I., Mitra, S., Ernst, P.B., Crowe, S.E.
Chang, C.L., Marra, G., Chauhan, D.P., Ha, H.T., Chang, D.K., Ricciardiello, L., Randolph, A., Carethers, J.M., Boland, C.R.
Lee, S.H., Chang, D.K., Goel, A., Boland, C.R., Bugbee, W., Boyle, D.L., Firestein, G.S.
Ekbom, A., Helmick, C., Zack, M., Adami, H.O.
Hatakeyama, M.
Yamada, N.A. and Farber, R.A.
Bavoux, C., Leopoldino, A.M., Bergoglio, V., O-Wang, J., Ogi, T., Bieth, A., Judde, J.G., Pena, S.D., Poupon, M.F., Helleday, T., Tagawa, M., Machado, C., Hoffmann, J.S., Cazaux, C.
Shacte, E., Williams, J.A., Lim, M., Levine, R.L.
Cabiscol, E., Piulats, E., Echave, P., Herrero, E., Ros, J.
Okazaki, T., Chung, U., Nishishita, T., Ebisu, S., Usuda, S., Mishiro, S., Xanthoudakis, S., Igarashi, T., Ogata, E.
Chung, U., Igarashi, T., Nishishita, T., Iwanari, H., Iwamatsu, A., Suwa, A., Mimori, T., Hata, K., Ebisu, S., Ogata, E., et al.
Kuninger, D.T., Izumi, T., Papaconstantinou, J., Mitra, S.
Izumi, T., Henner, W.D., Mitra, S.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments