





Abstract
GPAT and AIRC encode enzymes for steps one and six plus seven respectively in the pathway for de novo purine nucleotide synthesis in vertebrates. The human GPAT and AIRC genes are divergently transcribed from a 558 bp intergenic promoter region. Cis-acting sites and transcription factors important for bidirectional expression were identified. A cluster of sites between nt 215 and 260 are essential, although not sufficient, for expression of both genes. Two proteins from HepG2 cell nuclear extract, identified as NRF-1 and Sp1, bound to the promoter at sites within the 215–260 region. NRF-1 was required for stable binding of Sp1. Deletion of a 5′ promoter region including nt 215–260 resulted in decreased expression of GPAT and AIRC in transfected HepG2 cells. The decreased expression was accounted for by point mutations in an NRF-1 site and either of two flanking sites for Sp1. These transcription factors account in part for the coordinated expression of human GPAT and AIRC.
Introduction
De novo synthesis of purine nucleotides proceeds by a 10 step pathway to the branch point intermediate IMP. AMP and GMP are each derived in two steps from IMP. Six genes encode the enzymes for IMP synthesis. Three of these genes in vertebrates, GART, AIRC and IMPS, code for multifunctional enzymes (1). GPAT, which encodes the enzyme for step one and is the key regulatory enzyme of the pathway, has been found to be closely linked on human chromosome 4 to AIRC (steps six and seven) (2), whereas the remaining genes for the pathway are all on different chromosomes. GPAT and AIRC, along with GART and ADSS1 (3), are the only vertebrate genes of the pathway thus far cloned. GPAT and AIRC have been isolated from chicken (4), rat (5) and human (2). In these animals GPAT and AIRC are divergently transcribed from an intergenic promoter region of ∼230–625 bp. Whereas exon/intron organization and coding sequences are highly conserved in these mammalian and avian genes, there is only limited nucleotide sequence similarity in the intergenic region of the human and rat genes and there is no recognizable nucleotide sequence similarity between the comparable intergenic regions in the chicken and mammalian GPAT-AIRC. Yeast artificial chromosomes have been isolated that contain human GART, which encodes the trifunctional enzyme for steps two, three and five in the purine pathway (6). However, there is presently no further analysis of the GART gene.
In rat fibroblasts GPAT and AIRC mRNAs both showed ∼5- to 6-fold increases in the G1/S phase of the cell cycle (5), consistent with co-expression. There is no information currently about factors that are utilized to coordinate expression of genes for the de novo pathway. It is known, however, that one or a small number of cis-acting sites are sufficient for co-expression of other divergently transcribed genes (see for example 7–13) and thus could provide a mechanism to coordinate production of the enzymes encoded by GPAT and AIRC. In this report we have identified cis-acting sequences and transcription factors that are necessary for bidirectional transcription of the human GPAT-AIRC locus. This provides a starting point for determining how the six genes required for IMP synthesis are coordinately expressed.
Materials and Methods
GPAT transcription start site
The GPAT transcription start site was determined by RNase protection (14) using a kit from Ambion. Total RNA was isolated from HepG2 cells by the CsCl2 ultracentrifugation method (15). Two antisense RNA probes, nt 410–773 and 518–773 (Fig. 1), were synthesized in vitro by T7 RNA polymerase and radiolabeled with [α-32P]ATP.
Plasmids
The human GPAT-AIRC intergenic region nt 2–773 (Fig. 1) was cloned into the pLUC/CAT-3 bireporter promoter probe vector (16) to give the two possible orientations of the intergenic region in plasmids pHLC-1 and pHLC-2 (see Fig. 3). A series of deletions was introduced into the intergenic region as described previously (16). In the course of verifying the deletions both strands of the entire intergenic region were sequenced multiple times.
Transient transfection and reporter assays
HepG2 cells were grown to 60–80% confluency in Eagle's minimum essential medium (MEM) supplemented with 10% fetal bovine serum at 37°C with 5% CO2. Cells were transfected by the calcium phosphate procedure (17) using 8 µg CAT-LUC reporter plasmid and 2 µg RSV-lacZ plasmid (18). All transfections were repeated at least twice. Two thirds of the cells from a 35 mm dish were used to prepare extract for the chloramphenicol acetyltransferase (CAT) assay and one third for luciferase (LUC) and β-galactosidase assays. CAT was assayed by the liquid scintillation counting procedure (19). LUC and β-galactosidase activities were determined by chemiluminescent assays as described by the suppliers of reagents (Promega and Tropix). Light emission was measured as relative light units (RLUs) using a Monolight 2010 luminometer (Analytical Luminescence Laboratory). All enzyme assays were in duplicate and results averaged. Protein concentration was determined by the Bradford procedure (20). CAT and LUC specific activities from at least three separate transfections were normalized for β-galactosidase activity.
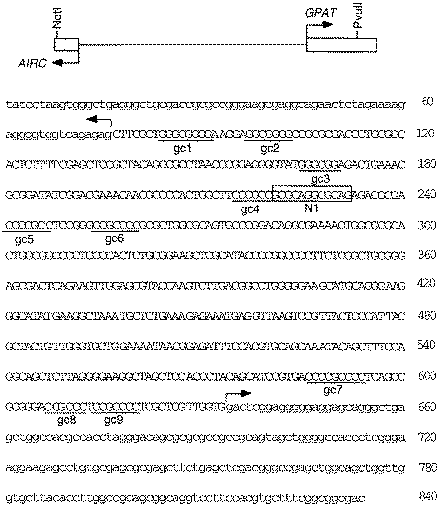
AIRC-GPAT intergenic region. A schematic diagram is given on top showing the region between transcription start sites (thin line) and 54UTRs (boxes) for AIRC and GPAT. The nucleotide sequence of 54UTRs is in lower case. A NotI site (not in the gene) was attached to nt 2 for cloning. GC boxes (gc-1 to gc-9) and recognition site 1 for NRF-1 (N1) are shown. The ATG initiation codon for AIRC and GPAT immediately precedes nt 1 and follows nt 840, but is not shown.
Protein-DNA binding
Incubations for gel shift assay of protein-DNA binding contained 25 mM HEPES, pH 7.6, 50 mM KCl, 0.1 mM EDTA, 0.1% NP40, 10% glycerol, 10 mM MgCl2, 2 µg BSA, 10 fmol 32P-labeled DNA probe, binding protein (25 µg nuclear extract, 2 ng affinity purified protein or 20 ng Escherichia coli extract) and 4 µg non-specific DNA [sonicated salmon sperm DNA or poly(dI·dC)]. The final volume was 20 µl. Sperm DNA was used as non-specific DNA for detection of binding to site N1 and poly(dI·dC) was used for binding to GC boxes. With purified DNA binding proteins, the same pattern of binding was obtained with or without non-specific DNA, thus non-specific DNA was omitted. The binding mixture was incubated for 15 min at room temperature. For supershift experiments, 1 µl antiserum or control serum was added after the 15 min incubation. The mixture was incubated for an additional 15 min at room temperature prior to electrophoresis on a 5% polyacrylamide gel (17). The following antisera were used: goat anti-NRF-1 raised against recombinant NRF-1 was a generous gift of Richard Scarpulla (Northwestern University Medical School); non-immune goat serum was from an unrelated goat; rabbit anti-TLS raised against recombinant TLS was provided by David Ron (Department of Medicine, New York University Medical Center).
DNase I footprinting was carried out according to standard procedures (17) with incubations for protein-DNA binding similar to those for gel shift. DNA probes of 167 (nt 180–346) or 276 (nt 2–277) bp containing GC boxes gc-4, gc-5, gc-6 and NRF-1 site N1 (Fig. 1) were made by PCR. The fragments were labeled with [32P]dCTP at one end.
Purification of HeLa cell DNA binding activity
HeLa cells were grown in spinner culture in MEM with 10% calf serum at 37°C to a cell density of ∼106 cells/ml. Nuclear extract was prepared from 15–20 l batches of cells (21) and stored at −80°C. For purification of the DNA binding activity, nuclear extract from a 20 l batch of cells was precipitated with 50% (NH4)2SO4 and the resulting pellet dissolved in 4 ml TM buffer containing 50 mM Tris-HCl, pH 7.9, 12.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10% glycerol. The entire solution was applied to a 2.5 × 88 cm column of Sephacryl S-300 equilibrated in TM buffer plus 0.1 M KCl. Fractions containing DNA binding activity were pooled, 200 µg/ml sonicated salmon sperm DNA added and the glycerol concentration increased to 20%. Aliquots were applied to three 1 ml DNA affinity columns equilibrated with buffer Z (25 mM HEPES, pH 7.6, 0.1 M KCl, 12.5 mM MgCl2, 1 mM DTT, 20% glycerol, 0.1% NP40). Each column was washed four times with 2 ml buffer Z containing 0.2 M KCl and proteins were then eluted batchwise with buffer Z containing increasing concentrations of KCl: 1 ml 0.3 M, 1 ml 0.4 M, 3 ml 0.5 M, 1 ml 0.6 M. Fractions eluted by 0.5 and 0.6 M KCl containing DNA binding activity were pooled and the salt concentration was reduced to 0.2 M KCl with buffer Z. Sonicated salmon sperm DNA was added to the diluted fraction as before, the solution was incubated on ice for 10 min and applied to the same three DNA affinity columns that had been used previously, stripped in buffer Z containing 1 M KCl and equilibrated with 0.2 M KCl. Elution was by the same batch method as the first time. Fractions with DNA binding activity in buffer Z plus 0.5 M KCl were pooled and stored at −80°C. To determine the protein concentration of the affinity purified binding proteins, 10% trichloroacetic acid was added to a 100 µl aliquot and frozen overnight at −80°C. After thawing, the precipitated protein was electrophoresed on an SDS−7.5% polyacrylamide gel with known amounts of bovine serum albumin alongside. After silver staining (17), the quantity of binding protein was estimated by comparison with protein standards. Data from three 15–20 l preparations were combined for the purpose of summarizing the results of protein purification, as given in Table 1.
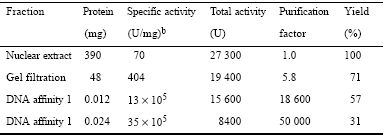
Purification of site N1 DNA binding activitya
aPurification was from ∼2.8 × 1010 cells obtained from 50 l growth medium.
bA unit of activity is defined as the protein required to shift 20% of 120 fmol labeled oligonucleotide under standard mobility shift conditions.
The DNA affinity columns were prepared by annealing 34mer oligonucleotides 5′-GATCCCCGCCGCGCAGGCGCAGAGACGCGACCCC and 5′-GATCGGGGTCGCGTCTCTGCGCCTGCGCGGCGGG, ligating the double-stranded oligomers end-to-end with T4 ligase and coupling the ligated dsDNA to CNBr-activated Sepharose by the method of Kadonaga (22).
Preparation and sequencing of peptides
Affinity purified protein was concentrated by centrifugation with a Centricon-10 membrane and precipitated with 10% trichloroacetic acid at −80°C overnight. Approximately 70 pmol protein purified from 100 l HeLa cells were electrophoresed on a SDS-7.5% acrylamide gel and stained with Coomassie blue. The stained protein band was excised, cut into pieces and digested with 0.6 ng protease Lys-C (Wako Quality Research Products) in 50 mM Tris-HCl, pH 9.0, 0.02% Tween 80 at 37°C for 15 h. Digested peptides were recovered by two extractions, each with 200 µl 60% acetonitrile, 0.1% trifluoroacetic acid at 37°C for 20 min. The combined extracts were concentrated to 50 µl in a rotary evaporator under vacuum and peptides separated by reverse phase HPLC using a C18 column and peptide detection at 214 nm. Peptide sequencing was carried out using an Applied Biosystems gas phase sequenator using standard operating procedures.
Recombinant transcription factors
A plasmid having full-length NRF-1 cDNA under control of the T7 promoter was provided by Richard Scarpulla (Northwestern University Medical School). Overexpression was obtained in E.coli B834(DE3) induced by 1 mM IPTG at 21°C in LB medium (23). Cells were grown for 6 h after induction. An extract was obtained by breaking cells in a French press and centrifugation at 27 000 g for 30 min. NRF-1 accounted for ∼10% of the soluble protein. Affinity-purified HeLa Sp1 was purchased from Promega. Binding of Sp1 was assayed by gel shift using a 25 bp synthetic oligonucleotide or with fragments of the GPAT-AIRC promoter.
Results
The GPAT transcription start site was estimated previously from the position of a pseudogene (2). We have now determined the 54-end of the GPAT mRNA by RNase protection using RNA from HepG2 cells and two different RNA probes. High expression of GPAT in liver (24) dictated the use of HepG2 cells. The probes correspond to nt 412–773 and 518–773 (Fig. 1). With each RNA probe a single protected fragment of 140 nt was obtained (Fig. 2), indicative of a transcription start site at nt 634. This transcription start site extends the GPAT 54 untranslated region (54UTR) 72 nt from that estimated previously from the pseudogene. The 558 bp intergenic region between start sites for transcription of AIRC and GPAT has a GC content of 66% and contains no TATA or CAAT boxes. The positions of nine GC boxes, potential sites for Sp1, are marked in Figure 1, along with N1, a binding site for the transcription factor NRF-1. Several errors in the previously reported sequence (accession no. U00239) were corrected.
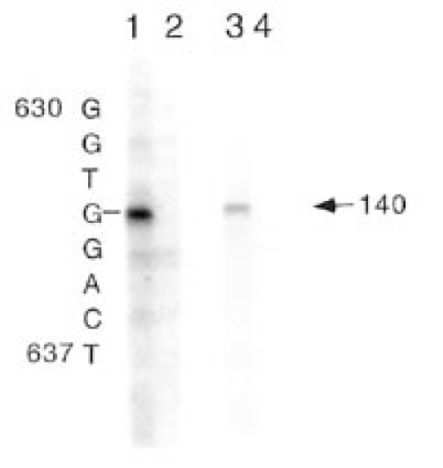
RNase protection to determine the GPAT transcription start site. Antisense RNA corresponding to nt 412–733 (lanes 1 and 2) and nt 518–733 (lanes 3 and 4) was hybridized to 20 µg HepG2 RNA (lanes 1 and 3) or 20 µg tRNA (lanes 2 and 4) and after RNase digestion was run alongside a sequencing ladder size standard on a 6% polyacrylamide sequencing gel. The arrow marks the position of the single 140 nt protected fragment. The size of the protected fragment defines the transcription start site, G at position 634.
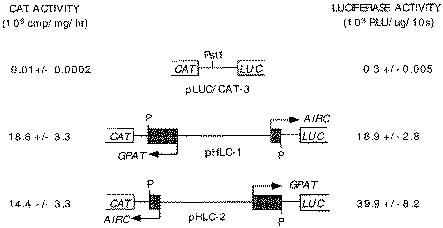
Constructs used to assay bidirectional promoter function. GPAT-AIRC nt 2–773 with added PstI adapters was inserted into the polylinker PstI site of pLUC/CAT-3. Values for CAT and LUC are the averages (± standard deviation) of at least three transfections, each assay done in duplicate. Results are normalized against β-galactosidase of a co-transfected RSV-β-gal plasmid. RLU, relative light units; P, PstI site.
Promoter analysis
To determine basal promoter function the intergenic region, nt 2–773 (Fig. 1) with PstI adapter sequences at each end was ligated in both orientations into the PstI site of the bidirectional promoter reporter vector pLUC/CAT-3 to give the CAT and LUC transcriptional fusions shown in Figure 3. Using this vector we assayed CAT and LUC reporter activities from the same extract, prepared from a single dish of transfected HepG2 cells. The results of CAT and LUC assays for the two promoter orientations are given in Figure 3. The data allow two independent assessments of relative promoter strength for transcription in the GPAT and AIRC directions. Expression of GPAT was 1.3-fold greater than that for AIRC based on comparison of the CAT reporter and 2.1-fold greater based on LUC. These results are consistent with the earlier finding of 3- to 4-fold greater expression in the GPAT direction relative to AIRC using a single reporter system (2).
One objective of this work was to identify cis-acting elements necessary for promoter function in order to understand the basis for bidirectional GPAT-AIRC transcription. As a first step, the intergenic region was scanned using a series of deletions similar to those used for the chicken GPAT-AIRC promoter (16). Plasmids with deletions were transfected into HepG2 cells and the two reporters were assayed to monitor transcription in the GPAT and AIRC directions. A deletion of nt 2–414 had the largest effect on bidirectional transcription. Transcription of AIRC was decreased to 26% and GPAT to 57% of the intact promoter (data not shown). The basis for decreased AIRC transcription was complicated, however, by the removal of the native AIRC transcription start site at nt 76 in the 2–414 deletion. Nevertheless, this result suggests that the region between nt 2 and 414 contains cis-acting sites required for transcription of GPAT and perhaps AIRC. The situation was clarified by identification of cis-acting sites required for protein binding in vitro and bidirectional expression in vivo described below.
Identification of a cis-acting protein binding site
A gel shift assay was used to search for protein binding from HepG2 nuclear extract. When the entire intergenic promoter was scanned, binding was detected with a DNA fragment containing positions 2–414 but not to one containing nt 415–773. Using subfragments, the binding was narrowed down to positions 2–277 (not shown). During the course of these experiments different patterns of protein binding were detected depending on the type of non-specific DNA that was included in the assays. With non-specific sperm DNA the gel shift was not competed by a 200-fold molar excess of unlabeled Sp1 oligonucleotide, indicating no binding to any of the six consensus GC motifs in this region (Fig. 1). With poly(dI·dC) non-specific DNA, smeared bands indicative of weak binding were obtained and these protein-DNA complexes were competed by a 100-fold molar excess of unlabeled Sp1 oligonucleotide (data not shown). DNase I footprinting was carried out to identify the protein binding sites. Footprints were obtained when sperm DNA was used to suppress non-specific binding, but not for the weaker binding obtained in the presence of poly(dI·dC). A DNase I footprint localized protein binding to the sequence between nt 218 and 244 (not shown, but see Fig. 8, lanes 1 and 2, for a related experiment). This region contains a 12 bp direct repeat, GCGCAG GCGCAG, and is flanked by Sp1 sites 4 and 5 (Fig. 1).
A 30 bp synthetic oligomer designated N1/gc-5, nt 218–247, was used to further characterize the protein-DNA interaction. A gel shift experiment with N1/gc-5 30mer and HepG2 nuclear extract is shown in Figure 4. Sperm DNA was used to inhibit non-specific binding in this experiment. Comparison of lane 1 with lane 3 shows a Mg2+ requirement for protein-DNA binding. Lane 2 demonstrates competition by a 50-fold molar excess of unlabeled 30mer. Lane 4 shows that a 200-fold excess of Sp1 oligonucleotide had no effect on protein binding, confirming that Sp1 does not bind in the presence of sperm DNA. Examination of protein binding to the N1/gc-5 DNA with a 2 bp replacement in the 12 bp repeat sequence provided further evidence for specificity. A CG→TT replacement abolished protein-DNA binding (Fig. 4, lane 5). The 12 bp repeat sequence was not found in a search of a transcription factor database (25,26) and was thus provisionally called site N1.
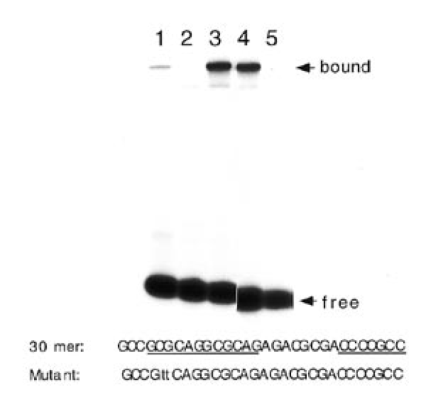
Protein binding to N1/gc-5 30mer. The nucleotide sequence of the 30mer is shown and positions of N1 and gc-5 are marked. The 2 bp N1 mutation is in lower case. Gel shift assay using 20 µg HepG2 nuclear extract with native (lanes 1–6) and mutant (lanes 7 and 8) 30mer: lane 1, Mg2+ omitted, lane 2, competition by 50-fold molar excess wild-type 30mer; lanes 3, no competitor DNA; lane 4, 200-fold excess unlabeled Sp1 oligomer; lane 5, mutant 30mer. The film was cut to remove irrelevant lanes.
Results similar to those shown in Figure 4, including the effects of non-specific DNA, were obtained with nuclear extract from HeLa cells (not shown). The gel shift with nuclear extract from HeLa and HepG2 cells was similar using the nt 2–277 and synthetic 30mer probes. In addition, virtually identical DNase I footprints were obtained.
Functional analysis of site N1 and neighboring gc boxes
In order to determine if N1 and gc sites 4, 5 and 6 have roles in transcription, we introduced mutations into these sites and examined their effect on bidirectional transcription in transfected HepG2 cells. The mutations, all 2 bp replacements, are outlined in Figure 5, together with the results of CAT and LUC reporter assays. An N1 site mutation reduced CAT expression to 20% of the native promoter activity and LUC expression was decreased by ∼30%. Thus, site N1 is seen in Figure 5 to have a role in bidirectional transcription. Individual mutations in each of the GC boxes resulted in modest decreases in bidirectional expression. The effects of the GC site mutations, although not large, were comparable for each direction of the promoter. When the site N1 and GC box mutations were combined the main effect was further reduction of GPAT expression by gc4 and gc6. There was no effect on AIRC beyond the decrease attributed to the N1 mutation except for gc6, in which case the mutation partially restored AIRC expression to the N1 mutant. This effect is unexplained. Overall, these results demonstrate that factors binding to this complement of sites are required for bidirectional transcription.
Purification and identification of site N1 protein(s)
In order to identify site N1 protein(s) we purified the binding activity assayed by gel shift. Since similar site N1 binding was detected from both HepG2 and HeLa cells, HeLa nuclei were used for the purification. A protein fraction was purified ∼50 000-fold and the results are summarized in Table 1 and Figure 6. Three purification steps, gel filtration and two cycles of DNA affinity chromatography led to a fraction with a single silver stained protein band of molecular mass ∼70 kDa. This fraction contained all of the recovered site N1 binding activity and the same DNase I footprint as obtained with HepG2 nuclear extract (data not shown). The purification was repeated two additional times, with results similar to those shown in Table 1 and Figure 6.

Inhibition of bidirectional transcription in HepG2 cells by N1, gc-4, gc-5 and gc-6 site mutations. Sites N1, gc-4, gc-5 and gc-6 in the bidirectional reporter vector pHLC-2 are shown schematically on top. Mutations are shown for each of the sites by lower case letters. CAT and LUC activities (± standard deviation) relative to the wild-type control are averages from at least three separate transfections of HepG2 cells. Each assay was done in duplicate and values were normalized against β-galactosidase from a co-transfected plasmid.
Protein sequencing was carried out to identify the binding activity in the purified fraction. No sequence information was obtained from ∼40 pmol intact protein extracted from a gel slice, suggesting a blocked N-terminus. For a second attempt, ∼70 pmol site N1 protein were electrophoresed on a SDS-7.5% polyacrylamide gel and the 70 kDa Coomassie Blue stained protein band was digested with Lys-C. A series of peptides was resolved by HPLC after extraction from the gel slice. Sequences were obtained for six peptides. Two peptide sequences, VFGAAPLQNVVRK and AFIPEMLK, correspond exactly with transcription factor NRF-1 (27). Two peptides of eight and nine residues and two 16 amino acid sequences correspond exactly to protein TLS (28). No other peptide sequences were obtained that might correspond to a third protein. Therefore, the purified fraction with site N1 binding activity contained two proteins, NRF-1 and TLS, each shown previously to migrate on SDS-polyacrylamide gels with an apparent mass of ∼70 kDa.
Binding of NRF-1 to site N1
The N1 12 bp motif corresponds to a consensus NRF-1 binding site determined previously, PyGCGCANGCGCPu (27). To confirm binding of NRF-1 to the GPAT-AIRC 12 bp motif, recombinant NRF-1 was overexpressed in E.coli and used for a gel shift assay with N1/gc-5 oligonucleotide and NRF-1 antiserum. The results are shown in Figure 7. A similar gel shift was obtained with HeLa nuclear extract (lane 1), affinity purified HeLa protein (lane 4) and recombinant NRF-1 (lane 7). NRF-1 antiserum supershifted the complex formed with each of the proteins (lanes 3, 6 and 9), whereas control serum was without effect (lanes 2, 5 and 8). The controls in lanes 10–12 show that there was no DNA binding by E.coli proteins nor was there a supershift by anti-NRF1 or control serum in the absence of NRF-1.
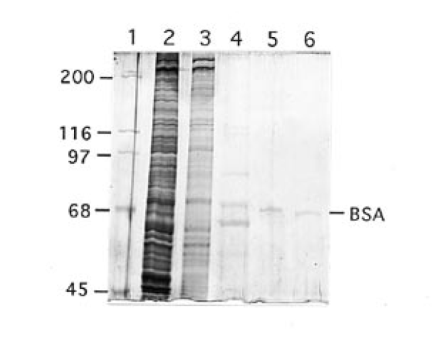
Silver stained SDS-PAGE of HeLa cell proteins at various stages of purification. Lane 1, molecular mass standards; lane 2, 4 µg nuclear extract; lane 3, 1 µg Sephacryl S-300 fraction; lane 4, ∼100 ng first DNA affinity fraction; lane 5, ∼15 ng second DNA affinity fraction; lane 6, 5 ng bovine serum albumin. It is not understood why apparently identical bands in lanes 4 and 5 are in slightly different positions.
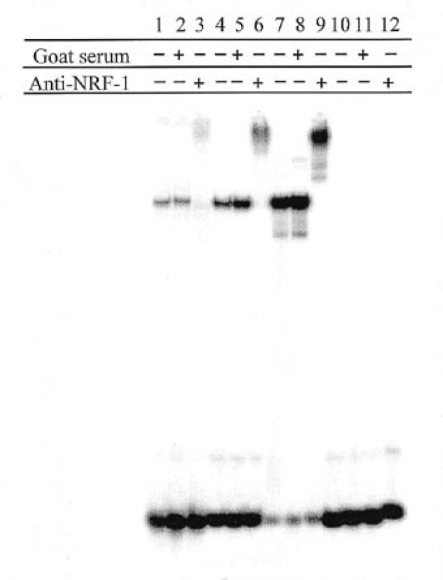
NRF-1 gel shift and supershift. Gel shift assay using: lanes 1–3, 20 µg HeLa nuclear extract; lanes 4–6, ∼4 ng affinity purified HeLa NRF-1; lanes 7–9, 20 ng E.coli extract containing recombinant NRF-1; lanes 10–12, 20 ng E.coli extract (vector control). The DNA was 32P-labeled 30mer. Following the binding reaction, 1 µl goat NRF-1 antiserum or 1 µl non-immune goat serum was added where indicated and the protein-DNA complexes were resolved by electrophoresis on a native 5% polyacrylamide gel. Lanes were counted for radioactivity with a Packard Instant Imager electronic autoradiography system and the figure is a printed display of the recorded image.
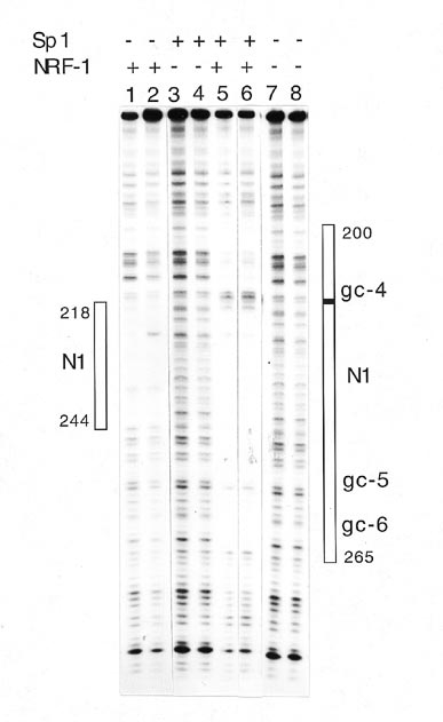
Footprints of NRF-1- and Sp1-promoter complexes. The promoter fragment, nt 180–346, labeled with 32P at its 34-end was incubated with 5 or 10 µg purified HeLa NRF-1 (lanes 1 and 2 respectively), 50 or 100 ng Sp1 (lanes 3 and 4 respectively) or 5 ng purified NRF-1 plus 50 or 100 ng Sp1 (lanes 5 and 6 respectively). No proteins were bound to DNA for lanes 7 and 8. Boxes to the left and right show boundaries of protected regions. A hypersensitive site is marked by a black box. The photograph was cut to remove irrelevant lanes.
We were unable to determine, using anti-TLS serum, whether TLS had a role in binding of NRF-1 to the oligonucleotide or was a contaminant. This is because anti-TLS cross-reacted with native and recombinant NRF-1 in gel shift experiments and increased the mobility of the NRF-1-DNA complex. A supershift was not observed. There was no cross-reaction of anti-TLS with denatured NRF-1 in a Western blot. Thus, the effect of anti-TLS on the NRF-1 gel shift is unexplained.
NRF-1-Sp1 interaction
In vivo mutation assay suggests that both NRF-1 and Sp1 have roles in transcription. It is important to determine if there is any interaction between these factors in vitro. Initial evidence for weak binding of Sp1 to the promoter was obtained from gel retardation experiments with HepG2 nuclear extract and a 167 bp fragment, nt 180–346. Two shifted species, obtained in low yield, were competed by a 100-fold molar excess of unlabeled Sp1 oligonucleotide, but not by the same molar excess of unlabeled N1/gc-5 30mer (not shown). Although this promoter DNA fragment contains sites gc4, gc5, gc6, N1 and N2, it was not possible to detect interactions of proteins from nuclear extract with Sp1 and NRF-1 sites because conflicting conditions were required to obtain specific binding to the two types of sites. Sperm DNA was most effective to detect specific binding of nuclear proteins to NRF-1 site N1 but inhibited binding of Sp1. Poly(dI·dC) was needed to detect specific binding of Sp1 but inhibited binding of nuclear proteins to site N1. It was, however, possible to demonstrate an NRF-1-Sp1-DNA interaction in experiments with purified Sp1, purified NRF-1 and promoter DNA carried out in the absence of non-specific competitor DNA. This interaction is shown by the DNase I footprints in Figure 8. There was no Sp1 footprint for the DNA/Sp1 mixture (lanes 3 and 4). However, Sp1 bound to gc4, gc5 and gc6 in the presence of NRF-1. As shown in Figure 8 (lanes 1 and 2), purified HeLa NRF-1 bound to nt 218–244, sequences that include NRF-1 site N1. With addition of Sp1 to the HeLa NRF-1/DNA mixture the site N1 footprint was expanded to include gc4, gc5 and gc6 (Fig. 8, lanes 5 and 6). This NRF-1/Sp1 footprint contains a DNase I hypersensitive site, marked in Figure 8, right side. NRF-1 thus increased the binding affinity of Sp1 to the three neighboring GC sites.
Discussion
The human GPAT and AIRC genes are divergently transcribed from a 558 bp intergenic promoter region. In previous work the human intergenic promoter region was isolated, transcription start sites for AIRC determined and promoter function estimated by cloning the promoter in both orientations into a single reporter vector (2). We have now determined the transcription start site for GPAT by RNase protection and identified cis-acting sites and transcription factors that are used for bidirectional transcription. Mutation of sites N1 and gc4, gc5 or gc6, between nt 214 and 260, resulted in decreased bidirectional expression. N1 is a key site for bidirectional transcription of these genes. It is the only site in the promoter for high affinity binding of nuclear proteins under the conditions that were used for gel shift and footprinting. Because N1 was not identified in a transcription database (25,26), it was necessary to purify the binding protein for identification as NRF-1. Two lines of evidence support a central role of NRF-1 in bidirectional expression. First, binding of NRF-1 to site N1 increased the affinity of Sp1 for flanking GC sites. Second, mutation of N1 indicates a major role of this site for AIRC transcription and a requirement together with gc4 or gc6 for concomitant GPAT transcription in transfected HepG2 cells. It should be noted, however, that the data in Figure 5, while supporting roles of NRF-1 and Sp1 for bidirectional expression in HepG2 cells, do not provide in vivo evidence to support the idea that Sp1 binding to GC sites is dependent upon NRF-1 binding to site N1. The data in Figure 5 show that interaction of Sp1 at gc4 and gc5 supported partial GPAT expression, although not AIRC expression, when binding of NRF-1 to site N1 was blocked by mutation. Therefore, the conclusion that Sp1 binding to GC sites is dependent upon NRF-1 is derived solely from in vitro gel shift and footprinting experiments. NRF-1 was affinity purified together with an RNA binding protein, TLS. Since TLS is not known to bind to DNA and was not required for binding of recombinant NRF-1 to the promoter, we assume that co-purification was a result of non-specific interactions.
A potential site for NRF-1 binding is also found in the rat GPAT-AIRC promoter and it remains to be determined whether NRF-1 has a role in co-expression of these genes in rat fibroblasts (5). An NRF-1 site is not found in the promoter region of the divergently transcribed chicken GPAT-AIRC genes (4). This may reflect different requirements for de novo purine nucleotide synthesis in birds and mammals. The pathway in mammals functions solely for biosynthesis of purine nucleotides, whereas in avian species there is the added function of synthesizing uric acid for excretion of excess nitrogen.
Analyses of cytochrome c and cytochrome oxidase promoters led to the previous identification of nuclear respiratory factors (NRF), one of which was designated nuclear respiratory factor 1 (NRF-1) (29,30). Functional NRF-1 sites have been identified in nuclear genes encoding a number of mitochondrial respiratory proteins, genes for mitochondrial DNA replication and transcription and genes encoding enzymes for protein synthesis and rate limiting enzymes in biosynthesis and catabolism (31). Thus NRF-1 is predicted to play a role in coordinating the expression of >50 mammalian nuclear and mitochondrial genes. To this list should be added the human GPAT and AIRC genes for de novo purine biosynthesis. GPAT-encoded glutamine PRPP amidotransferase is the key regulatory enzyme of the de novo purine nucleotide biosynthetic pathway. These results support the proposal by Scarpulla and co-workers that NRF-1 may help to coordinate respiratory metabolism with other biosynthetic and degradative pathways (32).
There is a potential functional link between mitochondrial respiration and GPAT-encoded glutamine PRPP amidotransferase. Regulation of glutamine PRPP amidotransferase turnover is linked to aerobic metabolism in Bacillus subtilis (33). According to the current model, decreased growth resulting from nutrient limitation leads to an elevated cellular oxygen level as a consequence of decreased respiratory chain activity. Oxidation of a labile glutamine PRPP amidotransferase Fe-S center initiates a change in conformation that triggers enzyme degradation. In this way enzyme turnover and purine nucleotide synthesis are regulated by the availability of nutrients and the capacity for growth. Human glutamine PRPP amidotransferase contains an Fe-S center and has the same properties of oxygen lability as the Bacillus enzyme (34). Although the detailed steps and signals surely differ in mammals, it will be interesting to evaluate the possibility of an NRF-1-mediated link between mitochondrial respiration and the rate limiting oxygen labile enzyme for purine biosynthesis. Chickens, which apparently do not utilize NRF-1 for GPAT expression, still contain a glutamine PRPP amidotransferase with an oxygen-labile Fe-S cluster, suggesting that this putative mechanism for enzyme regulation has been retained.
A number of examples of bidirectional transcription of closely linked genes in vertebrates have been described (see for example 7–13). In some of these cases both of the transcribed genes are known, whereas in others opposite strand transcription has been detected but the gene not identified. GPAT-AIRC and genes for human collagen type IV are the two best examples of divergent transcription of defined genes having closely related functions. The COL4A1 and COL4A2 genes code for the α1(IV) chains of collagen IV. A nuclear protein designated CTC box binding factor (CTCBF) binds to a CTC box within the 127 bp intergenic promoter and is required for bidirectional transcription (7). This transcription factor is homologous or identical to Ku antigen (35). Similar to the GPAT-AIRC locus, mutations in the CTC box reduce transcription only partially and to different extents in the two directions, suggesting that other elements contribute to promoter function. Indeed, CTC box motifs located within the first introns of COL4A1 and COL4A2, as well as intergenic CCAAT and GC boxes (36), may contribute to bidirectional transcription. Although the GPAT-AIRC promoter doesn't contain CCAAT or TATA motifs, additional cis-acting control elements in the intergenic promoter, as well as in downstream positions, likely have roles in expression which remain to be determined. NRF-1 and Sp1 binding to N1 and flanking gc sites are thus necessary, but not sufficient, for GPAT-AIRC bidirectional transcription. This report provides the first evidence for factors that are used to coordinate expression of genes for de novo purine synthesis in higher eukaryotes.
Acknowledgements
We thank Richard Scarpulla (Northwestern University Medical School) for providing NRF-1 antiserum and an NRF-1 cDNA clone, David Ron (New York University Medical Center) for TLS antiserum and other materials not used in the work described, as well as advice and stimulating discussions, Harry Charbonneau for expert advice on micro scale techniques for peptide isolation, Steven Broyles for advice on growth of HeLa cells and important discussions and Yongting Cai for technical assistance. Oligonucleotides were synthesized and peptides sequenced by Mary Bower and Alan Mahrenholz in the Purdue Laboratory for Macromolecular Structure, supported by the Diabetes Research and Training Center (NIH grant P60 DK20524). This research was supported by NIH grant GM 46466. Protein sequence and oligonucleotide synthesis were carried out by the Purdue Laboratory for Macromolecular Structure, supported by the Diabetes Research and Training Center (NIH grant P60 DK20524). This is journal paper number 15365 from the Purdue University Agricultural Research Station.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments