





Abstract
Developmental arrest in preimplantation embryos is one of the major causes of assisted reproduction failure. It is briefly defined as a delay or a failure of embryonic development in producing viable embryos during ART cycles. Permanent or partial developmental arrest can be observed in the human embryos from one-cell to blastocyst stages. These arrests mainly arise from different molecular biological defects, including epigenetic disturbances, ART processes, and genetic variants. Embryonic arrests were found to be associated with a number of variants in the genes playing key roles in embryonic genome activation, mitotic divisions, subcortical maternal complex formation, maternal mRNA clearance, repairing DNA damage, transcriptional, and translational controls. In this review, the biological impacts of these variants are comprehensively evaluated in the light of existing studies. The creation of diagnostic gene panels and potential ways of preventing developmental arrests to obtain competent embryos are also discussed.
Introduction
Preimplantation embryo development begins with the fertilization of a mature oocyte by a competent spermatozoon. Following that, the one-cell embryo undergoes mitotic divisions, namely sequential cleavage, to develop into two-cell, four-cell, and eight-cell morula, and blastocyst stage embryos (Shahbazi, 2020). To successfully complete this process, a number of genes essential for processes such as embryonic genome activation (EGA), cellular proliferation and differentiation must be timely and correctly expressed (Latham, 2023). Genetic variants in these genes’ coding or non-coding regions may result in developmental arrests during early development. Early embryonic arrest is a multifactorial condition whereby cellular division is halted for at least 24 h. This results in the failure to produce viable and good-quality embryos in ART cycles (Sfakianoudis et al., 2021). In other words, arrested embryos not only exhibit decreased metabolic activity but also cease cell division (Yang et al., 2022). Ultimately, their blastomeres undergo degeneration or lysis.
In ART cycles, the viability rate among human embryos is 40–70% with the remainder undergoing developmental arrest at different stages (Gardner and Lane, 1997; Schoolcraft et al., 2009). Indeed, ∼50% of embryos are unable to reach blastocyst stage (Gardner et al., 2000; French et al., 2010). More recent data indicate that at least one embryonic arrest occurs in ∼40% of ART cycles (ESHRE Special Interest Group of Embryology and Alpha Scientists in Reproductive Medicine, 2017). In more detail, 10–15% of ART-derived embryos exhibit permanent arrests commonly at the two- to four-cell stages during sequential mitotic divisions on Day 3, and no apoptosis indication was noted in these embryos (Betts and Madan, 2008). As in humans, the first four cleavages of bovine embryos are also prone to permanent cell cycle arrest and undergo blastomere death independent of apoptosis (Leidenfrost et al., 2011). An interesting study on bovine embryos by Favetta et al. (2004) revealed that 14.2% showed permanent arrest at the two- to four-cell stages if the first cleavage took place at 32 h post-insemination (hpi), and it was only 0.6% at 26–28 hpi (Favetta et al., 2004). These studies indicate that mammalian embryos display developmental arrests largely in the early periods, and the cleavage rate seems to be a determinant of this.
Some in vitro developed embryos that can be described as ‘transiently arrested’ exhibit no developmental progression for 24 h. Then, they progress up to the blastocyst stage (Sfakianoudis et al., 2021). Montjean et al. (2021) reported that transient embryonic arrests mostly occur in older women, more frequently in ICSI than conventional IVF (Montjean et al., 2021). This temporary event may lead to reduced implantation and delivery rates when compared to continuously developing embryos (Montjean et al., 2021). Thus, some embryos that cannot develop on time should be incubated in suitable culture conditions for at least 24 h. Permanent embryonic arrest is briefly defined as failure to develop to blastocyst stage 118 h post-ICSI. Distinguishing transient and permanent arrest types would help to determine the fate of developmentally delayed embryos.
In general, there are six potential reasons for permanent and transient arrests in preimplantation embryos: (i) the cessation of developmental progression in poor-quality embryos as a protective mechanism because they would be unable to produce highly competent blastocysts (Betts and Madan, 2008); (ii) in vitro culture conditions such as oxygen concentration and culture media may cause developmental arrests (Loutradis et al., 2000; McColLin et al., 2020); (iii) a change of development-related gene expression and small non-coding RNA levels (Betts and Madan, 2008); (iv) mitochondrial dysfunction and altered metabolism in embryos and even in oocytes (Thouas et al., 2004; Yang et al., 2022); (v) impaired/changed epigenetic landscapes, especially in DNA methylation and histone modifications, in oocytes and embryos (Yang et al., 2022); and (vi) genetic variants.
In the following sections, the potential relationship between genetic variants and the development of embryonic arrests in humans, and rarely in other mammalian species (stated in the corresponding parts) are comprehensively reviewed and discussed in the light of existing studies.
Genetic variants
Subcortical maternal complex members
The subcortical maternal complex (SCMC) structure consists of the following nine proteins: peptidyl arginine deiminase 6 (PADI6), TLE6, NLRP2, NLRP5, NLRP7, oocyte expressed protein (OOEP), KH domain containing 3 like (KHDC3L), zinc finger BED‐type containing 3 (ZBED3), and NLRP4F (Lu et al., 2017). This functionally conserved complex plays key physiological roles in oocyte-to-embryo transition, meiotic spindle positioning, maternal RNA metabolism, mitochondria redistribution, zygotic epigenetic reprogramming, translation regulation, and EGA (Bebbere et al., 2016; Lu et al., 2017). While the KHDC3L–OOEP–NLRP5–TLE6 subunit of SCMC participates in establishing correct spindle assembly, the OOEP–NLRP5–TLE6 subunit regulates spindle position through modulating F-actin dynamics to promote symmetrical cell division in mouse one-cell embryos (Zheng and Dean, 2009).
Peptidyl arginine deiminase 6
PADI6 (also known as ePAD and PAD6) is one of the Ca2+-dependent posttranslational modification enzymes that are required for oocyte maturation and preimplantation embryo development (Li et al., 2010; Xu et al., 2016). Basically, PADI6 mediates the citrullination process by transforming arginine residues with a positive charge into neutrally charged citrulline (Horibata et al., 2012). The PADI6 gene is highly expressed in the oocytes at germinal vesicle (GV), metaphase I (MI) and metaphase II (MII) stages, and eight-cell embryos, and minimally in the sperm, blastocysts, granulosa cells, and fetal somatic tissues such as those found in the human heart, liver, lungs, and spleen (Xu et al., 2016). The main task of PADI6 in mouse oocytes is to store ribosomal components within cytoplasmic lattices (Liu et al., 2017). Significant decrease of the RNA-binding protein MSY2 level in Padi66-null oocytes indicates that the mRNA, MSY2, and PADI6 complex may be formed in cytoplasmic lattices (Liu et al., 2017).
In the Padi6 knockout mouse models, while male mice were fertile, the females were found to be infertile due to embryonic arrest from pronuclear to two-cell stages (Esposito et al., 2007; Yurttas et al., 2008). As expected, the absence of PADI6 resulted in loss of citrullination in the MII oocytes, which led to dispersal of the cytoskeletal sheets (Esposito et al., 2007). Yurttas et al. (2008) demonstrated that PADI6 is essential to build cytoplasmic lattices for maternal ribosome storage. In addition, PADI6 takes part in regulating abundance and localization of the ribosomal components, de novo protein synthesis, and EGA in two-cell embryos. During this process, developmental arrest at early stages presumably results from impaired EGA, evidenced by decreased phosphorylated RNA polymerase II levels (a biomarker of transcriptional activity) (Yurttas et al., 2008). Defective organelle positioning and redistribution as well as suppressed acetylated microtubule profiles and disrupted stability of cytoplasmic microtubules were further observed in Padi6-null mouse oocytes (Kan et al., 2011). A recent study revealed that PADI6 also functions in establishing genomic imprinting marks during early development (Eggermann et al., 2021), which are essential for proper early embryogenesis.
In mutation analysis on the PADI6 gene in a consanguineous family, women with recurrent early embryonic arrest at the two- to four-cell stages on Day 3 had a homozygous nonsense variant (c.1141C>T, p.Q381∗) (Xu et al., 2016). The same study identified compound heterozygous variants (c.2009_2010del, p.E670Gfs∗48 and c.633T>A, p.H211Q or c.1618G>A, p.G540R and c.970C>T, p.Q324∗) in infertile patients with developmental arrest at the two- to five-cell stages on Day 3. A decrease of both phosphorylated RNA polymerase II and EGA-related gene expression levels were further defined in the arrested embryos (Xu et al., 2016). Other compound heterozygous missense variants (c.1793A>G, p.N598S and c.2045G>A, p.R682G) were discovered in infertile women showing early embryonic arrest at undefined stages (Qian et al., 2018).
An infertile woman who experienced embryonic arrest at the two- and seven-cell stages on Day 3 or Day 5 carried compound heterozygous missense variants (c.866C>T, p.P289L and c.1895C>T, p.P632L) (Wang et al., 2018). Furthermore, a homozygous frameshift mutation (c.1124dupT, p.L375Ffs*13) was discovered in an infertile patient with developmental arrest at the five- and six-cell stages on Day 3, or Day 5 embryos. On the other hand, Zheng et al. (2020a) found a homozygous frameshift mutation (c.1521dupC, p.S508Qfs*5) in a patient with developmental arrest at three- and four-cell stages. The same research group also found compound heterozygous missense mutations (c.A1117C, p.T373P and c.C1708T, p.R570C) in an infertile woman with embryonic arrest at the two- to five-cell stages.
Recent studies revealed a homozygous deletion (c.831_832del, p.S278Pfs*59) in the PADI6 gene of infertile women with arrested embryos at the two-cell, three-cell or other early stages on Day 3 (Liu et al., 2021; Zheng et al., 2021a). Homozygous missense (c.T487C, p.C163R) and stop-gain (c.G1425A, p.W475*) variants were also observed in infertile patients, showing embryonic arrest at one-cell or before eight-cell stages, respectively (Xu et al., 2022). Furthermore, Dong et al. (2022) found a number of variants in PADI6. A homozygous missense mutation (c.1756G>A, E586K) in an infertile woman with embryonic arrest at the two- to seven-cell stages on Day 3, and compound heterozygous variants (missense c.1006A>C, S336R and frameshift c.707dupT, L237A fs*24) were found in infertile patients who produced embryos arrested at the two- to five-cell stages. An infertile woman whose several embryos arrested at those stages carried compound homozygous variants (splicing c.294 + 5G>A and missense c.1247T>C, I416T). The compound heterozygous frameshift variants (c.5dupT, S3Qfs*65 and c.1996dupT, C666Lfs*53) were also discovered in an infertile woman with an arrested two-cell embryo on Day 3 (Dong et al., 2022).
Numerous variants have thus been defined in the PADI6 gene of women with embryonic arrest at distinct stages (Table 1). It is clear that the loss of the basic functions of PADI6 in cytoplasmic lattice formation, EGA, intracellular organelle distribution, and genomic imprinting may result in defective changes in the oocytes and embryos, changes that can entail early developmental arrests. As observed in the knockout mice, embryonic arrest at the two-cell stage indicates that PADI6 deficiency contributes to an adverse effect on the process of EGA. Embryonic arrests out of the two-cell stage may derive from divergent effects of those variants on the functional features of PADI6. These variants can be used in creating a diagnostic gene panel for women experiencing recurrent reproductive failure due to early embryonic arrests.
Main functions of defined variants and knockout phenotypes of the subcortical maternal complex (SCMC) genes.
| Gene | Main function(s) | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotypes (reference) |
|---|---|---|---|---|---|
| PADI6 |
| c.1141C>T (Nonsense, Hom) | Two- to four-cell | Xu et al. (2016) |
|
| c.2009_2010del and c.633T>A (Deletion and missense, Com Het) | Two- to five-cell | ||||
| c.1618G>A and c.970C>T (Missense and nonsense, Com Het) | |||||
| c.1793A>G and c.2045G>A (Missense mutations, Com Het) | Early embryos | Qian et al. (2018) | |||
| c.866C>T and c.1895C>T (Missense mutations, Com Het) | Two-cell, seven-cell, Day 5 embryos | Wang et al. (2018) | |||
| c.1124dupT (Frameshift, Hom) | Five-cell, six-cell, Day 5 embryos | ||||
| c.1521dupC (Frameshift, Hom) | Three-cell, four-cell | Zheng et al. (2020a ) | |||
| c.A1117C and c.C1708T (Missense mutations, Com Het) | Two- to five-cell | ||||
| c.831_832del (Deletion, Hom) | Two-cell, three-cell, early embryos | Zheng et al. (2021a ) and Liu et al. (2021) | |||
| c.T487C (Missense, Hom) | One-cell | Xu et al. (2022) | |||
| c.G1425A (Stop-gain, Hom) | Before eight-cell embryo | ||||
| c.1756G>A (Missense, Hom) | Two- to seven-cell | Dong et al. (2022) | |||
| c.1006A>C and c.707dupT (Missense and frameshift, Com Het) | Two- to five-cell | ||||
| c.294 + 5G>A and c.1247T>C (Splicing and missense, Com Het) | Two- to five-cell | ||||
| c.5dupT and c.1996dupT (Frameshift mutations, Com Het) | Two-cell | ||||
| TLE6 |
| c.1529C>A (Missense, Hom) | One-cell, two-cell, four-cell | Alazami et al. (2015) |
|
| c.1133delC (Frameshift, Hom) | Cleavage | Wang et al. (2018) | |||
| c.1621G>A (Missense, Hom) | Early embryos | Lin et al. (2020) | |||
| c.388G>A and c.1507G>A (Missense, Com Het) | Early embryos | ||||
| c.1631_1632delCA (Frameshift, Hom) | Two- to seven-cell | Zhang et al. (2021a) | |||
| c.475_476delCT (Frameshift, Hom) | Two- to nine-cell | ||||
| c.798_799insG sG and c.222G>C (Frameshift and Missense, Com Het) | Two- to six-cell | ||||
| c.222G>C, c.1564G>C, or c.893C>G (Missense, Hom) | Early embryos | Zheng et al. (2021a ) | |||
| c.222G>C and c.719C>G (Missense mutations, Com Het) | |||||
| c.1013G>A and c.1337G>A (Missense and stop-gain, Com Het) | |||||
| c.1245-2A>G (Splicing, Hom) | Two-cell, six-cell | Liu et al. (2021) | |||
| NLRP2 |
| c.1961C>A (Stop-gain, Hom) | Two- to three-cell | Mu et al. (2019) |
|
| c.773T>C and c.2254C>T (Missense and stop-gain, Com Het) | Two- to seven-cell | ||||
| (c.525G>C and c.2544A>T) (Missense mutations, Com Het) | Four-cell | ||||
| c.662C>T and c.1847A>T (Missense mutations, Com Het) | Five-cell, six-cell | ||||
| c.662C>T and c.1469C>T (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.343C>T (Stop-gain, Hom) | Early embryos | Zheng et al. (2021a) | |||
| NLRP5 |
| c.292C>T and c.2081C>T (Stop-gain and missense, Com Het) | Two- to seven-cell | Mu et al. (2019) |
|
| c.866G>A and c.3320C>T (Missense mutations, Com Het) | |||||
| c.1061C>T (Missense, Hom) | Before six-cell or eight-cell | Xu et al. (2020) | |||
| c.1903C>T, c.428G>C, or c.1384C>T (Missense mutations, Hom) | Early embryos | Zheng et al. (2021a) | |||
| c.2677T>A and c.3347T>G (Missense mutations, Com Het) | |||||
| c.1286_1289del (Frameshift, Hom) | Two-cell, three-cell | Huang et al. (2022) | |||
| c.971T>A and c.3341T>C (Missense mutations, Com Het) | Two-cell, three-cell | Tong et al. (2022) | |||
| c.1575_1576delAG and c.1830_1831delGT (Frameshift mutations, Com Het) | Two-cell, cleavage | ||||
| c.1202C>T and c.2378T>G (Missense mutations, Com Het) | Two- to eight-cell | ||||
| NLRP7 |
| c.2810 + 2T>G (Splice site, Hom) | Early embryos | Sills et al. (2017) | NA |
| OOEP |
| c.109C>G and c.110G>C (Missense mutations, Com Het) | Two- to six-cell | Tong et al. (2022) |
|
| KHDC3L |
| c.44delA (Frameshift, Hom) | Morula | Wang et al. (2018) |
|
| ZBED3 |
| NA |
| ||
| NLRP4F |
| NA |
| ||
| Gene | Main function(s) | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotypes (reference) |
|---|---|---|---|---|---|
| PADI6 |
| c.1141C>T (Nonsense, Hom) | Two- to four-cell | Xu et al. (2016) |
|
| c.2009_2010del and c.633T>A (Deletion and missense, Com Het) | Two- to five-cell | ||||
| c.1618G>A and c.970C>T (Missense and nonsense, Com Het) | |||||
| c.1793A>G and c.2045G>A (Missense mutations, Com Het) | Early embryos | Qian et al. (2018) | |||
| c.866C>T and c.1895C>T (Missense mutations, Com Het) | Two-cell, seven-cell, Day 5 embryos | Wang et al. (2018) | |||
| c.1124dupT (Frameshift, Hom) | Five-cell, six-cell, Day 5 embryos | ||||
| c.1521dupC (Frameshift, Hom) | Three-cell, four-cell | Zheng et al. (2020a ) | |||
| c.A1117C and c.C1708T (Missense mutations, Com Het) | Two- to five-cell | ||||
| c.831_832del (Deletion, Hom) | Two-cell, three-cell, early embryos | Zheng et al. (2021a ) and Liu et al. (2021) | |||
| c.T487C (Missense, Hom) | One-cell | Xu et al. (2022) | |||
| c.G1425A (Stop-gain, Hom) | Before eight-cell embryo | ||||
| c.1756G>A (Missense, Hom) | Two- to seven-cell | Dong et al. (2022) | |||
| c.1006A>C and c.707dupT (Missense and frameshift, Com Het) | Two- to five-cell | ||||
| c.294 + 5G>A and c.1247T>C (Splicing and missense, Com Het) | Two- to five-cell | ||||
| c.5dupT and c.1996dupT (Frameshift mutations, Com Het) | Two-cell | ||||
| TLE6 |
| c.1529C>A (Missense, Hom) | One-cell, two-cell, four-cell | Alazami et al. (2015) |
|
| c.1133delC (Frameshift, Hom) | Cleavage | Wang et al. (2018) | |||
| c.1621G>A (Missense, Hom) | Early embryos | Lin et al. (2020) | |||
| c.388G>A and c.1507G>A (Missense, Com Het) | Early embryos | ||||
| c.1631_1632delCA (Frameshift, Hom) | Two- to seven-cell | Zhang et al. (2021a) | |||
| c.475_476delCT (Frameshift, Hom) | Two- to nine-cell | ||||
| c.798_799insG sG and c.222G>C (Frameshift and Missense, Com Het) | Two- to six-cell | ||||
| c.222G>C, c.1564G>C, or c.893C>G (Missense, Hom) | Early embryos | Zheng et al. (2021a ) | |||
| c.222G>C and c.719C>G (Missense mutations, Com Het) | |||||
| c.1013G>A and c.1337G>A (Missense and stop-gain, Com Het) | |||||
| c.1245-2A>G (Splicing, Hom) | Two-cell, six-cell | Liu et al. (2021) | |||
| NLRP2 |
| c.1961C>A (Stop-gain, Hom) | Two- to three-cell | Mu et al. (2019) |
|
| c.773T>C and c.2254C>T (Missense and stop-gain, Com Het) | Two- to seven-cell | ||||
| (c.525G>C and c.2544A>T) (Missense mutations, Com Het) | Four-cell | ||||
| c.662C>T and c.1847A>T (Missense mutations, Com Het) | Five-cell, six-cell | ||||
| c.662C>T and c.1469C>T (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.343C>T (Stop-gain, Hom) | Early embryos | Zheng et al. (2021a) | |||
| NLRP5 |
| c.292C>T and c.2081C>T (Stop-gain and missense, Com Het) | Two- to seven-cell | Mu et al. (2019) |
|
| c.866G>A and c.3320C>T (Missense mutations, Com Het) | |||||
| c.1061C>T (Missense, Hom) | Before six-cell or eight-cell | Xu et al. (2020) | |||
| c.1903C>T, c.428G>C, or c.1384C>T (Missense mutations, Hom) | Early embryos | Zheng et al. (2021a) | |||
| c.2677T>A and c.3347T>G (Missense mutations, Com Het) | |||||
| c.1286_1289del (Frameshift, Hom) | Two-cell, three-cell | Huang et al. (2022) | |||
| c.971T>A and c.3341T>C (Missense mutations, Com Het) | Two-cell, three-cell | Tong et al. (2022) | |||
| c.1575_1576delAG and c.1830_1831delGT (Frameshift mutations, Com Het) | Two-cell, cleavage | ||||
| c.1202C>T and c.2378T>G (Missense mutations, Com Het) | Two- to eight-cell | ||||
| NLRP7 |
| c.2810 + 2T>G (Splice site, Hom) | Early embryos | Sills et al. (2017) | NA |
| OOEP |
| c.109C>G and c.110G>C (Missense mutations, Com Het) | Two- to six-cell | Tong et al. (2022) |
|
| KHDC3L |
| c.44delA (Frameshift, Hom) | Morula | Wang et al. (2018) |
|
| ZBED3 |
| NA |
| ||
| NLRP4F |
| NA |
| ||
Various variants discovered in these genes were associated with development of embryonic arrests in infertile women. NA, not available. All other abbreviations are given in the text.
Main functions of defined variants and knockout phenotypes of the subcortical maternal complex (SCMC) genes.
| Gene | Main function(s) | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotypes (reference) |
|---|---|---|---|---|---|
| PADI6 |
| c.1141C>T (Nonsense, Hom) | Two- to four-cell | Xu et al. (2016) |
|
| c.2009_2010del and c.633T>A (Deletion and missense, Com Het) | Two- to five-cell | ||||
| c.1618G>A and c.970C>T (Missense and nonsense, Com Het) | |||||
| c.1793A>G and c.2045G>A (Missense mutations, Com Het) | Early embryos | Qian et al. (2018) | |||
| c.866C>T and c.1895C>T (Missense mutations, Com Het) | Two-cell, seven-cell, Day 5 embryos | Wang et al. (2018) | |||
| c.1124dupT (Frameshift, Hom) | Five-cell, six-cell, Day 5 embryos | ||||
| c.1521dupC (Frameshift, Hom) | Three-cell, four-cell | Zheng et al. (2020a ) | |||
| c.A1117C and c.C1708T (Missense mutations, Com Het) | Two- to five-cell | ||||
| c.831_832del (Deletion, Hom) | Two-cell, three-cell, early embryos | Zheng et al. (2021a ) and Liu et al. (2021) | |||
| c.T487C (Missense, Hom) | One-cell | Xu et al. (2022) | |||
| c.G1425A (Stop-gain, Hom) | Before eight-cell embryo | ||||
| c.1756G>A (Missense, Hom) | Two- to seven-cell | Dong et al. (2022) | |||
| c.1006A>C and c.707dupT (Missense and frameshift, Com Het) | Two- to five-cell | ||||
| c.294 + 5G>A and c.1247T>C (Splicing and missense, Com Het) | Two- to five-cell | ||||
| c.5dupT and c.1996dupT (Frameshift mutations, Com Het) | Two-cell | ||||
| TLE6 |
| c.1529C>A (Missense, Hom) | One-cell, two-cell, four-cell | Alazami et al. (2015) |
|
| c.1133delC (Frameshift, Hom) | Cleavage | Wang et al. (2018) | |||
| c.1621G>A (Missense, Hom) | Early embryos | Lin et al. (2020) | |||
| c.388G>A and c.1507G>A (Missense, Com Het) | Early embryos | ||||
| c.1631_1632delCA (Frameshift, Hom) | Two- to seven-cell | Zhang et al. (2021a) | |||
| c.475_476delCT (Frameshift, Hom) | Two- to nine-cell | ||||
| c.798_799insG sG and c.222G>C (Frameshift and Missense, Com Het) | Two- to six-cell | ||||
| c.222G>C, c.1564G>C, or c.893C>G (Missense, Hom) | Early embryos | Zheng et al. (2021a ) | |||
| c.222G>C and c.719C>G (Missense mutations, Com Het) | |||||
| c.1013G>A and c.1337G>A (Missense and stop-gain, Com Het) | |||||
| c.1245-2A>G (Splicing, Hom) | Two-cell, six-cell | Liu et al. (2021) | |||
| NLRP2 |
| c.1961C>A (Stop-gain, Hom) | Two- to three-cell | Mu et al. (2019) |
|
| c.773T>C and c.2254C>T (Missense and stop-gain, Com Het) | Two- to seven-cell | ||||
| (c.525G>C and c.2544A>T) (Missense mutations, Com Het) | Four-cell | ||||
| c.662C>T and c.1847A>T (Missense mutations, Com Het) | Five-cell, six-cell | ||||
| c.662C>T and c.1469C>T (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.343C>T (Stop-gain, Hom) | Early embryos | Zheng et al. (2021a) | |||
| NLRP5 |
| c.292C>T and c.2081C>T (Stop-gain and missense, Com Het) | Two- to seven-cell | Mu et al. (2019) |
|
| c.866G>A and c.3320C>T (Missense mutations, Com Het) | |||||
| c.1061C>T (Missense, Hom) | Before six-cell or eight-cell | Xu et al. (2020) | |||
| c.1903C>T, c.428G>C, or c.1384C>T (Missense mutations, Hom) | Early embryos | Zheng et al. (2021a) | |||
| c.2677T>A and c.3347T>G (Missense mutations, Com Het) | |||||
| c.1286_1289del (Frameshift, Hom) | Two-cell, three-cell | Huang et al. (2022) | |||
| c.971T>A and c.3341T>C (Missense mutations, Com Het) | Two-cell, three-cell | Tong et al. (2022) | |||
| c.1575_1576delAG and c.1830_1831delGT (Frameshift mutations, Com Het) | Two-cell, cleavage | ||||
| c.1202C>T and c.2378T>G (Missense mutations, Com Het) | Two- to eight-cell | ||||
| NLRP7 |
| c.2810 + 2T>G (Splice site, Hom) | Early embryos | Sills et al. (2017) | NA |
| OOEP |
| c.109C>G and c.110G>C (Missense mutations, Com Het) | Two- to six-cell | Tong et al. (2022) |
|
| KHDC3L |
| c.44delA (Frameshift, Hom) | Morula | Wang et al. (2018) |
|
| ZBED3 |
| NA |
| ||
| NLRP4F |
| NA |
| ||
| Gene | Main function(s) | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotypes (reference) |
|---|---|---|---|---|---|
| PADI6 |
| c.1141C>T (Nonsense, Hom) | Two- to four-cell | Xu et al. (2016) |
|
| c.2009_2010del and c.633T>A (Deletion and missense, Com Het) | Two- to five-cell | ||||
| c.1618G>A and c.970C>T (Missense and nonsense, Com Het) | |||||
| c.1793A>G and c.2045G>A (Missense mutations, Com Het) | Early embryos | Qian et al. (2018) | |||
| c.866C>T and c.1895C>T (Missense mutations, Com Het) | Two-cell, seven-cell, Day 5 embryos | Wang et al. (2018) | |||
| c.1124dupT (Frameshift, Hom) | Five-cell, six-cell, Day 5 embryos | ||||
| c.1521dupC (Frameshift, Hom) | Three-cell, four-cell | Zheng et al. (2020a ) | |||
| c.A1117C and c.C1708T (Missense mutations, Com Het) | Two- to five-cell | ||||
| c.831_832del (Deletion, Hom) | Two-cell, three-cell, early embryos | Zheng et al. (2021a ) and Liu et al. (2021) | |||
| c.T487C (Missense, Hom) | One-cell | Xu et al. (2022) | |||
| c.G1425A (Stop-gain, Hom) | Before eight-cell embryo | ||||
| c.1756G>A (Missense, Hom) | Two- to seven-cell | Dong et al. (2022) | |||
| c.1006A>C and c.707dupT (Missense and frameshift, Com Het) | Two- to five-cell | ||||
| c.294 + 5G>A and c.1247T>C (Splicing and missense, Com Het) | Two- to five-cell | ||||
| c.5dupT and c.1996dupT (Frameshift mutations, Com Het) | Two-cell | ||||
| TLE6 |
| c.1529C>A (Missense, Hom) | One-cell, two-cell, four-cell | Alazami et al. (2015) |
|
| c.1133delC (Frameshift, Hom) | Cleavage | Wang et al. (2018) | |||
| c.1621G>A (Missense, Hom) | Early embryos | Lin et al. (2020) | |||
| c.388G>A and c.1507G>A (Missense, Com Het) | Early embryos | ||||
| c.1631_1632delCA (Frameshift, Hom) | Two- to seven-cell | Zhang et al. (2021a) | |||
| c.475_476delCT (Frameshift, Hom) | Two- to nine-cell | ||||
| c.798_799insG sG and c.222G>C (Frameshift and Missense, Com Het) | Two- to six-cell | ||||
| c.222G>C, c.1564G>C, or c.893C>G (Missense, Hom) | Early embryos | Zheng et al. (2021a ) | |||
| c.222G>C and c.719C>G (Missense mutations, Com Het) | |||||
| c.1013G>A and c.1337G>A (Missense and stop-gain, Com Het) | |||||
| c.1245-2A>G (Splicing, Hom) | Two-cell, six-cell | Liu et al. (2021) | |||
| NLRP2 |
| c.1961C>A (Stop-gain, Hom) | Two- to three-cell | Mu et al. (2019) |
|
| c.773T>C and c.2254C>T (Missense and stop-gain, Com Het) | Two- to seven-cell | ||||
| (c.525G>C and c.2544A>T) (Missense mutations, Com Het) | Four-cell | ||||
| c.662C>T and c.1847A>T (Missense mutations, Com Het) | Five-cell, six-cell | ||||
| c.662C>T and c.1469C>T (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.343C>T (Stop-gain, Hom) | Early embryos | Zheng et al. (2021a) | |||
| NLRP5 |
| c.292C>T and c.2081C>T (Stop-gain and missense, Com Het) | Two- to seven-cell | Mu et al. (2019) |
|
| c.866G>A and c.3320C>T (Missense mutations, Com Het) | |||||
| c.1061C>T (Missense, Hom) | Before six-cell or eight-cell | Xu et al. (2020) | |||
| c.1903C>T, c.428G>C, or c.1384C>T (Missense mutations, Hom) | Early embryos | Zheng et al. (2021a) | |||
| c.2677T>A and c.3347T>G (Missense mutations, Com Het) | |||||
| c.1286_1289del (Frameshift, Hom) | Two-cell, three-cell | Huang et al. (2022) | |||
| c.971T>A and c.3341T>C (Missense mutations, Com Het) | Two-cell, three-cell | Tong et al. (2022) | |||
| c.1575_1576delAG and c.1830_1831delGT (Frameshift mutations, Com Het) | Two-cell, cleavage | ||||
| c.1202C>T and c.2378T>G (Missense mutations, Com Het) | Two- to eight-cell | ||||
| NLRP7 |
| c.2810 + 2T>G (Splice site, Hom) | Early embryos | Sills et al. (2017) | NA |
| OOEP |
| c.109C>G and c.110G>C (Missense mutations, Com Het) | Two- to six-cell | Tong et al. (2022) |
|
| KHDC3L |
| c.44delA (Frameshift, Hom) | Morula | Wang et al. (2018) |
|
| ZBED3 |
| NA |
| ||
| NLRP4F |
| NA |
| ||
Various variants discovered in these genes were associated with development of embryonic arrests in infertile women. NA, not available. All other abbreviations are given in the text.
Transducin-like enhancer of split 6
The transducin-like enhancer of split 6 (TLE6) protein is encoded by a maternal effect gene and functions in protein kinase A-mediated meiotic progression in oocytes and first cleavage of one-cell embryos (Duncan et al., 2014). As expected, Tle6 is expressed in mouse oocytes during oogenesis, but not in early embryos (Li et al., 2008; Duncan et al., 2014). Nevertheless, TLE6 was detected in preimplantation embryos toward the blastocyst stage as well as in the cortex of oocytes undergoing meiotic resumption. It is important to recognize that TLE6 undergoes phosphorylation following meiotic resumption by the action of cAMP-dependent protein kinase (Duncan et al., 2014). This phosphorylation may help TLE6 to bind the remaining SCMC complex components (Lin et al., 2020). TLE6 thus participates in stabilizing SCMC that associates with the F-actin assembly regulator, cofilin, so that symmetric division in early embryos is ensured (Yu et al., 2014). The human TLE6 protein notably exhibits 44% identity with mouse homolog and is similarly detected in the subcortex of human oocytes and early embryos (Zhu et al., 2015).
The loss of Tle6 in mice causes infertility, delayed embryonic development to two-cell stage, defective early development at E2.5 and E3.5, fragmentation in blastomeres, and failure of blastocyst formation (Yu et al., 2014). However, fertilization and cleavage processes took place normally. Qin et al. (2019) also reported that TLE6 deficiency entailed disruption of cytoplasmic lattices in mouse oocytes. Taken together, these knockout studies revealed that defective early embryo development most likely occurs due to impaired cytoplasmic lattice formation especially in oocytes since this formation is associated with protein synthesis and microtubule organization (Kan et al., 2011).
Numerous variants were identified in the TLE6 gene of infertile women with embryonic arrest. Two sisters showing primary infertility due to developmental arrest at the one-cell, two-cell, or four-cell stage had a homozygous substitution (c.1529C>A, p.S510Y) (Alazami et al., 2015). This variant disrupted the phosphorylation status of TLE6 and its interaction with SCMC (Alazami et al., 2015). Another infertile patient with arrested cleavage-stage embryos on Day 3 carried a homozygous frameshift mutation (c.1133delC, p.A378Efs*75) (Wang et al., 2018). This mutation resulted in the generation of truncated protein which was unable to perform basic functions such as EGA and meiotic progression.
More recently, Lin et al. (2020) discovered a homozygous missense (c.1621G>A, p.E541K) or compound heterozygous missense mutations (c.388G>A, p.D130N and c.1507G>A, p.V503I) in two infertile women with embryonic arrest. A homozygous frameshift variant (c.1631_1632delCA, p.P544Rfs*5 in exon 17) was also noted in a woman with embryonic arrest at the two- to seven-cell stages, whereas another infertile woman whose embryos mostly arrested at the two- to nine-cell stages had a homozygous frameshift variant (c.475_476delCT, p.L159Dfs*14 in exon 7) (Zhang et al., 2021a). The compound heterozygous mutations (frameshift c.798_799insG sG, p.Q267Afs*54 and missense c.222G>C, p.Q74H) were discovered in an infertile patient with arrested embryos from two- to six-cell stages (Zhang et al., 2021a). Furthermore, Zheng et al. observed the following variants in the TLE6 gene in primary infertile women: homozygous missense (c.222G>C, p.Q74H; c.1564G>C, p.D522H, or c.893C>G, p.T298R), compound heterozygous (c.222G>C, p.Q74H and c.719C>G, p.A240G or c.1013G>A, p.R338H and c.1337G>A, p.W446*) (Zheng et al., 2021a). An infertile patient with embryonic arrest at two-cell and six-cell on Day 3 possessed a homozygous splicing mutation (c.1245-2A>G) (Liu et al., 2021).
The TLE6 gene can contain a wide range of novel variants that lead to embryonic arrest from the two- to seven-cell stages. This is similar to findings in knockout mouse models (Table 1). The divergence of the embryonic arrest stages may arise from distinct effects of these variants on the functional features of TLE6. Since TLE6 interacts with SCMC components such as NLRP5, KHDC3L, and OOEP through its specific domains (Bebbere et al., 2016) and undergoes phosphorylation, these variants may impact these actions at different levels. Thus, functional reflections of these variants on the TLE6 domains and phosphorylated residues should be examined in more detail.
The NLR family, pyrin domain containing genes
The nucleotide-binding domain and leucine-rich repeat (NLR) family members take part in establishing genomic imprints and other DNA methylation landscapes during post-zygotic periods (Amoushahi et al., 2019). There are 14 members of this family including NLRP1, 2, 4, 5, 7–9, and 11–14, which are expressed in oocytes at GV, MI, and MII stages as well as in the early embryos at Days 2, 3, and 5 (Zhang et al., 2008). In humans, NLRP2 mRNA was found at low levels in oocytes and Day 3 embryos, and then at increased levels in Day 5 embryos (Zhang et al., 2008).
The NLR family pyrin domain containing 2 (NLRP2, also known as NALP2, PAN1, and PYPAF2) is a maternal effect gene. It encodes NLRP2 protein that localizes in the subcortical region of oocytes and early embryos as well as in granulosa cells during folliculogenesis. While NLRP2 mRNA levels remarkably decrease in mouse embryos before EGA, its protein expression is present from early embryos toward blastocyst stage (Peng et al., 2012). In a more detailed analysis using immunogold electron microscopy in the same study, NLRP2 was detected in the cytoplasm, the nucleus and in proximity to nuclear pores of the oocytes as well as in the surrounding granulosa cells. Consistent with intracellular distributions, NLRP2 plays roles in cell cycle progression, apoptosis, and cell viability through modulating the MAPK signaling pathway (Zhang et al., 2019b).
In accordance with these basic roles, loss or decreased NLRP2 expression leads to various functional deficiencies in germ cells. While mouse GV oocytes with knocked down Nlrp2 expression could progress to the MII stage, parthenogenetic activation of these MII oocytes resulted in embryonic arrest at the two-cell stage (Peng et al., 2012). This is possibly due to impaired interaction between the NLRP2 and FAF1 proteins. The latter, Fas-associated protein factor 1, is implicated in successfully completing second meiotic division (Peng et al., 2018). Similarly, maternal depletion of Nlrp2 in one-cell embryos contributed to early embryonic arrest, but its overexpression permitted normal embryonic development but increased apoptosis in blastocysts (Peng et al., 2012). These findings indicate that holding NLRP2 at normal levels in oocytes is important to allow normal function during early development.
Nlrp2 null mice showed decreased reproductive rates with advancing age, suggesting that NLRP2 plays a role in protecting oocyte quality (Kuchmiy et al., 2016). Activated Nlrp2-null oocytes exhibited a slow developmental progression and could not reach blastocyst stage. Litters of knockout females were predominantly smaller than litters of wild type counterparts (Kuchmiy et al., 2016). Knocking out Nlrp2 in female mice led to subfertility, smaller litters with birth defects, growth anomalies in offspring, and embryonic arrest before blastocyst stage upon in vitro culturing of produced embryos (Mahadevan et al., 2017). Additionally, altered DNA methylation landscapes were observed in the embryos and offspring. Germline mutation in the human NLRP2 gene consistently resulted in impaired imprinting establishment, which caused the familial imprinting disorder known as the Beckwith–Wiedemann syndrome (Meyer et al., 2009). In parallel with the previous studies, Arian et al. (2021) revealed that upon transfer of the embryos from Nlrp2-deficient female mice to a wild type uterus, various types of phenotypes such as smaller litters, stillbirths, and offspring with birth defects and growth abnormalities were observed.
As a result of mutation screening in infertile women, a number of autosomal recessive novel variants were discovered in the NLRP2 gene which decreased its protein levels in human oocytes and embryos. An infertile patient with embryonic arrest at the two- to three-cell stage on Day 3 had a stop-gain mutation (c.1961C>A, p.Ser654* in exon 6) that resulted in the production of truncated protein (Mu et al., 2019). Also, compound heterozygous mutations (missense c.773T>C, p.F258S and stop-gain c.2254C>T, p.R752*) in the NLRP2 gene were detected in an infertile woman who had embryonic arrest at the two- to seven-cell stages on Day 3. Infertile women who produced a four-cell arrested embryo and other embryos that failed to form a blastocyst exhibited compound heterozygous missense mutations (c.525G>C, p.W175C and c.2544A>T, p.E848D). In the same study, other compound heterozygous missense variants (c.662C>T, p.T221M and c.1847A>T, p.E616V) were observed in an infertile patient with embryonic arrest at the five-cell and six-cell stages on Day 3. It is important to note that a healthy baby was subsequently conceived in the third ICSI application for this patient (Mu et al., 2019). The other infertile patients with embryonic arrest at the two- to six-cell stages had compound heterozygous missense mutations (c.662C>T, p.T221M and c.1469C>T, p.R490C) in the NLRP2 gene. It is important to note that this patient gave birth to live full-term infants in later attempts (Mu et al., 2019). These issues indicate that embryonic arrest cannot be observed in all sibling embryos. Recently, Zheng et al. defined a homozygous stop-gain variant (c.343C>T, p.Arg115*) in an autosomal recessive manner in the NLRP2 gene of an infertile woman with early embryonic arrest (Zheng et al., 2021a). Overall, as two infertile patients carrying the compound heterozygous mutations experienced healthy births, the remaining variants may have led to embryonic arrests accompanied by other molecular biological defects, which was not examined in these patients. This interesting issue merits further investigation.
Another NLRP family member, NLRP5 (also known as MATER) is a maternally encoded protein that participates in regulating mitochondrial function (Fernandes et al., 2012). NLRP5 includes a pyrin domain at the N-terminus, a central NACHT domain, and a leucine-rich repeat region at the C-terminus. Zhang et al. (2008) demonstrated that NLRP5 expression was at lower levels in Day 1 abnormal embryos, but at higher levels in Days 3 and 5 arrested embryos when compared to normal counterparts in humans (Zhang et al., 2008).
Upon knocking out the Nlrp5 gene, female mice exhibited sterility while males were fertile (Tong et al., 2000). This study also revealed that NLRP5 is required for normal embryonic development beyond the two-cell stage. In Nlrp5-null ovulated mouse oocytes and embryos, there were aberrant mitochondrial localization and increased mitochondrial activation for providing required ATP, which results in enhanced reactive oxygen species (ROS) levels and, consequently, mitochondrial depletion due to increased cellular stress (Fernandes et al., 2012). In parallel with the finding in mice, knockdown of NLRP5 expression in one-cell embryos of sows resulted in early embryonic arrest (Peng et al., 2015). Similarly, the depletion of NLRP5 in the oocytes of Rhesus macaque monkeys caused embryonic arrest primarily between the eight-cell and sixteen-cell stages (Wu, 2009). These studies revealed that partial or complete loss of NLRP5 is associated with developing early embryonic arrest in various species.
Infertile sisters with developmental arrest at the two- to seven-cell stages on Day 3 carried compound heterozygous variants (stop-gain c.292C>T, p.Q98* and missense c.2081C>T, p.T694I) in the NLRP5 gene (Mu et al., 2019). Further compound heterozygous missense variants (c.866G>A, p.G289E and c.3320C>T, p.T1107I) were found in an infertile patient with embryonic arrest at the two- to seven-cell stages on Day 3 (Mu et al., 2019). Studies in HeLa cells, oocytes, and embryos revealed that these variants not only decreased NLRP5 levels in vitro, but also impaired its stability. A novel homozygous missense variant (c.1061C>T, p.P354L) was discovered in an infertile patient from a consanguineous Chinese family whose embryos mostly arrested before the six-cell or eight-cell stage on Day 3 (Xu et al., 2020). On the other hand, in women with early embryonic arrests, the NLRP5 gene included homozygous missense variants (c.1903C>T, p.R635C; c.428G>C, p.R143P or c.1384C>T, p.R462C) or compound heterozygous missense variants (c.2677T>A, p.S893T and c.3347T>G, p.L1116W) (Zheng et al., 2021a).
In a subsequent study, it was reported that there was a homozygous frameshift variant (c.1286_1289del, p.V429Efs*30) in an infertile patient with embryonic arrest at the two- to three-cell stage on Day 3 in NLRP5 (Huang et al., 2022). This mutation was associated with an oocyte maturation abnormality due to the loss of a large part of the NACHT domain and the entire LRR domain at the C-terminus, which is required for maintaining the functional features of NLRP5. Moreover, Tong et al. (2022) demonstrated that an infertile woman with embryonic arrest at the two-cell or three-cell stage, including severe fragmentation, had compound heterozygous missense mutations (c.971T>A, p.M324K and c.3341T>C, p.L1114P). Another infertile patient with embryonic arrest at the one- to eight-cell stages carried different compound heterozygous frameshift mutations (c.1575_1576delAG, p.R525Sfs*17 and c.1830_1831delGT, p.Y611Rfs*2). Sisters with embryonic arrests also possessed compound heterozygous missense variants (c.1202C>T, p.P401L and c.2378T>G, p.L793R) in NLRP5 (Tong et al., 2022).
The above-mentioned studies have revealed a variety of variants in the NLRP5 gene that lead to embryonic arrest at different stages (Table 1). These variants at distinct domains of NLRP5 may contribute to loss of the functions related to sustaining mitochondrial activity and SCMC stability. Further studies are therefore needed to determine altered intracellular mechanisms that were caused by these variants and resulted in developmental arrest at preimplantation stages. Nevertheless, the aforementioned variants may be screened in infertile women displaying recurrent embryonic arrest from the two- to eight-cell stages.
NLRP7 (also known as NALP7) belongs to the CATERPILLAR protein family and is implicated in the processes of inflammation and apoptosis. The NLRP7 gene likely originated from the evolutionary duplication of NLRP2 in humans, but it does not exist in the murine genome (Duenez-Guzman and Haig, 2014). It is important to note that NLRP7 localizes together with KHDC3L to the cytoskeleton, the cortical region of growing oocytes, and embryos toward blastocyst stage (Akoury et al., 2015). Also, NLRP7 co-localizes with PADI6 in the subcortical regions of human oocytes and early embryos (Qian et al., 2018). Human NLRP7 mRNA levels are reduced from oocytes to Day 3 embryos and increase again in Day 5 embryos (Zhang et al., 2008). Although NLRP7 expression decreases in the abnormal embryos at Days 1 and 5, it increases in the arrested embryos at Day 3. This finding suggests that the NLRP7 mRNA profile may be used as a biomarker for defining embryos that would undergo embryonic arrest.
Based on an analysis of the literature, although there is no mouse knockout model for the Nlrp7 gene, its knockdown in ovine preimplantation embryos was found to adversely affect embryonic development (Li et al., 2019). Several studies revealed that functional loss of this maternal-effect NLRP7 gene results in recurrent spontaneous abortions, stillbirths, and intrauterine growth retardation as well as recurrent hydatidiform moles and reproductive wastage in humans (Murdoch et al., 2006; Qian et al., 2018). Further studies are required, however, to understand its potential relationship with embryonic development. Loss of NLRP7 roles in the processes of apoptosis and SCMC formation may cause emergence of these phenotypic impairments.
A mutation screening on an infertile woman with recurrent hydatidiform mole and embryonic arrest in her IVF embryos before or at blastocyst stage revealed a pathogenic homozygous variant (c.2810 + 2T>G at the invariant donor splice site) in NLRP7 (Sills et al., 2017). It is notable that these embryos remained in an arrested state after 144 h of in vitro culture. A recent study further found a homozygous truncating variant (c.1857_1858del, p.L619Afs*18) in the NLRP7 gene of an infertile patient with recurrent hydatidiform mole, fertilization failure, and three pronuclei in fertilized oocytes (Maddirevula et al., 2020). Since NLRP7 plays key roles in early embryonic development by participating in the SCMC and apoptosis process, mutation screening should especially be performed in infertile patients with oocyte-to-embryo transition and early development failures.
Oocyte expressed protein
Oocyte expressed protein (OOEP; also known as FLOPED, and MOEP in mice) is a member of SCMC, which resides in the subcortex of oocytes and early embryos (Li et al., 2008). Deficiency of OOEP in female mice led to infertility, embryonic arrest at the two-cell or four-cell stage, delayed first cleavage, asymmetrical divisions, and absence of cytoplasmic lattices in oocytes (Tashiro et al., 2010). Ooep null mouse blastocysts exhibited a significantly reduced differentiation rate of embryonic stem cells (Zhao et al., 2013). Taken together, as a key component of SCMC, OOEP participates in normal development of oocytes and early embryos.
A survey of the available literature shows that there is a limited number of studies that examined possible mutations in the OOEP gene of infertile patients with embryonic arrests. A recent investigation by Tong et al. (2022) reported that an infertile woman with embryonic arrest at the two- to six-cell stages on Day 3 with 10–80% fragmentation rates had compound heterozygous missense variants (c.109C>G, p.R37G and c.110G>C, p.R37P) in OOEP. At the same time, no significant changes were noted for oocyte maturation and fertilization rates in the affected patient. This variant caused reduced levels and instability of OOEP in vitro, and mutant OOEP was unable to activate transcription of many genes, some of which participate in EGA. This issue is related to early embryo development after EGA.
In addition to the basic role of OOEP in SCMC, it is also implicated in regulating development-related gene expression and establishing DNA methylation as revealed by Begemann et al. (2018). This group further demonstrated that a homozygous c.109C>T mutation in OOEP was associated with multilocus imprinting disturbances, defined in a child whose mother carried this mutation. The limited knowledge on the relationship between OOEP and embryonic arrest has prompted researchers to screen potential variants in infertile patients having embryonic arrests due to impaired SCMC formation and aberrant DNA methylation.
KH domain containing 3 like
KH domain containing 3 like (KHDC3L; also known as C6ORF221, ECAT1, and FILIA) is another basic component of SCMC and has a role in early embryo development (Zhu et al., 2015). KHDC3L is also involved in spindle assembly in cooperation with the OOEP–NLRP5–TLE6 complex (Monk et al., 2017). Furthermore, there is a physical interaction between KHDC3L and NLRP5 in cleavage-stage mouse embryos (Ohsugi et al., 2008). In human early embryonic cells, the association of KHDC3L with PARP1 induces its activity so that HR-based DNA repair takes part on double-strand break (DSB) sites (Zhang et al., 2019a). Importantly, to perform these functions, KHDC3L undergoes phosphorylation of the threonine (Thr) 145 and Thr156 residues by ataxia telangiectasia mutated (ATM). Related research has revealed that in infertile women with recurrent pregnancy loss, two deletions (p.E150_V160del and p.E150_V172del) caused a loss of phosphorylation on the Thr156 residue, thereby disrupting PARP1 activation and HR-based DSB repair (Zhang et al., 2019a).
Maternal mutant Khdc3l in mice led to 50% fertility loss, inactivation of spindle assembly checkpoint, chromosome misalignment, mitotic delay, and increased aneuploidy incidence in early embryos (Zheng and Dean, 2009). In contrast, deficiency of KHDC3L in human embryonic stem cells did not cause any abnormal changes in morphologic appearance, expression of pluripotency markers, proliferation rate, cell cycle distribution, and long-term survival (Zhang et al., 2019a). These studies indicate that KHDC3L specifically influences mitotic division-related events during early embryo development, but has no effect on embryonic stem cells.
There is a limited number of studies defining several variants in the human KHDC3L gene. An infertile woman with embryonic arrest at morula stage on Day 5 or Day 6 carried a novel homozygous frameshift mutation (c.44delA, p.Q15Rfs*25) (Wang et al., 2018). The biallelic variants such as c.1A>G, c.3G>T, c.322_325delGACT, c.1A>G and c.322_325delGACT were also discovered in patients with recurrent hydatidiform moles (Parry et al., 2011; Nguyen et al., 2018; Qian et al., 2018; Fallahi et al., 2020) or familial biparental hydatidiform moles (Parry et al., 2011). Although a close relationship was established between the KHDC3L variants and hydatidiform mole development, only one study explored possible variants in a woman who produced an arrested embryo. Transfer of early-stage preimplantation embryos including KHDC3L variants may result in implantation in the uterus at the morula stage, which would result in the formation of pathological conceptus.
Zinc finger BED‐type containing 3
The zinc finger BED‐type containing 3 (ZBED3) protein is a novel maternal component of SCMC and contributes to modulating the distribution of organelles. Maternal loss of Zbed3 in mice decreased fecundity of females due to delayed embryonic development, asymmetric mitotic division, impaired organelle distributions (e.g., endoplasmic reticulum and mitochondria), and abnormalities in microtubule dynamics and cytoplasmic lattice formation in the oocytes and zygotes (Gao et al., 2018). Although ZBED3 plays key roles in various cellular events in oocytes and early embryos possibly by participating in WNT/Beta-catenin signaling (Chen et al., 2009), no study has examined potential variants in infertile women with embryonic arrest. Since there was no phenotype detected related to embryonic arrest in the knockout mouse model, potential variants in ZBED3 may show its effect on embryonic arrest along with other variants in the SCMC genes.
Another oocyte-specific SCMC protein NLR family, the pyrin domain containing 4F (NLRP4F) functions in forming a cytoplasmic lattice and distributing organelles. As expected, depletion of Nlrp4f in mice resulted in reduced fecundity and delayed early embryo development as well as impaired organelle distribution in the oocytes and early embryos (Qin et al., 2019). Additionally, there was abnormal cytoplasmic lattice formation, but no change was detected in the interaction of NLRP4F with other SCMC proteins. Functional features and phenotypic reflections of NLRP4F deficiency on oocyte maturation and early embryo development suggest that potential variants might be discovered in infertile women with embryonic arrests.
So far, I have focused on the defined variants in the components of SCMC, which functions in the processes of spindle organization, EGA, and epigenetic reprogramming. Consistent with mouse knockout phenotypes, a variety of variants in the SCMC-related genes, such as PADI6, TLE6, NLRP2 and 5, OOEP, and KHDC3L were found to be associated with embryonic arrest from one-cell to morula stages (Fig. 1). These findings offer the promising suggestion that a diagnostic gene panel for these genes can be created for infertile women with embryonic arrest at a specific stage.
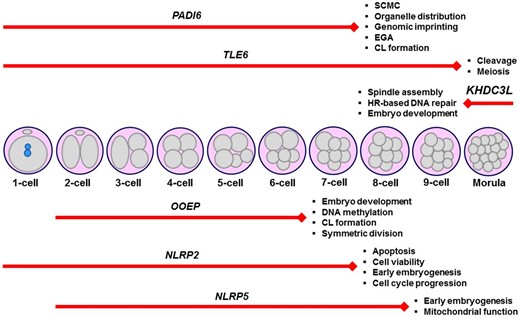
Schematic representation of the embryonic arrest stages in infertile women carrying various variants in the subcortical maternal complex (SCMC) genes, including PADI6, TLE6, KHDC3L, OOEP, NLRP2, and NLRP5. It is noteworthy that genetic variants (Table 1) that lead to arresting embryos at specific stages were only shown herein. Additionally, the main functions of the corresponding genes are given at the end of each red line. The length of the red lines covers embryonic arrest stages. CL, cytoplasmic lattice; EGA, embryonic genome activation; HR, homologous recombination.
Spindle assembly and chromosome segregation-related genes
Correct chromosome segregation is largely dependent on proper and timely assembly of microtubules. In this process, centrosome-based cytoplasmic microtubule-organizing centers (MTOCs) together with the tubulin beta 8 class VIII (TUBB8), centromere protein H (CENPH), and cell division cycle 20 (CDC20) proteins play an important role in creating spindles in a normal structure (Nigg and Raff, 2009).
Tubulin beta 8 class VIII
The primate-specific TUBB8 gene encodes a β-tubulin isotype, which is exclusively expressed in human oocytes and embryos (Feng et al., 2016; Sang et al., 2021). Basically, TUBB8 aids the assembly of meiotic and mitotic spindles. A number of studies have been done to establish the relationship between TUBB8 variants and embryonic arrest. The first study on this subject by Feng et al. (2016) reported that infertile women who carried one of these heterozygous missense mutations (c.A628G, p.I210V; c.C713T, p.T238M, or c.A1043G, p.N348S) were able to yield early embryos. However, these embryos failed to develop beyond the four-cell or eight-cell stage likely due to not having properly assembled spindles for mitotic progression. Indeed, these variants contributed to absent (p.I210V), disorganized (p.T238M), or quite low level of spindles (p.N348S).
A woman with primary infertility produced a few embryos arrested at the four-cell stage and possessed a homozygous missense mutation (c.209C>T, P70L) in the TUBB8 gene (Chen et al., 2017a). Infertile sisters with embryonic arrest at an early stage also had a heterozygous missense mutation (c.990G>A, M330I). The other heterozygous missense variants (c.1057G>A, V353I or c.613G>A, E205K) were discovered in infertile patients with early embryonic arrest. A compound heterozygous missense mutation (c.580G>A, E194K and c.1245G>A, M415I) was likewise detected in an infertile patient who produced embryos arrested at early stages on Day 3. There was a homozygous deletion in exons 1–4 of the TUBB8 gene in an infertile patient with embryonic arrest at the one-cell stage (Chen et al., 2017a). On the other hand, in a woman with embryonic arrest at the one-cell stage due to cleavage failure, Yuan et al. (2018) identified a homozygous missense variant (c.322G>A, p.E108K). Later, Chen et al. (2019) found the following additional variants in the TUBB8 gene: heterozygous (c.10A>C, p.I4L; c.1000C>G, p.Q334E; c.1171C>T, p.R391C; c.181C>A, p.P61T; c.1057G>A, p.V353I; c.1249G>T, p.D417Y; c.763G>A, p.V255M; c.883G>C, p.D295H, or c.600T>G, p.F200L) and homozygous (c.721C>T, p.R241C) ones in infertile patients with embryonic arrests.
In recent years, an infertile patient with arrested embryos at cleavage stage was found to carry a heterozygous missense mutation (c.1041C>A, p.N347K) in TUBB8 (Jia et al., 2020). A number of additional TUBB8 variants were also found in infertile women with embryonic arrest: heterozygous (c.1172G>A, p.R391H; c.1139G>A, p.R380H; c.236G>A, p.G79E; c.743C>T, p.A248V; c.940G>T, p.A314S; c.728C>G, p.P243R; c.497C>T, p.T166I; c.736C>G, p.L246V; c.925C>T, p.R309C), homozygous (c.1076G>A, p.R359Q), and compound heterozygous (c.82C>T, p.H28Y and c.398T>C, p.F133S) missense variants as well as a heterozygous nonsense variant (c.400C>T, p.Q134*) (Zhao et al., 2020a). The other heterozygous missense variants, including c.G6T, p.R2S; c.C43A, p.Q15K; c.C124G, p.L42V; c.G262C, p.D88H; c.G421C, p.G141R; c.C544T, p.P182S; c.G586A, p.A196T; c.A604C, p.I202L; c.A694T, p.T232S; c.A898T, p.M300L; c.G904A, p.A302T; c.G959A, p.R320H; c.C1055T, p.A352V; c.C1171T, p.R391C; c.G1172T, p.R391L; c.G1172A, p.R391H, and c.C1232A, p.A411D, were defined in infertile patients who suffered from early embryonic arrest (Zheng et al., 2021b). Other infertile women with early embryonic arrest had a homozygous (c.161C>T, p.A54V in exon 2) or a heterozygous (c.959G>A, p.R320H in exon 4) missense variant in the TUBB8 gene (Cao et al., 2021). Further heterozygous missense variants (c.1045G>A, p.V349I; c.10A>C, p.I4L; c.938C>T, p.A313V; c.136C>T, p.R46C, or c.893A>G, p.N298S) were identified in infertile patients who produced arrested embryos (Yang et al., 2021b).
A novel heterozygous missense variant (c.524T>C, p.V175A) was defined in an infertile patient who generated zygotes with multiple pronuclei that arrested before first cleavage (Sha et al., 2021). Another heterozygous variant (c.10_12delins CTT, p.I4L) was found in an infertile patient with zygotes possessing multiple pronuclei and arrested embryos at the two- to four-cell stages on Day 3 and before the first cleavage. Also, a c.1045G>A, p.V349I missense variant was found in an infertile woman with three pronuclei zygotes and embryonic arrest at the two- to five-cell or seven-cell stage on Day 3 (Sha et al., 2021). On the other hand, Lu et al. (2021) revealed that an infertile patient with embryonic arrest at the one-cell, two-cell or three-cell stage carried a heterozygous missense variant (c.208C>A, p.P70T) in TUBB8. Other heterozygous missense variants (c.907T>C, p.C303R or c.C916T, p.R306C) were defined in infertile patients who had several embryos that arrested at the cleavage stage. Embryonic arrest at the eight-cell or cleavage stage was observed in the same infertile woman who had a heterozygous missense variant (c.173G>A, p.R58K). Moreover, there was another heterozygous missense variant (c.326G>T, p.G109V) in an infertile patient with embryonic arrest at the cleavage stage (Lu et al., 2021).
Collectively, while several variants (for example, S176L, R214H, A393D, G98R, G98E, I368T, and D128Gfs*27) in the human TUBB8 gene caused loss of spindle appearance, the remaining ones led to aberrant spindle morphology and distribution (Fei and Zhou, 2022). This finding suggests that TUBB8 deficiency reveals itself through disrupting dynamic spindle organization, which results in the arrest of meiotic or mitotic progression. Thus, it seems reasonable to screen these variants in infertile women with spindle defects either in arrested oocytes or early preimplantation embryos. Importantly, as presented in Table 2, a great number of variants were defined in the human TUBB8 gene. These need further functional evaluation to understand their biological effects on early development. Determining the most common deleterious variants can help to create a more reliable diagnostic gene panel for infertile women with embryonic arrest at specific stages.
Main functions of defined genetic variants and knockout phenotypes of the genes playing roles in spindle assembly, chromosome segregation, meiotic recombination, maternal mRNA clearance, and other cellular processes in mammalian oocytes and early embryos.
| Gene | Main functions | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotype (reference) |
|---|---|---|---|---|---|
| TUBB8 |
| c.A628G, c.C713T, or c.A1043G (Missense mutations, Het) | Early embryos | Feng et al. (2016) | NA |
| c.209C>T (Missense, Hom) | Four-cell | Chen et al. (2017a ) | |||
| c.990G>A (Missense, Het) | Early embryos | ||||
| c.1057G>A (Missense, Het) | |||||
| c.613G>A (Missense, Het) | |||||
| c.580G>A and c.1245G>A (Missense, Com Het) | |||||
| Deletion (Hom) | One-cell | ||||
| c.1041C>A (Missense, Hom) | One-cell | Yuan et al. (2018) | |||
| c.10A>C, c.1000C>G, c.1171C>T, c.181C>A, c.1057G>A, c.1249G>T, c.763G>A, c.883G>C, or c.600T>G (Missense mutations, Het) c.721C>T (Missense, Hom) | Early embryos | Chen et al. (2019) | |||
| c.1041C>A (Missense, Het) | Early cleavage | Jia et al. (2020) | |||
| c.1172G>A, c.1139G>A, c.236G>A, c.743C>T, c.940G>T, c.728C>G, c.497C>T, c.736C>G, c.925C>T (Missense mutations, Het) c.1076G>A (Missense, Hom) c.82C>T and c.398T>C (Missense, Com Het) c.400C>T (Nonsense, Het) | Early embryos | Zhao et al. (2020a ) | |||
| c.G6T, c.C43A, c.C124G, c.G262C, c.G421C, c.C544T, c.G586Ac.A604C, c.A694T, c.A898T, c.G904A, c.G959A, c.C1055T, c.C1171T, c.G1172T, c.G1172A, c.C1232A (Missense mutations, Het) | Early embryos | Zheng et al. (2021b) | |||
| c.161C>T (Missense, Hom) | Early embryos | Cao et al. (2021) | |||
| c.959G>A (Missense, Het) | |||||
| c.1045G>A, c.10A>C, c.938C>T, c.136C>T, or c.893A>G (Missense mutations, Het) | Early embryos | Yang et al. (2021b) | |||
| c.524T>C (Missense, Het) | One-cell | Sha et al. (2021) | |||
| c.10_12delinsCTT (Missense, Het) | One-, two- to four-cell | ||||
| c.1045G>A (Missense, Het) | Two- to five-cell and seven-cell | ||||
| c.208C>A (Missense, Het) | One-cell, two-cell, or three-cell | Lu et al. (2021) | |||
| c.173G>A (Missense, Het) | Eight-cell or cleavage | ||||
| c.326T>G (Missense, Het) | Cleavage | ||||
| c.907T>C (Missense, Het) | |||||
| c.916C>T (Missense, Het) | |||||
| CENPH |
| c.635T>C (Missense, Hom) | Cleavage | Maddirevula et al. (2020) | NA |
| CDC20 |
| c.965G>A and c.1176_1179del TCTG (Missense and frameshift deletion, Com Het) | Early embryos | Zhao et al. (2020b) |
|
| c.683A>G (Missense, Hom) | |||||
| c.544C>T and c.965G>A (Nonsense and missense, Com Het) | |||||
| c.964C>T and c.965G>A (Stop-gain and missense, Com Het) | Early embryos | Zhao et al. (2021) | |||
| c.330 + 1G>A and c.1155G>C (Splicing and missense, Com Het) | |||||
| c.784C>T and c.631G>A (Nonsense and missense, Com Het) | Four-cell, five-cell | Huang et al. (2021) | |||
| FBXO43 |
| c.1490_1497dup (Duplication, Hom) | Two-/three- to five-cell | Wang et al. (2021) |
|
| c.1747C>T (Nonsense, Hom) | Two-cell | ||||
| c.154delG (Deletion, Hom) | Three-cell | ||||
| MEI1 |
| c.1792 + 1G>T (Splicing, Hom) | Early embryos | Dong et al. (2021) |
|
| c.1585T>A (Missense, Hom) | |||||
| c.186G>C and c.734-2A>C (Missense and splicing, Com Het) | |||||
| c.745C>T (Nonsense, Hom) | |||||
| REC114 |
| c.397T>G (Missense, Hom) | Two- to six-cell | Wang et al. (2020) |
|
| c.546 + 5G>A (Splicing, Hom) | Early embryos | ||||
| TRIP13 |
| c.592A>G and c.739G>A (Missense mutations, Com Het) | One-cell or before eight-cell | Zhang et al. (2020) |
|
| c.1141G>A (Missense, Hom) | One-cell, three-cell | Hu et al. (2022) | |||
| c.77A>G and c.1258A>G (Missense mutations, Com Het) | One-cell | ||||
| BTG4 |
| c.73C>T (Nonsense, Hom) | One-cell | Zheng et al. (2020b) |
|
| c.1A>G (Start-lost, Hom) | |||||
| c.475_478del (Frameshift, Hom) | |||||
| c.166G>A (Missense, Hom) | |||||
| ZFP36L2 |
| c.922_930del (Non-frameshift deletion, Hom) | Early embryos | Zheng et al. (2022) |
|
| (c.922_930del and c.922T>G) (Non-frameshift deletion and Missense, Com Het) | |||||
| PATL2 |
| c.953T>C and c.839G>A (Missense mutations, Com Het) | Early embryos | Chen et al. (2017b) |
|
| c.649T>A and c.566T>G (Missense mutations, Com Het) | |||||
| c.223-14_223-2del and c.1224þ2T>C (Splicing mutations, Com Het) | |||||
| c.877G>T and c.223-14_223-2del (Missense and splicing, Com Het) | Early embryos | Wu et al. (2019) | |||
| |||||
| c.778G>A and c.223-1_223-2del (Missense and Deletion) | |||||
| c.890delC (Frameshift, Hom) | Two-cell | Huang et al. (2022) | |||
| MOS |
| c.285C>A (Missense, Hom) | Two- to seven-cell | Zhang et al. (2021b) |
|
| c.416T>C and c.737G>A (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.960C>A (Nonsense, Hom) | Two-, three-, or five-cell | ||||
| c.791C>G (Missense, Hom) | Two- to six-cell | Zhang et al. (2022) | |||
| c.875C>T and c.596A>T (Missense mutations, Com Het) | Two- to eight-cell or three- to four-cell | ||||
| c.285C>A (Missense, Hom) | Early embryos | Zeng et al. (2022) | |||
| c.467delG and c.956G>A (Frameshift and missense, Com Het) | Early embryos | ||||
| TBPL2 |
| c.788 + 3A>G (Splicing, Hom) | Early embryos | Yang et al. (2021a ) |
|
| Gene | Main functions | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotype (reference) |
|---|---|---|---|---|---|
| TUBB8 |
| c.A628G, c.C713T, or c.A1043G (Missense mutations, Het) | Early embryos | Feng et al. (2016) | NA |
| c.209C>T (Missense, Hom) | Four-cell | Chen et al. (2017a ) | |||
| c.990G>A (Missense, Het) | Early embryos | ||||
| c.1057G>A (Missense, Het) | |||||
| c.613G>A (Missense, Het) | |||||
| c.580G>A and c.1245G>A (Missense, Com Het) | |||||
| Deletion (Hom) | One-cell | ||||
| c.1041C>A (Missense, Hom) | One-cell | Yuan et al. (2018) | |||
| c.10A>C, c.1000C>G, c.1171C>T, c.181C>A, c.1057G>A, c.1249G>T, c.763G>A, c.883G>C, or c.600T>G (Missense mutations, Het) c.721C>T (Missense, Hom) | Early embryos | Chen et al. (2019) | |||
| c.1041C>A (Missense, Het) | Early cleavage | Jia et al. (2020) | |||
| c.1172G>A, c.1139G>A, c.236G>A, c.743C>T, c.940G>T, c.728C>G, c.497C>T, c.736C>G, c.925C>T (Missense mutations, Het) c.1076G>A (Missense, Hom) c.82C>T and c.398T>C (Missense, Com Het) c.400C>T (Nonsense, Het) | Early embryos | Zhao et al. (2020a ) | |||
| c.G6T, c.C43A, c.C124G, c.G262C, c.G421C, c.C544T, c.G586Ac.A604C, c.A694T, c.A898T, c.G904A, c.G959A, c.C1055T, c.C1171T, c.G1172T, c.G1172A, c.C1232A (Missense mutations, Het) | Early embryos | Zheng et al. (2021b) | |||
| c.161C>T (Missense, Hom) | Early embryos | Cao et al. (2021) | |||
| c.959G>A (Missense, Het) | |||||
| c.1045G>A, c.10A>C, c.938C>T, c.136C>T, or c.893A>G (Missense mutations, Het) | Early embryos | Yang et al. (2021b) | |||
| c.524T>C (Missense, Het) | One-cell | Sha et al. (2021) | |||
| c.10_12delinsCTT (Missense, Het) | One-, two- to four-cell | ||||
| c.1045G>A (Missense, Het) | Two- to five-cell and seven-cell | ||||
| c.208C>A (Missense, Het) | One-cell, two-cell, or three-cell | Lu et al. (2021) | |||
| c.173G>A (Missense, Het) | Eight-cell or cleavage | ||||
| c.326T>G (Missense, Het) | Cleavage | ||||
| c.907T>C (Missense, Het) | |||||
| c.916C>T (Missense, Het) | |||||
| CENPH |
| c.635T>C (Missense, Hom) | Cleavage | Maddirevula et al. (2020) | NA |
| CDC20 |
| c.965G>A and c.1176_1179del TCTG (Missense and frameshift deletion, Com Het) | Early embryos | Zhao et al. (2020b) |
|
| c.683A>G (Missense, Hom) | |||||
| c.544C>T and c.965G>A (Nonsense and missense, Com Het) | |||||
| c.964C>T and c.965G>A (Stop-gain and missense, Com Het) | Early embryos | Zhao et al. (2021) | |||
| c.330 + 1G>A and c.1155G>C (Splicing and missense, Com Het) | |||||
| c.784C>T and c.631G>A (Nonsense and missense, Com Het) | Four-cell, five-cell | Huang et al. (2021) | |||
| FBXO43 |
| c.1490_1497dup (Duplication, Hom) | Two-/three- to five-cell | Wang et al. (2021) |
|
| c.1747C>T (Nonsense, Hom) | Two-cell | ||||
| c.154delG (Deletion, Hom) | Three-cell | ||||
| MEI1 |
| c.1792 + 1G>T (Splicing, Hom) | Early embryos | Dong et al. (2021) |
|
| c.1585T>A (Missense, Hom) | |||||
| c.186G>C and c.734-2A>C (Missense and splicing, Com Het) | |||||
| c.745C>T (Nonsense, Hom) | |||||
| REC114 |
| c.397T>G (Missense, Hom) | Two- to six-cell | Wang et al. (2020) |
|
| c.546 + 5G>A (Splicing, Hom) | Early embryos | ||||
| TRIP13 |
| c.592A>G and c.739G>A (Missense mutations, Com Het) | One-cell or before eight-cell | Zhang et al. (2020) |
|
| c.1141G>A (Missense, Hom) | One-cell, three-cell | Hu et al. (2022) | |||
| c.77A>G and c.1258A>G (Missense mutations, Com Het) | One-cell | ||||
| BTG4 |
| c.73C>T (Nonsense, Hom) | One-cell | Zheng et al. (2020b) |
|
| c.1A>G (Start-lost, Hom) | |||||
| c.475_478del (Frameshift, Hom) | |||||
| c.166G>A (Missense, Hom) | |||||
| ZFP36L2 |
| c.922_930del (Non-frameshift deletion, Hom) | Early embryos | Zheng et al. (2022) |
|
| (c.922_930del and c.922T>G) (Non-frameshift deletion and Missense, Com Het) | |||||
| PATL2 |
| c.953T>C and c.839G>A (Missense mutations, Com Het) | Early embryos | Chen et al. (2017b) |
|
| c.649T>A and c.566T>G (Missense mutations, Com Het) | |||||
| c.223-14_223-2del and c.1224þ2T>C (Splicing mutations, Com Het) | |||||
| c.877G>T and c.223-14_223-2del (Missense and splicing, Com Het) | Early embryos | Wu et al. (2019) | |||
| |||||
| c.778G>A and c.223-1_223-2del (Missense and Deletion) | |||||
| c.890delC (Frameshift, Hom) | Two-cell | Huang et al. (2022) | |||
| MOS |
| c.285C>A (Missense, Hom) | Two- to seven-cell | Zhang et al. (2021b) |
|
| c.416T>C and c.737G>A (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.960C>A (Nonsense, Hom) | Two-, three-, or five-cell | ||||
| c.791C>G (Missense, Hom) | Two- to six-cell | Zhang et al. (2022) | |||
| c.875C>T and c.596A>T (Missense mutations, Com Het) | Two- to eight-cell or three- to four-cell | ||||
| c.285C>A (Missense, Hom) | Early embryos | Zeng et al. (2022) | |||
| c.467delG and c.956G>A (Frameshift and missense, Com Het) | Early embryos | ||||
| TBPL2 |
| c.788 + 3A>G (Splicing, Hom) | Early embryos | Yang et al. (2021a ) |
|
All these variants were found to cause embryonic arrests at early stages in humans. NA, not available. All other abbreviations are given in the text.
Main functions of defined genetic variants and knockout phenotypes of the genes playing roles in spindle assembly, chromosome segregation, meiotic recombination, maternal mRNA clearance, and other cellular processes in mammalian oocytes and early embryos.
| Gene | Main functions | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotype (reference) |
|---|---|---|---|---|---|
| TUBB8 |
| c.A628G, c.C713T, or c.A1043G (Missense mutations, Het) | Early embryos | Feng et al. (2016) | NA |
| c.209C>T (Missense, Hom) | Four-cell | Chen et al. (2017a ) | |||
| c.990G>A (Missense, Het) | Early embryos | ||||
| c.1057G>A (Missense, Het) | |||||
| c.613G>A (Missense, Het) | |||||
| c.580G>A and c.1245G>A (Missense, Com Het) | |||||
| Deletion (Hom) | One-cell | ||||
| c.1041C>A (Missense, Hom) | One-cell | Yuan et al. (2018) | |||
| c.10A>C, c.1000C>G, c.1171C>T, c.181C>A, c.1057G>A, c.1249G>T, c.763G>A, c.883G>C, or c.600T>G (Missense mutations, Het) c.721C>T (Missense, Hom) | Early embryos | Chen et al. (2019) | |||
| c.1041C>A (Missense, Het) | Early cleavage | Jia et al. (2020) | |||
| c.1172G>A, c.1139G>A, c.236G>A, c.743C>T, c.940G>T, c.728C>G, c.497C>T, c.736C>G, c.925C>T (Missense mutations, Het) c.1076G>A (Missense, Hom) c.82C>T and c.398T>C (Missense, Com Het) c.400C>T (Nonsense, Het) | Early embryos | Zhao et al. (2020a ) | |||
| c.G6T, c.C43A, c.C124G, c.G262C, c.G421C, c.C544T, c.G586Ac.A604C, c.A694T, c.A898T, c.G904A, c.G959A, c.C1055T, c.C1171T, c.G1172T, c.G1172A, c.C1232A (Missense mutations, Het) | Early embryos | Zheng et al. (2021b) | |||
| c.161C>T (Missense, Hom) | Early embryos | Cao et al. (2021) | |||
| c.959G>A (Missense, Het) | |||||
| c.1045G>A, c.10A>C, c.938C>T, c.136C>T, or c.893A>G (Missense mutations, Het) | Early embryos | Yang et al. (2021b) | |||
| c.524T>C (Missense, Het) | One-cell | Sha et al. (2021) | |||
| c.10_12delinsCTT (Missense, Het) | One-, two- to four-cell | ||||
| c.1045G>A (Missense, Het) | Two- to five-cell and seven-cell | ||||
| c.208C>A (Missense, Het) | One-cell, two-cell, or three-cell | Lu et al. (2021) | |||
| c.173G>A (Missense, Het) | Eight-cell or cleavage | ||||
| c.326T>G (Missense, Het) | Cleavage | ||||
| c.907T>C (Missense, Het) | |||||
| c.916C>T (Missense, Het) | |||||
| CENPH |
| c.635T>C (Missense, Hom) | Cleavage | Maddirevula et al. (2020) | NA |
| CDC20 |
| c.965G>A and c.1176_1179del TCTG (Missense and frameshift deletion, Com Het) | Early embryos | Zhao et al. (2020b) |
|
| c.683A>G (Missense, Hom) | |||||
| c.544C>T and c.965G>A (Nonsense and missense, Com Het) | |||||
| c.964C>T and c.965G>A (Stop-gain and missense, Com Het) | Early embryos | Zhao et al. (2021) | |||
| c.330 + 1G>A and c.1155G>C (Splicing and missense, Com Het) | |||||
| c.784C>T and c.631G>A (Nonsense and missense, Com Het) | Four-cell, five-cell | Huang et al. (2021) | |||
| FBXO43 |
| c.1490_1497dup (Duplication, Hom) | Two-/three- to five-cell | Wang et al. (2021) |
|
| c.1747C>T (Nonsense, Hom) | Two-cell | ||||
| c.154delG (Deletion, Hom) | Three-cell | ||||
| MEI1 |
| c.1792 + 1G>T (Splicing, Hom) | Early embryos | Dong et al. (2021) |
|
| c.1585T>A (Missense, Hom) | |||||
| c.186G>C and c.734-2A>C (Missense and splicing, Com Het) | |||||
| c.745C>T (Nonsense, Hom) | |||||
| REC114 |
| c.397T>G (Missense, Hom) | Two- to six-cell | Wang et al. (2020) |
|
| c.546 + 5G>A (Splicing, Hom) | Early embryos | ||||
| TRIP13 |
| c.592A>G and c.739G>A (Missense mutations, Com Het) | One-cell or before eight-cell | Zhang et al. (2020) |
|
| c.1141G>A (Missense, Hom) | One-cell, three-cell | Hu et al. (2022) | |||
| c.77A>G and c.1258A>G (Missense mutations, Com Het) | One-cell | ||||
| BTG4 |
| c.73C>T (Nonsense, Hom) | One-cell | Zheng et al. (2020b) |
|
| c.1A>G (Start-lost, Hom) | |||||
| c.475_478del (Frameshift, Hom) | |||||
| c.166G>A (Missense, Hom) | |||||
| ZFP36L2 |
| c.922_930del (Non-frameshift deletion, Hom) | Early embryos | Zheng et al. (2022) |
|
| (c.922_930del and c.922T>G) (Non-frameshift deletion and Missense, Com Het) | |||||
| PATL2 |
| c.953T>C and c.839G>A (Missense mutations, Com Het) | Early embryos | Chen et al. (2017b) |
|
| c.649T>A and c.566T>G (Missense mutations, Com Het) | |||||
| c.223-14_223-2del and c.1224þ2T>C (Splicing mutations, Com Het) | |||||
| c.877G>T and c.223-14_223-2del (Missense and splicing, Com Het) | Early embryos | Wu et al. (2019) | |||
| |||||
| c.778G>A and c.223-1_223-2del (Missense and Deletion) | |||||
| c.890delC (Frameshift, Hom) | Two-cell | Huang et al. (2022) | |||
| MOS |
| c.285C>A (Missense, Hom) | Two- to seven-cell | Zhang et al. (2021b) |
|
| c.416T>C and c.737G>A (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.960C>A (Nonsense, Hom) | Two-, three-, or five-cell | ||||
| c.791C>G (Missense, Hom) | Two- to six-cell | Zhang et al. (2022) | |||
| c.875C>T and c.596A>T (Missense mutations, Com Het) | Two- to eight-cell or three- to four-cell | ||||
| c.285C>A (Missense, Hom) | Early embryos | Zeng et al. (2022) | |||
| c.467delG and c.956G>A (Frameshift and missense, Com Het) | Early embryos | ||||
| TBPL2 |
| c.788 + 3A>G (Splicing, Hom) | Early embryos | Yang et al. (2021a ) |
|
| Gene | Main functions | Variant (type, zygosity) | Embryonic arrest stage | Reference | Knockout phenotype (reference) |
|---|---|---|---|---|---|
| TUBB8 |
| c.A628G, c.C713T, or c.A1043G (Missense mutations, Het) | Early embryos | Feng et al. (2016) | NA |
| c.209C>T (Missense, Hom) | Four-cell | Chen et al. (2017a ) | |||
| c.990G>A (Missense, Het) | Early embryos | ||||
| c.1057G>A (Missense, Het) | |||||
| c.613G>A (Missense, Het) | |||||
| c.580G>A and c.1245G>A (Missense, Com Het) | |||||
| Deletion (Hom) | One-cell | ||||
| c.1041C>A (Missense, Hom) | One-cell | Yuan et al. (2018) | |||
| c.10A>C, c.1000C>G, c.1171C>T, c.181C>A, c.1057G>A, c.1249G>T, c.763G>A, c.883G>C, or c.600T>G (Missense mutations, Het) c.721C>T (Missense, Hom) | Early embryos | Chen et al. (2019) | |||
| c.1041C>A (Missense, Het) | Early cleavage | Jia et al. (2020) | |||
| c.1172G>A, c.1139G>A, c.236G>A, c.743C>T, c.940G>T, c.728C>G, c.497C>T, c.736C>G, c.925C>T (Missense mutations, Het) c.1076G>A (Missense, Hom) c.82C>T and c.398T>C (Missense, Com Het) c.400C>T (Nonsense, Het) | Early embryos | Zhao et al. (2020a ) | |||
| c.G6T, c.C43A, c.C124G, c.G262C, c.G421C, c.C544T, c.G586Ac.A604C, c.A694T, c.A898T, c.G904A, c.G959A, c.C1055T, c.C1171T, c.G1172T, c.G1172A, c.C1232A (Missense mutations, Het) | Early embryos | Zheng et al. (2021b) | |||
| c.161C>T (Missense, Hom) | Early embryos | Cao et al. (2021) | |||
| c.959G>A (Missense, Het) | |||||
| c.1045G>A, c.10A>C, c.938C>T, c.136C>T, or c.893A>G (Missense mutations, Het) | Early embryos | Yang et al. (2021b) | |||
| c.524T>C (Missense, Het) | One-cell | Sha et al. (2021) | |||
| c.10_12delinsCTT (Missense, Het) | One-, two- to four-cell | ||||
| c.1045G>A (Missense, Het) | Two- to five-cell and seven-cell | ||||
| c.208C>A (Missense, Het) | One-cell, two-cell, or three-cell | Lu et al. (2021) | |||
| c.173G>A (Missense, Het) | Eight-cell or cleavage | ||||
| c.326T>G (Missense, Het) | Cleavage | ||||
| c.907T>C (Missense, Het) | |||||
| c.916C>T (Missense, Het) | |||||
| CENPH |
| c.635T>C (Missense, Hom) | Cleavage | Maddirevula et al. (2020) | NA |
| CDC20 |
| c.965G>A and c.1176_1179del TCTG (Missense and frameshift deletion, Com Het) | Early embryos | Zhao et al. (2020b) |
|
| c.683A>G (Missense, Hom) | |||||
| c.544C>T and c.965G>A (Nonsense and missense, Com Het) | |||||
| c.964C>T and c.965G>A (Stop-gain and missense, Com Het) | Early embryos | Zhao et al. (2021) | |||
| c.330 + 1G>A and c.1155G>C (Splicing and missense, Com Het) | |||||
| c.784C>T and c.631G>A (Nonsense and missense, Com Het) | Four-cell, five-cell | Huang et al. (2021) | |||
| FBXO43 |
| c.1490_1497dup (Duplication, Hom) | Two-/three- to five-cell | Wang et al. (2021) |
|
| c.1747C>T (Nonsense, Hom) | Two-cell | ||||
| c.154delG (Deletion, Hom) | Three-cell | ||||
| MEI1 |
| c.1792 + 1G>T (Splicing, Hom) | Early embryos | Dong et al. (2021) |
|
| c.1585T>A (Missense, Hom) | |||||
| c.186G>C and c.734-2A>C (Missense and splicing, Com Het) | |||||
| c.745C>T (Nonsense, Hom) | |||||
| REC114 |
| c.397T>G (Missense, Hom) | Two- to six-cell | Wang et al. (2020) |
|
| c.546 + 5G>A (Splicing, Hom) | Early embryos | ||||
| TRIP13 |
| c.592A>G and c.739G>A (Missense mutations, Com Het) | One-cell or before eight-cell | Zhang et al. (2020) |
|
| c.1141G>A (Missense, Hom) | One-cell, three-cell | Hu et al. (2022) | |||
| c.77A>G and c.1258A>G (Missense mutations, Com Het) | One-cell | ||||
| BTG4 |
| c.73C>T (Nonsense, Hom) | One-cell | Zheng et al. (2020b) |
|
| c.1A>G (Start-lost, Hom) | |||||
| c.475_478del (Frameshift, Hom) | |||||
| c.166G>A (Missense, Hom) | |||||
| ZFP36L2 |
| c.922_930del (Non-frameshift deletion, Hom) | Early embryos | Zheng et al. (2022) |
|
| (c.922_930del and c.922T>G) (Non-frameshift deletion and Missense, Com Het) | |||||
| PATL2 |
| c.953T>C and c.839G>A (Missense mutations, Com Het) | Early embryos | Chen et al. (2017b) |
|
| c.649T>A and c.566T>G (Missense mutations, Com Het) | |||||
| c.223-14_223-2del and c.1224þ2T>C (Splicing mutations, Com Het) | |||||
| c.877G>T and c.223-14_223-2del (Missense and splicing, Com Het) | Early embryos | Wu et al. (2019) | |||
| |||||
| c.778G>A and c.223-1_223-2del (Missense and Deletion) | |||||
| c.890delC (Frameshift, Hom) | Two-cell | Huang et al. (2022) | |||
| MOS |
| c.285C>A (Missense, Hom) | Two- to seven-cell | Zhang et al. (2021b) |
|
| c.416T>C and c.737G>A (Missense mutations, Com Het) | Two- to six-cell | ||||
| c.960C>A (Nonsense, Hom) | Two-, three-, or five-cell | ||||
| c.791C>G (Missense, Hom) | Two- to six-cell | Zhang et al. (2022) | |||
| c.875C>T and c.596A>T (Missense mutations, Com Het) | Two- to eight-cell or three- to four-cell | ||||
| c.285C>A (Missense, Hom) | Early embryos | Zeng et al. (2022) | |||
| c.467delG and c.956G>A (Frameshift and missense, Com Het) | Early embryos | ||||
| TBPL2 |
| c.788 + 3A>G (Splicing, Hom) | Early embryos | Yang et al. (2021a ) |
|
All these variants were found to cause embryonic arrests at early stages in humans. NA, not available. All other abbreviations are given in the text.
Centromere protein H
The CENPH protein functions in organizing the kinetochore throughout the cell cycle (Alonso et al., 2007). Although no mouse model exists for Cenph knockout, its mutation in infertile women was examined in one study. In an affected individual whose embryos mostly showed arrest at different cleavage stages, a homozygous missense variant (c.635T>C, p.I212T) was defined (Maddirevula et al., 2020). As kinetochores play key roles in chromosome segregation during meiotic and mitotic divisions (Potapova and Gorbsky, 2017), further variants are likely to be found in infertile patients with embryonic arrest due especially to various types of aneuploidy.
Cell division cycle 20
The CDC20 protein is highly expressed in oocytes and serves as a coactivator of anaphase promoting complex/cyclosome (APC/C) during mitosis, and modulates spindle assembly checkpoints (Musacchio and Hardwick, 2002). CDC20 also contributes to the suppression of apoptosis by promoting BIM (also known as BCL2L11 (BCL2 like 11)) degradation (Wan et al., 2014). Loss of CDC20 in mice led to embryogenesis failure, arrest at the metaphase stage of two-cell embryos, and increasing cyclin B1 levels in the arrested embryos (Li et al., 2007). Accordingly, Cdc20-null mouse embryonic and adult cells displayed a permanent mitotic arrest until the occurrence of cellular death (Manchado et al., 2010). Jin et al. (2010) further created a hypomorphic female mouse model for Cdc20, which exhibited infertility or subfertility, failure in the first few embryonic divisions, production of aneuploid oocytes and embryos, prolonged MI timing, inability to develop into blastocysts, disruption in degradation of mitotic cyclins and securin in MI oocytes. These investigations indicate that CDC20 plays crucial roles in oocyte maturation and early embryogenesis through modulating APC/C and spindle assembly processes.
In mutation screening, an infertile woman with embryos arrested at early stages possessed compound heterozygous variants (missense c.965G>A, p.R322Q and frameshift deletion c.1176_1179delTCTG, p.C392*) in the CDC20 gene (Zhao et al., 2020b). Another woman with early embryonic arrest had a homozygous missense mutation (c.683A>G, p.Y228C). The compound heterozygous variants, including nonsense c.544C>T, p.R182* and missense c.965G>A, p.T322Q were discovered in an affected individual with early embryonic arrest (Zhao et al., 2020b). A more recent study revealed that compound heterozygous CDC20 mutations (stop-gain c.964C>T, p.R322* and missense c.965G>A, p.R322Q) were defined in an infertile patient who produced embryos arrested at early stages (Zhao et al., 2021). Another affected woman with early embryo arrest carried compound heterozygous variants (splicing c.330 + 1G>A, p.E111Ifs*36 and missense c.1155G>C, p.W385C). A patient with early embryonic arrest at the four- to five-cell stage on Day 3 had compound heterozygous variants (nonsense c.784C>T, p.R262* and missense c.631G>A, p.A211T) in CDC20 (Huang et al., 2021). Accompanying the functions of CDC20 in meiotic and mitotic progression and emerging phenotypes when knocked out in mice, embryonic arrest at distinct stages was identified in infertile patients. Thus, these variants can be included to a specific diagnostic gene panel designed for infertile women with early preimplantation embryo arrests owing to defects in spindles and cell divisions.
F-box protein 43
F-box protein 43 (FBXO43) (also known as EMI2, and XErp1) is implicated in inhibiting APC/C complex and cell cycle regulation in early mouse embryos and MII oocytes as a component of cytostatic factor (Madgwick et al., 2006). In other words, FBXO43 modulates meiotic and mitotic progression as a regulator of cell-cycle machinery. Its encoding gene expression is restricted to ovaries and testes in mice (Shoji et al., 2006). A more detailed study in Xenopus oocytes revealed that FBXO43 inhibits APC/C ubiquitin ligase activity (Isoda et al., 2011). At fertilization, the calmodulin-dependent protein kinase II (CAMKII alpha (CAMK2A)), a downstream target of Ca2+ oscillations, phosphorylates FBXO43, and WEE1B so that the M-phase promoting factor (MPF) is destroyed. This results in triggering meiotic resumption (Sanders and Swann, 2016). Possibly while undergoing dynamic phosphorylation, FBXO43 functions in regulating APC/C activity, maintaining oocyte arrest, and modulating early embryo development (Tischer et al., 2012).
Knockout of Fbxo43 caused sterility in both mouse sexes possibly due to defective meiosis, but no abnormal changes were noted in embryonic development and mitotic cell division (Gopinathan et al., 2017). Additionally, there was defective morphology in MII oocytes, and some oocytes exhibited parthenogenetic division. In instances of knockdown of Fbxo43 by using 5’ UTR siRNA in mouse oocytes, parthenogenetic activation, and pronucleus formation were observed (Wang et al., 2021). Both studies revealed that FBXO43 contributes to regulating meiotic and mitotic divisions during oocyte maturation and early embryonic development, respectively.
In mutation screening on the FBXO43 gene, the homozygous variants, c.1490_1497dup, p.E500Sfs*2; c.1747C>T, p.Q583*, or c.154delG, p.D52Tfs*30, were defined in three infertile women, from three independent families, with early embryonic arrest at two-/three- to five-cell, two-cell, or three-cell stages on Day 3, respectively (Wang et al., 2021). Further analysis revealed that all these variants not only decreased FBXO43 and its downstream target Cyclin B1 levels in HEK293T cells, but also reduced FBXO43’s ability to rescue parthenogenetic activation (Wang et al., 2021). The emergence of different embryonic arrest types dependent on various variants in FBXO43 (Table 2) may originate from a decreased level and/or loss of specific domains such as the zinc-binding region (ZBR) fragment that allows the performance of basic functions, including inhibition of APC/C complex and cell cycle regulation (Shoji et al., 2014).
Meiotic recombination
Successful meiotic recombination that necessitates physiologically formed DSBs is essential to produce competent embryos. There are many proteins implicated in this event. One of these proteins is the vertebrate-specific meiotic double-strand break formation protein 1 (MEI1), which is known as a chromosome synapsis-related protein. It participates in forming genetically programmed DSBs and thereby, in initiating meiotic recombination (Reinholdt and Schimenti, 2005). The Mei1 gene is exclusively expressed in mouse gonads, where it helps to form normal meiotic chromosome synapsis (Libby et al., 2003). Expectedly, Mei1 mutant female mice were found to be infertile and unable to produce litters. They also had certain ovarian defects such as small sizes, disorganized structure, absence of normal follicles, and reduced follicle numbers (Libby et al., 2002). Aberrant chromosomal arrangement was likewise discovered in the oocytes, most of which were arrested at the MI stage. These findings suggest that MEI1 not only plays a role in meiotic recombination, which occurs largely at prenatal periods in oocytes, but also participates in regulating chromosome segregation during meiosis.
To the best of my knowledge, there is only a limited number of studies analyzing potential variants in the human MEI1 gene. In infertile women with arrest at early embryonic stages, novel biallelic variants (splicing c.1792 + 1G>T; missense c.1585T>A, p.F529I; compound heterozygous missense c.186G>C, p.K62N and splicing c.734-2A>C; protein truncating nonsense c.745C>T, p.Q249*) were discovered (Dong et al., 2021). Some MEI1 mutations were also associated with azoospermia due to meiotic arrest (Ben Khelifa et al., 2018) and recurrent implantation failure (Dong et al., 2021). Definition of only a few variants in MEI1 gene in infertile women with embryonic arrest prevents researchers from creating a diagnostic gene panel for this gene. Thus, further evaluation must be performed in infertile patients owing to the defects in physiological DSB formation, chromosome synapsis, and meiotic recombination events.
Meiotic recombination-related protein REC114
REC114 contributes to forming DSBs, which are essential to initiate homologous chromosome recombination. The evolutionarily conserved REC114 protein resides on the axis of meiotic chromosomes and creates a complex with MEI4 and interactor of HORMAD1 1 (IHO1) to promote DSB formation (Kumar et al., 2018). Deficiency of REC114 in mice resulted in sterility in both sexes. In both spermatocytes and oocytes, there was an impairment in the formation of meiotic DSBs. As a result, DSB and DMC1 foci levels were dramatically reduced (Kumar et al., 2018). As has been demonstrated, REC114 is a critical protein in comprising meiotic recombination in oocytes, which is also a determinant of successful early embryonic development.
When screening mutations in the REC114 gene, a woman with primary infertility exhibited developmental arrest at the two- to six-cell stages on Day 3, and had a homozygous missense mutation (c.397T>G, p.C133G in an autosomal recessive manner) (Wang et al., 2020). This mutation reduced REC114 levels and disrupted its interaction with the MEI4 protein. To define new variants in REC114, further screening should be performed in infertile patients with recombination-related defects, and even in patients from different ethnicities due to possibility of genetic differences among humans (Mountain and Risch, 2004; Huang et al., 2015). These different genetic backgrounds may reveal distinct phenotypes despite having the same specific mutations.
Thyroid hormone receptor interaction protein 13
As with REC114, thyroid hormone receptor interaction protein 13 (TRIP13) participates in dissociating mitotic checkpoint complexes (Wang et al., 2014) and meiotic recombination through the formation of DSBs (Li and Schimenti, 2007). For this purpose, TRIP13 uses own AAA–ATPase activity, which is essential for modulating the HORMA proteins (implicated in both meiotic checkpoint signaling and meiotic recombination) and regulating DNA repair process (Li and Schimenti, 2007). In other words, TRIP13 removes HORMAD1 and 2 from synapsed chromosome axes in oocyte meiosis (Biswas et al., 2021). Although TRIP13 is unable to discern synapsis, it contributes to detecting unrepaired DNA damage sites (Li and Schimenti, 2007). As a result, TRIP13 functions in cell division processes such as spindle assembly checkpoint and DNA repair (Capalbo et al., 2022). Its loss in female mice resulted in infertility, severe impairment in ovarian reserve due to oocyte loss around the time of birth, severely decreased follicle numbers, and meiotic defects in the oocytes (Li and Schimenti, 2007). These phenotypes most likely arise from loss of the actions of TRIP13 in DSB formation for meiotic recombination, DNA repair, and spindle assembly organization.
In an infertile woman whose embryos arrested at the one-cell or before the eight-cell stage, there were compound heterozygous missense mutations (c.592A>G, p.I198V and c.739G>A, p.V247M) in the TRIP13 gene (Zhang et al., 2020). Based on in vitro and in vivo experiments, the following variants in TRIP13: p.H26R, p.E303K, and p.V247M were found to reduce either TRIP13 or deposition of the downstream target HORMAD2 levels, which impaired DNA repair efficiency (Zhang et al., 2020). In a more recent study, a homozygous missense mutation (c.1141G>A, p.E381K) was discovered in infertile patients with embryonic arrest at the one-cell or three-cell stage (Hu et al., 2022). Another infertile patient with embryonic arrest at the one-cell stage carried compound heterozygous missense variants (c.77A>G, p.H26R and c.1258A>G, p.K420E) (Hu et al., 2022). Taken together, different variants in the TRIP13 gene led to embryonic arrest at early preimplantation stages. This issue indicates that these variants may cause loss of the functions related to meiotic recombination, DNA repair, and spindle assembly checkpoint.
The discussions in this article of the defined variants and functional importance of several meiotic recombination-related genes have been based on a limited number of studies (Table 2). As meiotic recombination is a central process that contributes to producing competent embryos, further analyses should be performed especially on the other recombination-linked genes such as SPO11, DMC1, RAD51, and MEIOB (Hunter, 2015). After defining all the possible variants in these genes, a diagnostic gene panel may be created for infertile women with embryonic arrests due to meiotic recombination defects in oocytes and the resulting early embryos.
Maternal mRNA clearance
Defects in the clearance of maternal mRNAs through maternal- and zygotic-decay pathways often result in embryonic arrest at the zygote or eight-cell embryo stage in humans (Sha et al., 2020).
B-cell translocation gene 4
The B-cell translocation gene 4 (BTG4) protein acts as an adaptor protein for the CCR4-NOT deadenylase complex to regulate maternal mRNA decay (Yu et al., 2016). BTG4 also contributes to inhibiting spontaneous oocyte activation by modulating deadenylation of maternal mRNAs (Pasternak et al., 2016). Thus, BTG4 is accepted as a licensing factor for maternal-to-zygotic transition and functions in maternal mRNA decay during early embryogenesis (Liu et al., 2016; Yu et al., 2016). Deficiency of BTG4 in female mice led to infertility due to embryonic arrest at the one-cell or two-cell stage, but males were fertile (Yu et al., 2016). Decreased primordial and secondary follicle numbers, impaired homologous synapsis and DSB formation (Kumar et al., 2018) as well as aberrant EGA, are among the additional phenotypes that disrupted maternal mRNA clearance and translational activation failure in the oocytes (Yu et al., 2016; Brachova et al., 2021). As has been demonstrated, BTG4 takes part in various cellular events during folliculogenesis and early development. Molecular details for introducing the roles of BTG4 in each event merit further investigation.
An analysis of the literature reveals that only one study has examined potential variants in the human BTG4 gene. In infertile Chinese women having zygotic arrest due to cleavage failure, the homozygous nonsense (c.73C>T, p.Q25Ter in the BTG domain), start-lost (c.1A>G, p.? in the BTG domain), and frameshift (c.475_478del, p.I159LfsTer15 in the C-terminal domain) variants were defined. They led to complete absence of the full-length form (Zheng et al., 2020b). While no BTG4 protein was detected in HeLa cells transfected with the plasmids that carried the c.73C>T or c.1A>G variant, the c.475_478del variant caused generation of truncated BTG4 protein by using the same experiment model (Zheng et al., 2020b). In another affected individual with embryonic arrest before the first cleavage stage, a homozygous missense variant (c.166G>A, p.A56T in the BTG domain) was identified. Although this variant did not influence temporal distribution and localization of the BTG4 protein, it abolished the interaction between BTG4 and the deadenylase CNOT7. Notably, all these variants showed a Mendelian recessive inheritance pattern (Zheng et al., 2020b). These findings suggest that the BTG domain-encoding region of the human BTG4 gene seems to be more prone to genetic changes. This merits further investigation to characterize the key role of this domain in maternal mRNA regulation.
Zinc finger protein 36-like 2
Zinc finger protein 36-like 2 (ZFP36L2) is an RNA-binding protein that functions in decaying maternal mRNAs through an AU-rich element-dependent way in mice (Sha et al., 2019). Conditional knockout of Zfp36l2 in mouse oocytes caused prevention of global transcriptional silencing, defects in both oocyte maturation and fertilization, and complete infertility in females (Chousal et al., 2018). Mouse models lacking the 29 N-terminal amino acid residues of ZFP36L2 exhibited infertility, blocked ovulation, and embryonic arrest at the two-cell stage (Ramos, 2012; Ball et al., 2014) possibly due to impairment in timely clearance of maternal mRNAs.
In infertile women with recurrent preimplantation embryo arrests, a homozygous non-frameshift deletion (c.922_930del, p.S308_S310del) or compound heterozygous variants (non-frameshift deletion c.922_930del, p.S308_S310del and missense c.922T>G, p.S308A) were discovered (Zheng et al., 2022). As expected, these variants repressed maternal mRNA decay in human zygotes, and in HeLa cells as well. Further variants should be screened in infertile women with early embryonic arrest, especially at the two-cell stage, to determine precise relationship between ZFP36L2 variants and embryonic arrest development. As many genes play key roles in the process of maternal mRNA decay, closely related-genes such as CCR4, ERK1/2, YTHDF, PABPC1, PARN, and CNOT6L that contribute to regulating maternal mRNA clearance (Sha et al., 2019) can also be screened in infertile patients with impaired maternal mRNA decay.
Other cellular processes
Translational control during oocyte and embryo development is an essential process for providing required proteins in a time-dependent manner. As transcriptional activity usually ceases before the resumption of meiosis in GV oocytes, translational regulation of maternal mRNAs at the later part of oocyte maturation and early embryogenesis contributes to generating competent oocytes and embryos.
PAT1 homolog 2 (PATL2)
PAT1 homolog 2 (PATL2) is an RNA-binding protein that plays key roles in translation control in cooperation with its partner cytoplasmic polyadenylation element-binding protein 1 (CPEB1) in oocytes (Christou-Kent et al., 2018). In other words, PATL2 inhibits premature translational activation by establishing a complex with CPEB1 (Biswas et al., 2021). Lack of Patl2 in mice resulted in female infertility, aberrant oocyte maturation owing to meiotic deficiency, decreased translational repression, and an increased number of MI-arrested oocytes, and it reduced both litter size and oocyte quality (Christou-Kent et al., 2018). Additionally, there were morphological and developmental defects in the oocytes and zygotes as well as dysregulated expression of the genes that function in oocyte maturation and early embryo development. All these phenotypes emerging upon loss of PATL2 suggest that this protein plays a role in various cellular events from meiotic progression to early embryonic development, possibly until EGA.
Upon mutation analysis in PATL2 gene in women suffering from primary infertility, compound heterozygous missense variants (c.953T>C, p.I318T and c.839G>A, p.R280Q) were observed in an affected individual with embryonic arrest at early stages (Chen et al., 2017b). Another infertile woman, one of whose two embryos underwent early embryonic arrest, had compound heterozygous missense variants (c.649T>A, p.Y217N and c.566T>G, p.L189R). Further, an infertile patient whose embryos mostly arrested at early stages carried compound heterozygous splicing variants (c.223-14_223-2delCCCTCCTGTTCCA, p.R75Vfs*21 and c.1224 + 2T>C, p.Q411Wfs*3) (Chen et al., 2017b).
A recent study by Wu et al. (2019) reported that there were compound heterozygous variants (missense c.877G>T, p.D293Y and splicing c.223-14_223-2del, p.R75Vfs*21) in the PATL2 gene in an affected patient with several embryos that exhibited early developmental arrest. Further compound heterozygous variants (frameshift deletion c.716delA, p.N239fs*9 and splicing c.223-14_223-2del, p.R75Vfs*21) were discovered in an infertile woman with early embryonic arrest. In the same study, an affected individual whose embryos all underwent early developmental arrest was found to have compound heterozygous variants (missense c.778G>A, p.V260M and splicing c.223-1_223-2del, p.R75Vfs*21) (Wu et al., 2019). Huang et al. (2022) recently defined a novel homozygous frameshift variant (c.890delC, p.A297Efs*20) in the PATL2 gene of an infertile woman who produced embryos arrested at the two-cell stage. As stated above, various variants were detected in the human PATL2 gene, which contribute to forming different embryonic arrest types (Table 2). This issue may derive from different impacts of these variants on the functional domains of the PATL2 protein, such as the conserved PAT1 domain. Also, possible changes in other molecular biological events such as cell cycle regulation might contribute to different arrest types.
As translational control is a critical step in ensuring proper oocyte maturation and early embryogenesis, a number of genes take part in these processes. In addition to PATL2, the remaining translation-linked genes such as EPAB, PABPC1, eIF4G, eIF4E, PAP, CPEB1, etc. (Ozturk and Uysal, 2017; Ozturk, 2019) should also undergo mutation screening to create a large-scale diagnostic gene panel for translation-related genes.
Moloney sarcoma oncogene
The Moloney sarcoma oncogene (MOS) protein acts as a serine/threonine kinase to activate the ERK signaling cascade through phosphorylating mitogen-activated protein kinase (MEK, also known as MAPK) during oocyte maturation (Verlhac et al., 2000). The MOS–ERK pathway functions in cytoskeleton assembly, mRNA clearance, mitochondrial homeostasis (Wang and Sang, 2021), and up-regulation of MPF activity at the late stages of oocyte maturation (Heikinheimo and Gibbons, 1998). The Mos gene is expressed in vertebrate oocytes at high levels and serves as a cytostatic factor during MII arrest. Its levels decrease after fertilization (Watanabe et al., 1991; Verlhac et al., 2000). In the case of MOS deficiency in mice, although males were fertile, the females displayed sterility, failure of meiotic arrest, ovarian cysts and teratomas, spontaneous parthenogenetic activation of unfertilized oocytes, inability of MAPK activation, spindle defects, and disrupted asymmetric division in oocytes (Colledge et al., 1994; Hashimoto et al., 1994; Choi et al., 1996). As MOS does not show cytostatic factor activity in early mouse embryos (Kashima et al., 2007), these phenotypes seem to arise from the functional loss of MOS in the oocytes during oogenesis.
An infertile patient who produced embryos mostly arrested from the two- to seven-cell stages carried a homozygous missense mutation (c.285C>A, p.N95K) in the MOS gene (Zhang et al., 2021b). Another infertile individual with arrested embryos from the two- to six-cell stages had missense compound heterozygous variants (c.416T>C, p.M139T and c.737G>A, p.R246H). The homozygous nonsense variant (c.960C>A, p.C320Ter) was also discovered in an infertile woman with embryonic arrest at the two-cell, three-cell, or five-cell stage (Zhang et al., 2021b). In the same study, a series of in vitro experiments demonstrated that these MOS variants caused inactivation of ERK in both HEK293 cells and mouse oocytes. Indeed, the phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) level in mouse oocytes could not be reversed due to MOS insufficiency.
A recent study also reported that the MOS gene included a homozygous missense variant (c.791C>G, p.S264C) in an infertile woman with embryonic arrest at the two- to six-cell stages, and compound heterozygous missense variants (c.875C>T, p.A229V and c.596A>T, p.H199L) in infertile patients having embryos arrested at the two- to eight-cell or three- to four-cell stages (Zhang et al., 2022). In addition, Zeng et al. (2022) discovered a homozygous missense variant (c.285C>A, p.A95K) in MOS in an infertile patient with embryonic arrest at an early stage. The compound heterozygous mutations (frameshift deletion c.467delG, p.G156Afs*18 and missense c.956G>A, p.A319H) were identified in an infertile woman who generated arrested early embryos (Zeng et al., 2022). These variants led to reduced MOS levels and its inability to phosphorylate ERK1/2 in HEK293T cells (Zeng et al., 2022). Additionally, there were impairments in both MII exit capacity and the formation of F-actin cytoskeleton structures in mouse oocytes. As a result, all the variants in the human MOS gene, which are associated with embryonic arrest at different stages, can be used in creating a diagnostic gene panel after elucidating the functional reflection(s) of each variant.
TATA-box-binding protein like 2
As a TATA-binding protein (TBP) homolog, TATA-box-binding protein like 2 (TBPL2) is an oocyte-specific transcription factor and is only expressed in vertebrate oocytes (Gazdag et al., 2007). In other words, TBPL2 mediates transcriptional initiation by way of RNA polymerase II action (Fei and Zhou, 2022), and interacts with TATA-box and the transcription factors TFIIA, TFIIB, and other factors (Mishal and Luna-Arias, 2022). The depletion of transcription factor IIb (TfIIb) in mouse oocytes notably resulted in early embryonic arrest at the two-cell stage in more than 85% of embryos (Liu et al., 2013). The embryonic arrest at a high rate may be caused by an impaired transcriptional process because TFIIB, as a transcriptional activator, contributes to pre-initiation transcription complex formation (Greber and Nogales, 2019). The Tbpl2 null female mice exhibited sterility due to defective folliculogenesis (i.e. no production of fully grown GV oocytes), changed chromatin organization, and disrupted transcriptional activity of key oocyte-specific genes (Gazdag et al., 2009). These defective changes indicate that TBPL2 plays roles in modulating transcription during oocyte development.
An analysis of the literature revealed only one study that found an infertile woman with early embryonic arrest who had a homozygous splicing variant (c.788 + 3A>G in intron 4, p.R233X) in the TBPL2 gene (Yang et al., 2021a). This variant caused impairment in the integrity of TBPL2 mRNA. Thus, further variants should be examined in infertile patients with embryonic arrest due to transcription-related failures.
So far, significant findings have been presented on a number of variants in the genes playing roles in different cellular events such as mitochondrial functions, transcriptional, and translational control (Table 2 and Fig. 2). In some genes, only a few gene variants have been defined in infertile women with embryonic arrest at distinct stages. After making comprehensive mutation screening on these genes and determining functional reflection of the ones that are defined, a diagnostic gene panel may be created for each cellular event. Following identification of genetic variants in infertile patients, potential arrest-preventing strategies can be utilized for producing competent embryos that would be capable of creating a healthy pregnancy.
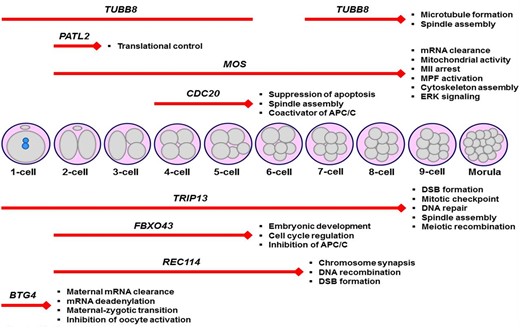
Schematic representation of the embryonic arrest stages in infertile women carrying various variants in the genes. These genes play crucial roles in a wide range of cellular events, such as translational control, meiosis, spindle organization, and cell cycle regulation, in mammalian oocytes and early embryos. It is noteworthy that the genetic variants (Table 2) that lead to arresting embryos at specific stages were only shown herein. Additionally, the main functions of the corresponding genes are given at the end of each red line. The length of the red lines covers the embryonic arrest stages. DSB, DNA double-strand break; MII, metaphase II; MPF, M-phase promoting factor.
Possible future treatment options for overcoming embryonic arrest
Although the molecular determinants of developmental arrest in early embryos are not fully elucidated yet, there are a few potential treatments for overcoming embryonic arrests. It has been demonstrated that decreasing P66Shc (66 kDa isoform of Src homology 2 domain-containing (Shc)) levels in bovine embryos by using knockdown technology resulted in a remarkable reduction of the rates of permanent embryo arrests at the two- to four-cell stages (Betts et al., 2014). The positive effect of this treatment may originate from decreasing intracellular ROS levels, DNA damage, and apoptosis in those embryos (Betts et al., 2014). Expression levels and functional features of P66Shc merit further analysis in human embryos to evaluate their potentiality in preventing arrested embryos.
Another option is to activate arrested zygotes with a calcium Ionophore, which has resulted in increased clinical pregnancy and live birth rates in humans (Xi et al., 2020; Shan et al., 2021). This treatment also contributed to higher fertilization, blastocyst formation, and implantation rates, with no significant changes in high-quality embryo proportion, cleavage rates, congenital birth defects, and neonatal sex ratios. This chemical molecule most likely induces intracellular calcium-dependent signaling cascades to provide cell cycle progression. An interesting study by Liu et al. (2011) revealed that microinjection of recombinant BCL-XL (recBCL-XL) protein with an automated robotic system into mouse zygotes prevented cell cycle arrest of embryos arising from suboptimal culture conditions. It was observed that recBCL-XL was capable of inhibiting ROS accumulation and promoting repair of mitochondrial bioenergetics (Liu et al., 2011; Grindler and Moley, 2013). It is unknown whether this kind of intervention may increase abnormal embryo rates in a short or a long period of developmental progression because blocking the normal apoptotic process is required for removing unhealthy cells. All these interventions can be implemented only at certain development periods and for specific proteins that play roles in a narrow window of early development. Also, it seems to be reasonable to perform these applications in one-cell embryos since there are more embryonic cells in the later embryonic stages. For this purpose, further studies are required to optimize these interventions and understand their long-term effects in mammalian model organisms (such as mice) before attempting them in human embryos.
An experimental study designed to prevent developmental arrest in mouse embryos used a replacement of the cytoplasms of compromised oocytes with normal ones or nuclear transfer. However, cytoplasmic transfer does not provide significant improvement in reproductively aged human oocytes while nuclear transfer does (Tang et al., 2020). For this issue, nuclear transfer to enucleated young oocytes (Tanaka and Watanabe, 2019) might become an alternative approach since women have chosen to delay childbearing in recent decades (Martin et al., 2018). Thus, further studies are essential for understanding the short- and long-term effects of nuclear transfer in the resulting embryos and even in healthy pregnancy rates.
A limited number of studies on a few mammalian species have examined potential ways of preventing embryonic arrests. After performing more investigations on the short- and long-term outcomes of using these embryos, similar strategies can be evaluated in human discarded embryos. As is known, some women are able only to generate arrested embryos. Therefore, inducing embryonic developmental progression may help these patients to conceive a healthy baby. The important point is that if there may be defined mutations in infertile patients, inducing embryonic progression would not be a reasonable approach. In these patients, genetic based interventions for these embryos should be considered. However, there are strict regulations and limitations for using genetic editing in human embryos for reproductive purposes (Lovell-Badge et al., 2021).
Conclusion
Numerous genetic variants in the genes playing key roles in SCMC, mitotic division, maternal mRNA clearance, DNA repair, transcriptional and translational processes have been characterized in infertile women with developmental arrest in preimplantation embryos (Figs 1 and 2). Further analyses would help to identify more variants in the related genes when screened in a large group of infertile women. It is important that the functional reflection of each variant in human embryos should also be evaluated. After that, creating a diagnostic gene panel for each cellular defect may help to rapidly determine the genetic background of women producing arrested embryos. Thus, highly sophisticated technologies such as CRISPR-Cas9 can be used in the context of ethical permissions to treat these variants in order to produce normal embryos.
Data availability
No new data were generated or analyzed in support of this article.
Acknowledgements
The author thanks Robert Glen (PhD) for helpful comments and corrections on the article.
Authors’ roles
This review was researched, written, and revised by S.O. only.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Conflict of interest
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.

{kind=link}
{kind=link}