






Abstract
Ataxia-telangiectasia (AT) and MRE11-defective Ataxia-telangiectasia-like disorder (ATLD) patients show progressive cerebellar ataxia. ATM, mutated in AT, can be activated in response to oxidative stress as well as DNA damage, which could be linked to disease-related neurodegeneration. However, the role of MRE11 in oxidative stress responses has been elusive. Here, we showed that MRE11 could participate in ATM activation during oxidative stress in an NBS1/RAD50-independent manner. Importantly, MRE11 was indispensable for ATM activation. We identified FXR1 as a novel MRE11-binding partner by mass spectrometry. We confirmed that FXR1 could bind with MRE11 and showed that both localize to the cytoplasm. Notably, MRE11 and FXR1 partly localize to the mitochondria, which are the major source of cytoplasmic reactive oxygen species (ROS). The contribution of FXR1 to DNA double-strand break damage responses seemed minor and limited to HR repair, considering that depletion of FXR1 perturbed chromatin association of homologous recombination repair factors and sensitized cells to camptothecin. During oxidative stress, depletion of FXR1 by siRNA reduced oxidative stress responses and increased the sensitivity to pyocyanin, a mitochondrial ROS inducer. Collectively, our findings suggest that MRE11 and FXR1 might contribute to cellular defense against mitochondrial ROS as a cytoplasmic complex.
INTRODUCTION
Endogenous and exogenous genotoxic stresses cause different types of DNA damage. Among them, DNA double-strand breaks (DSB), which are often generated by exposure to ionizing radiation (IR), are among the most serious for mammalian cells. Hence, cells have developed DNA damage responses (DDRs) such as cell cycle checkpoints and DNA repair mechanisms. DSBs are mainly repaired by non-homologous end-joining (NHEJ) and homologous recombination (HR) in eukaryotes, with NHEJ used preferentially in higher eukaryotes such as mammals [1]. Radiation-hypersensitive disorders have been useful to identify the underlying mechanisms of DSB damage responses following IR. One of the best known of these is ataxia-telangiectasia (AT). Cells derived from AT patients commonly show radiation hypersensitivity, radio-resistant DNA synthesis (RDS) and chromosome aberrations [2, 3]. The gene responsible for AT is ATM, whose product ATM is a protein kinase [2, 3]. ATM is an inactive homodimer in the absence of DNA damage, but DSBs trigger its auto-phosphorylation and subsequent dimer dissociation. An important DDR complex, MRE11/RAD50/NBS1 (MRN), binds to ATM monomer and promotes its recruitment to DSB damage sites. ATM then phosphorylates several DDR factors, such as p53, Chk2 and SMC1, in turn activating cell cycle checkpoints [3]. The typical RDS phenotype in AT is due to a defect of the cell cycle checkpoint in S phase.
Genetic defects in NBS1 or MRE11 also show radiation hyper-sensitive phenotypes [3]. NBS1 is mutated in Nijmegen breakage syndrome (NBS), while MRE11 is mutated in ataxia-telangiectasia-like disorder (ATLD). These genetic disorders show cellular phenotypes similar to AT, suggesting common roles for DDR following IR. The product of NBS1 has several direct-interaction domains to DDR proteins like ATM and MRE11, thus facilitating DDRs. The product of MRE11 possesses DNA nuclease activity, which is important for the initial step of HR repair that might lead to chromosome aberrations if defective. Thus, research on these radiation-hypersensitive disorders revealed that the underlying cause of their cellular phenotype was DDR.
Although AT, NBS and ATLD share a similar cellular phenotype, some of their clinical manifestations are distinct, particularly the neurodegeneration phenotypes. AT and ATLD patients show progressive cerebellar ataxia, whereas almost all NBS patients show microcephaly [3]. However, the genes involved in cell cycle checkpoints and DSB repair are not accountable for the neurodegeneration phenotypes. Ataxia–oculomotor apraxia (AOA) is a phenotype related to cerebellar ataxia and is also found in AOA1, AOA2 and AOA3 along with AT and ATLD [4]. Aprataxin, the product of the gene causing AOA1, participates in DNA single-strand break repair [5, 6] while senataxin, the gene product responsible for AOA2, is crucial to resolve DNA–RNA hybrid formation (R-loop) in transcript-related DNA damage [7]. In AOA3, cytochrome b is mutated, and the patient-derived cells show abnormal mitochondrial dynamics [8, 9]. Although ATM kinase activation is dependent on DSB damage generation, it can also occur following oxidative stress [3, 10]. Guo et al. reported that oxidative stress by H2O2 treatment causes ATM activation in vitro and in vivo, likely due to the formation of disulfide bonds causing conformational change of the ATM dimers [10]. Neural stem cells are known to be sensitive to oxidative stress, and neural cells from ATM-deficient mice show decreased viability. However, radical scavenger treatment recovers the viability of ATM-deficient cells [11, 12], suggesting that ATM function is pivotal to resist oxidative stress in vitro but might be indispensable for the viability of neural cells in the cerebellum. Furthermore, we reported that AOA3-patient cells showed excessive accumulation of ROS (reactive oxygen species), particularly the mitochondria-related ROS superoxide, which perturbed ATM-dependent phosphorylation [8, 9]. We also showed that induction of superoxide by pyocyanin treatment repressed ATM-dependent phosphorylation. Collectively, evidence suggests that the oxidative stress caused by ATM function defects might lead to neurodegeneration phenotypes in AOA3 cells.
As ATLD and AT patients show similar neurodegeneration phenotypes, we hypothesized that ATLD cells harbor defects in ATM kinase and that MRE11 may be important for ATM activation upon oxidative stress. We showed here that MRE11 participates in ATM regulation in response to H2O2- or pyocyanin-induced oxidative stress. We identified FXR1 as a novel cytoplasmic MRE11-binding partner and showed that it also participates in the oxidative stress response. Finally, we discuss the role of MRE11 and FXR1 in cellular response against oxidative stress.
MATERIALS AND METHODS
Cells and culture
HeLa, U2OS, hTERT-immortalized human fibroblasts (48BR), SV40-transformed normal fibroblast (MRC5SV), SV40-transformed ATLD patient-derived fibroblasts (ATLD2SV and HMfibroSV) and SV40-transformed AT patient-derived fibroblasts (AT5BIVA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and antibiotics [9, 13, 14]. Mutated MRE11 in ATLD2SV has a nonsense mutation and expresses C-terminal-truncated MRE11 protein, and mutated MRE11 in HMfibroSV has a missense mutation, in which MRE11 protein is unstable. Normal donor, AT, ATLD and NBS patient-derived lymphoblastoid cells were cultured in RPMI1640 (Sigma-Aldrich) supplemented with 10% FBS (Invitrogen) and antibiotics [9].
Generation of GFP-FXR1-expressing cells
Human FXR1 (FBL, NM_005087.4) and MRE11 (MRE11, NM_005591) were amplified from a human fetal cDNA library (Clontech) by PCR with Pyrobest DNA polymerase (TAKARA). Then, FXR1 cDNA was inserted into pEGFP-C1 (Clontech) vectors, and MRE11 cDNA was inserted into pCMV-Tag2B-FLAG (Promega) vectors; the insertions were confirmed by DNA sequencing.
siRNA knockdown experiments
Sub-confluent cells were plated on culture dishes for 24 h and then transfected with siRNAs targeting MRE11 (B-Bridge International Inc.), NBS1 (B-Bridge International Inc.), RAD50 (B-Bridge International Inc.) and FXR1 (Qiagen Co.), or with negative control siRNA (B-Bridge International Inc.) using Lipofectamine RNAiMax (Invitrogen Life Technology). Cell were re-plated after 1 day and used for each experiment or immunofluorescence the next day.
Antibodies
The following antibodies were used in this study: mouse monoclonal anti-γH2A histone family X (H2AX) [#05–636], anti-RPA32 [NA-19 L6], anti-RPA70 [NA-18] and rabbit polyclonal anti-H2B [#07–371] (Merck Millipore); rabbit polyclonal anti-FXR1 [A300-892A], anti-phospho-RPA32 [A300-246A], anti-phospho-SMC1 (S966) [A300-050A], anti-SMC1 [A300-055A], anti-RAD50 [A300-184A] and anti-phospho-KAP1 [A300-767A] (Bethyl Laboratories Inc.); rabbit polyclonal anti-phospho-Chk2 (T68) [#2661], anti-phospho-Chk1 (S317) [#2344], anti-phospho-pRad17 (S645) [#6981], anti-phospho-p38MAPK [#4511] and rabbit polyclonal anti-TOMO20 [11802–1-AP] (Proteintech); mouse monoclonal anti-p53-pS15 [#9286] (Cell Signaling Technology); rabbit polyclonal anti-NBS1 [NB100–143] and anti-MRE11 [NB100–142] (Novus Biologicals); rabbit polyclonal anti-phospho-ATM [ab8192] (Abcam); mouse monoclonal anti-KAP1 [GTX49179], anti-NBS1 [GTX70222], anti-MRE11 [GTX70212] and anti-RAD50 [GTX70282] (GeneTex); mouse monoclonal anti-beta-actin [A53169] (Sigma-Aldrich); mouse monoclonal anti-p53 [sc-126] and anti-ATM [sc-23992] (Santa Cruz); mouse polyclonal anti-RAD51 [70–001] (Bioacademia); and mouse monoclonal anti-GST [27–4577-01] (GE Healthcare).
Immunoprecipitation, western blotting and immunofluorescence
Immunoprecipitation was performed as reported previously [13] by incubating the samples with rabbit polyclonal anti-MRE11 and anti-NBS1 antibodies (Novus) or anti-FXR1 rabbit polyclonal antibody (Bethyl). Co-immunoprecipitated proteins were detected by western blot analysis.
Western blotting of whole cell extract was performed as described previously [13]. Target proteins were detected with the primary antibodies listed above and secondary horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (GE Healthcare) and visualized using the ECL plus chemiluminescence system (GE Healthcare).
Immunofluorescence staining was carried out as described previously [15] using Alexa-488-conjugated anti-rabbit or Alexa-594-conjugated anti-mouse IgG (Molecular Probes) to visualize the localization of target proteins.
ATM kinase assay
The ATM kinase assay was carried out as described previously [9]. ATM was immunoprecipitated from whole-cell extracts using an anti-ATM antibody (Calbiochem). The phosphorylation of p53 (substrate) by immunoprecipitated ATM was estimated by western blot analysis using anti-phospho-p53 (S15) antibody (Cell Signaling) and mouse monoclonal anti-GST (GE Healthcare).
HR activity analysis
HR activity analysis was carried out as described previously [9,13]. To measure the HR repair of I-SceI-generated DSBs, 50 μg of the I-SceI expression vector (pCBASce) was introduced into 106 U2OS-DRGFP cells by electroporation (GenePulser; Bio-Rad). To determine the amount of HR repair activity, the percentage of GFP-positive cells was quantified 3 days post-electroporation by using a flow cytometer (FACSCalibur; Becton Dickinson).
Mass spectrometry analysis
293E cells were transfected with the generated pCMV-Tag2B-FLAG MRE11 plasmids using FugeneHD (Promega) and harvested after 2 days. Cytoplasmic and nuclear fractions were extracted from the harvested cells and were used for immunoprecipitation with anti-FLAG antibody (Sigma). The candidate MRE11-binding proteins in immunoprecipitates were identified by a HPLC-mass spectrometer (AB SCIEX Co.).
Propidium iodide staining to detect cell cycle distribution and apoptosis
Cells were treated with camptothecin (CPT). At the time points indicated in Fig. 3D, cells were harvested, fixed using 70% ethanol, and then incubated at −20°C overnight. Fixed cells were treated with RNase (5 mg/ml) and stained with propidium iodide (PI; 50 μg/ml). The apoptotic fractions (sub-G1) were quantified using a flow cytometer.
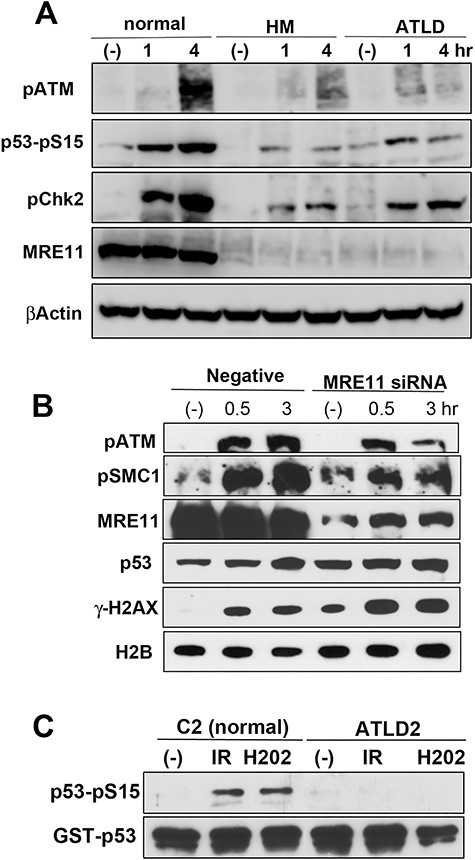
MRE11 participates in ATM activation following oxidative stress. (A) MRC5SV (normal), MRE11-defective HMfibroSV (HM) and ATLD2SV (ATLD) cells were treated with H2O2 (100 μM) for the indicated times and analysed using the indicated antibodies. (B) U2OS cells were transfected with MRE11 siRNA. After 2 days, these cells were treated with H2O2 (100 μM) for the indicated times and analyzsd using the indicated antibodies. (C) Normal (C2ABR) or ATLD2 (ATLD2ABR) lymphoblastoid cells were γ-ray-irradiated (10 Gy) or treated with 100 μM H2O2. After 1 h, cells were harvested and a kinase activity assay was measured using the immunocomplexes with an anti-ATM antibody. Phosphorylation was detected by western blot analysis using an anti-phospho-p53 antibody. The amount of substrate in the reaction was confirmed using an anti-GST antibody.
RESULTS
MRE11 participates in ATM activation following oxidative stress
To clarify the interplay of ATM and MRE11 in oxidative stress responses, we firstly investigated ATM-dependent phosphorylation following H2O2 treatment in MRE11-defective ATLD patient fibroblasts (Fig. 1 and S1A, see online supplementary material). H2O2 treatment induced auto-phosphorylation of ATM in normal cells and increased the phosphorylation of ATM substrates SMC1, Chk2 and p53 (Fig. 1A and S1A). However, γ-H2AX, the marker of DNA DSBs, did not increase remarkably (Fig. S1A), suggesting that this treatment did not markedly cause the generation of DSB. Intriguingly, in ATLD-patient cells (HMfibroSV and ATLD2SV cells), ATM auto-phosphorylation and phosphorylation of ATM substrates (SMC1, Chk2 and p53) did not markedly increase after the treatment (Fig. 1A). Next, we examined the effect of MRE11 knockdown (Fig. 1B). MRE11-depleted cells showed repression of ATM auto-phosphorylation and SMC1 phosphorylation. These results suggest that ATM activation in response to oxidative stress is defective in MRE11-lacking cells. In contrast, NBS1 might be dispensable for ATM activation, because ATM-related phosphorylation increased normally in NBS1-defective patient cells after H2O2 treatment, but not in AT patient cells (Fig. S1B). Knockdown of RAD50 did not perturb ATM-dependent phosphorylation, showing that RAD50 is also dispensable for ATM activation (Fig. S1C). To elucidate the functional role of MRE11 in ATM activation upon oxidative stress, we tested ATM activity in vitro by analysing phosphorylation of the substrate p53 (Fig. 1C). Anti-ATM immunoprecipitates from normal cells after irradiation or H2O2 treatment showed full activity, while immunoprecipitates from ATLD2 cells did not, suggesting that MRE11 might be indispensable for ATM activation following oxidative stress. Mitochondria generate and accumulate superoxide, and ATM is known to localize in mitochondria to function in oxidative stress response [16]. Hence, we investigated whether MRE11 is localized to mitochondria. We prepared the mitochondrial fraction using a Mitochondria Isolation Kit (Thermo Scientific) and confirmed the successful purification with the antibody against the mitochondrial marker TOM20 (Fig. S1D; left panel). Fig. S1D (right panel) shows that MRE11 was detected in the mitochondrial fraction in normal and AT cells. Taken together, MRE11 might partly localize to the mitochondria, independently of ATM, and function in ATM activation upon oxidative stress.
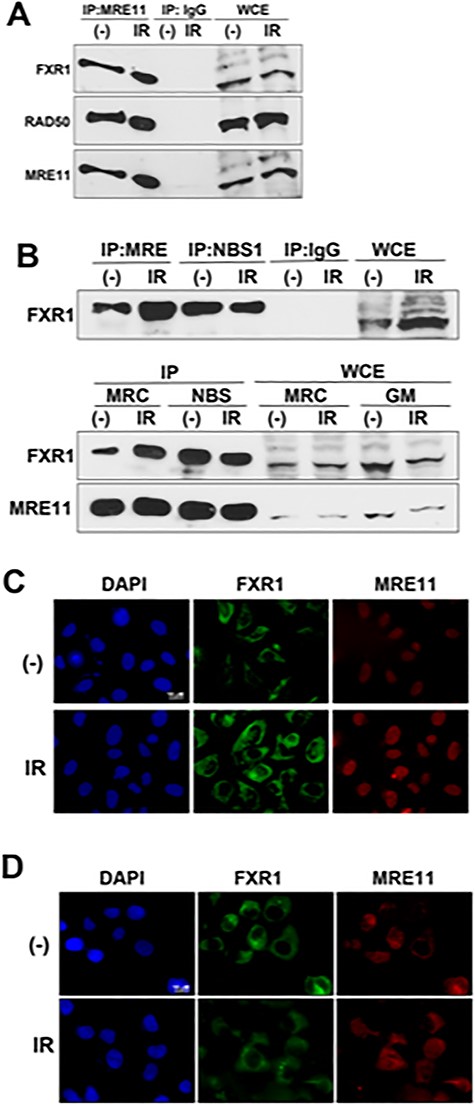
FXR1 interacts with MRE11. Extracts from C3ABR (A and B: upper panel), or MRC5SV (B: lower panel) and NBS1-defective NBS cells (B: lower panel) without (−) or with 10 Gy of IR were immunoprecipitated with anti-MRE11 antibody (Novus), anti-NBS1 antibody (Novus) or normal IgG, and then the immuno-complexes were detected by western blot analysis using the indicated antibodies. WCE = Whole cell extract. (C and D) Cytoplasmic distribution of FXR1. U2OS cells (C) or NBS cells (D) were irradiated with 5 Gy of IR. After 0.5 h, the cells were fixed and immuno-staining was performed using anti-FXR1 (Bethyl) and anti-MRE11 antibody (GeneTex).
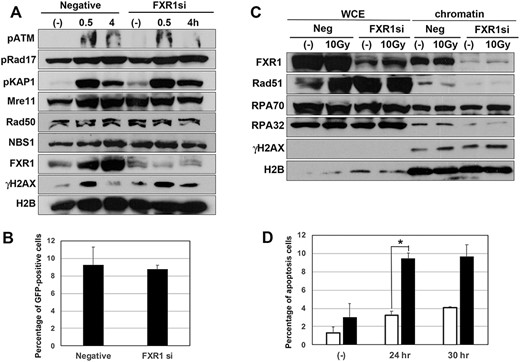
FXR1 may be dispensable for DSB damage responses. (A) HeLa cells were transfected by FXR1 siRNA or negative control siRNA, and after 2 days these cells were treated with 5 Gy of IR and analysed by western blot using the indicated antibodies. (B) U2OS-DRGFP cells were transfected by FXR1 siRNA or negative control siRNA, and after 2 days DR-GFP assay was performed to measure HR activity. HR activity level was measured as the percentage of GFP-positive cells by flow cytometery. (C) FXR1 may play a role in chromatin accumulation of HR factors. U2OS cells were transfected with FXR1 siRNA or negative control siRNA, and after 2 days these cells were irradiated with 10 Gy of γ-ray. After 4 h their cells were harvested and their chromatin fractions were prepared. Chromatin accumulation of DDR proteins was detected by western blot analysis using the indicated antibodies. WCE = Whole cell extract. (D) HeLa cells were transfected with FXR1 siRNA (closed column) or negative control siRNA (open column), and after 2 days their cells were treated with camptothecin (10 μM), then the percentage of apoptosis in cells was measured by flow cytometery; *P < 0.05.
FXR1 is a novel MRE11-binding partner protein
MRE11 functions in DDRs such as HR repair and ATM-dependent cell cycle checkpoints as a component of the MRN complex, but NBS1 might be dispensable for ATM activation following oxidative stress (Fig. S1B). Hence, we speculated that MRE11 forms another complex for oxidative stress responses and we tried to identify novel binding partners for MRE11 using mass spectrometry. To enrich for MRE11-containing complexes, we performed immunoprecipitation from cytoplasmic and nuclear fractions of 293E cells, transfected with either wild-type MRE11 (WT), or a mutated MRE11 (329 T) that was expressed in the ATLD patient cell line (HMfibroSV) [14]. Mass spectrometry analysis identified >30 candidate MRE11-binding proteins from the cytoplasmic or nuclear fractions (Fig. S2A, see online supplementary material). As ROS are mainly generated from mitochondria in the cytoplasm, we focused on a candidate (of ~60 kDa) from the cytoplasmic fraction (Fig. S2B). The identified protein is FXR1, with an almost unknown role in the stress responses against ROS. We next verified the interaction between FXR1 and MRE11 with immunoprecipitation and western blot analysis. As shown in Fig. 2A, anti-MRE11 antibody precipitated FXR1 as well as RAD50, a component of the MRN complex, independently on irradiation. Anti-NBS1 antibody also precipitated FXR1 (Fig. 2B; upper panel). However, interaction between MRE11 and FXR1 was also observed in NBS1-lacking patient cells, and seemed higher than in normal cells (Fig. 2B; lower panel), suggesting that NBS1 might be dispensable for this interaction. We next examined the localization of FXR1 with immunofluorescence. MRE11 is known to localize mostly in the nucleus [17]. FXR1 localized in the cytoplasm independently of irradiation and was not detected in the nucleus in normal cells (Fig. 2C). FXR1 remained cytoplasmic following H2O2 or pyocyanin treatment also (Fig. S3A, see online supplementary material). While NBS1 possesses a nuclear localization signal (NLS), MRE11 does not. Thus, MRE11 localizes to the nucleus upon formation of the MRN complex [17]. Consistently, NBS1-lacking patient cells showed localization of most MRE11 in the cytoplasm, as well as FXR1, independently of irradiation (Fig. 2D). Moreover, oxidative stress treatment did not influence cytoplasmic localization of FXR1 in NBS cells (Fig. S3B). These findings suggest that FXR1 might interact with MRE11 in the cytoplasm independently of NBS1. However, as anti-NBS1 antibody co-precipitated FXR1 with MRE11 (Fig. 2B upper panel) and as a small portion of NBS1 is known to distribute into cytoplasm in normal cells, NBS1 may participate in the FXR1/MRE11 complex.
FXR1 may be dispensable for DSB damage responses
As MRE11 is crucial for HR repair and ATM activation, we next investigated the role of FXR1 in these DDRs. We verified the efficiency of FXR1 knockdown in U2OS (Fig. S4A, see online supplementary material) and HeLa cells (Fig. 3A) and showed that effective reduction of FXR1 did not alter either MRE11 or RAD50 expression in these cell lines. FXR1-depleted HeLa cells showed normal ATM auto-phosphorylation, ATM-dependent phosphorylation of KAP1 and γ-H2AX 0.5 h after irradiation, and ATR-dependent phosphorylation of Rad17. Similar results were observed in U2OS cells (Fig. S4B). These findings suggest that FXR1 is dispensable for ATM- and ATR-dependent phosphorylation. We next investigated HR repair activity in the knocked-down cells using a DR-GFP assay. Transfection of I-SceI expression plasmid to U2OS-DRGFP cells induces GFP expression with about 10% positive cells. As shown in Fig 3B, FXR1 depletion did not influence the percentage of GFP-positive cells. As a marker of HR activation, we examined the accumulation of HR factors by western blotting (Fig. 3C). The accumulation of HR factors such as Rad51 and RPA did not increase after irradiation, as most of DNA damage by IR is repaired by NHEJ; however, depletion of FXR1 abolished the accumulation of Rad51 and RPA32, both with and without irradiation. Treatment with CPT, a topoisomerase I inhibitor, causes single-strand breaks and subsequent replication stress in S phase [18]. The replication stress could lead to the generation of DSBs. Many such DSBs are repaired by HR and HR-defective cells could show sensitivity to CPT. Here, CPT treatment increased cell death in FXR1 knocked-down cells (Fig. 3D). Both the abnormality in chromatin accumulation of HR factors and the sensitization to CPT in FXR1-depleted cells suggests that FXR1 may partially contribute to HR repair.
FXR1 participates in the oxidative stress response
We next investigated the role of FXR1 in the oxidative stress response. As ROS are mainly generated by mitochondria, we used the mitochondrial electron transfer inhibitor pyocyanin to induce oxidative stress [9]. In control cells, the oxidative stress pathway was activated, as shown by increased phosphorylation of p38MAPK 4 h after addition of pyocyanin, and markers associated with ATM-dependent phosphorylation, such as p53 and γH2AX, also showed normal induction (Fig. 4A). However, these responses were repressed in FXR1-depleted cells, suggesting that FXR1 might be necessary for oxidative stress responses including the ATM-dependent pathway. We then examined ATLD cells treated with pyocyanin and found that dysfunction of MRE11 perturbed ATM-dependent phosphorylation as well as phosphorylation of p38MAPK (Fig. 4B). However, depletion of NBS1 by siRNA did not abolish ATM-dependent phosphorylation in pyocyanin-treated cells (Fig. S5A, see online supplementary material). Finally, if FXR1 is involved in oxidative stress responses, depleting it may sensitize the cells to pyocyanin treatment. To verify this, we analysed FXR1-depleted U2OS cells treated with pyocyanin (Fig. S5B). As shown, the viability of FXR1-depleted cells was reduced to almost 60% after 24 h of treatment, while in control cells it remained unchanged. From these results, we infer that FXR1 might play a role in cellular responses against oxidative stress.
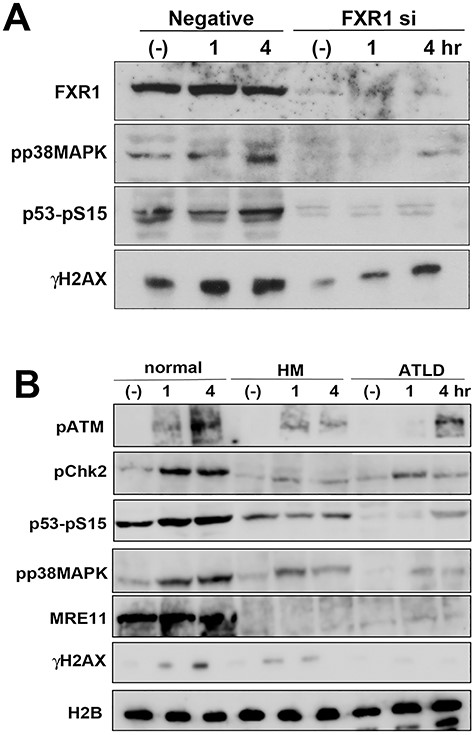
FXR1 participates in the oxidative stress responses. (A) U2OS cells were transfected with FXR1 siRNA or negative control siRNA, and after 2 days these cells were treated with 50 μM of pyocyanin for the indicated times. Whole cell extracts from the harvested cells were analysed by western blot using the indicated antibodies. (B) MRC5SV (normal) and MRE11-defective ATLD cells (HMfibroSV or ATLD2SV) were treated with 50 μM pyocyanin for the indicated times and whole cell extracts from the harvested cells were analysed by the indicated antibodies.
DISCUSSION
Both AT and ATLD patients show a similar neurodegeneration phenotype, such as progressive cerebellar ataxia, but the molecular mechanisms of pathogenesis have been elusive. ATM is activated in response to H2O2-induced oxidative stress [10], and defects in its activity were suggested to sensitize neural cells to oxidative stress, leading to ataxia. The role of MRE11 in DDR such as HR repair and the ATM-dependent cell cycle checkpoint has been studied in detail. However, whether MRE11 is associated with oxidative stress responses along with ATM was unknown. Here, we showed that MRE11 is indispensable for ATM activation and ATM-dependent phosphorylation during oxidative stress (Fig. 1). Notably, both NBS1 and RAD50 were dispensable for ATM activation (Fig. S1B, C). We also identified FXR1 as a novel MRE11-binding partner (Fig. S2A and B) and showed that FXR1 could bind with MRE11 in an NBS1-independent manner (Fig. 2A and B). Furthermore, we also found that they localize in the cytoplasm and accumulate in the mitochondrial fraction (Fig. 2C and D and Fig. S1D, S3A and B). Although MRE11 is an important factor for DNA damage responses, particularly HR repair, we found that FXR1 may partially contribute to HR repair, but not to ATM activation in response to DSB damage (Figs 3 and S4).
Until now, the functions of MRE11 in the cytoplasm were less characterized. Kondo et al. reported that cytoplasmic MRE11 functions as a sensor of cytosolic double-strand DNAs (dsDNAs), which are caused by viral or bacterial infection and activate an interferon β-related inflammatory response through the STING pathway [19]. NBS patient cells also show amplification of these responses, suggesting that NBS1 might not be necessary for detection of cytosolic dsDNA. As NBS cells show cytoplasmic localization of MRE11 [17], the functions of cytoplasmic MRE11 could be specific and distinct from its nuclear functions, such as regulation of HR repair and ATM activation. Remarkably, AT patients and Atm-knockout mice show oxidative stress-related abnormalities in neural cells [11, 12, 20], and ATLD patients show oxidative stress accumulation in several brain cell types [21]. We show here that MRE11 localizes to mitochondria (Fig. S1D), where most cytoplasmic ROS are generated. Together, these data support a novel role of MRE11 in the cytoplasm against oxidative stress, through ATM activation.
FXR1 is an RNA-binding protein with high homology to FMR1, the main factor responsible for fragile X syndrome [22]. Both FXR1 and FMR1 directly bind to the Cdc42 effector PAK1 through their KH(2) domain [23]. Cdc42 was reported to participate in the oxidative stress response in cultured neuronal cells [24]. Fragile X syndrome patients show various brain dysfunctions including mental retardation, Parkinson’s disease, progressive cerebellar ataxia and cognition disorder [25]. Moreover, Fmr1-knockout mice show mitochondrial fragmentation by impaired mitochondrial fusion and increased oxidative stress in immature neurons [26]. Hence, Fragile X syndrome-related FMR1 and FXR1 might play a role in defense against oxidative stress, possibly through mitochondria, in brain cells. Importantly, we found that depletion of FXR1 reduced oxidative stress-related responses (Fig. 4A) and increased sensitivity to pyocyanin treatment (Fig. S5B). Moreover, MRE11 might also be indispensable for ATM activation in response to mitochondrial ROS accumulation (Fig. 4B). Therefore, complex formation between MRE11 and FXR1 may be indispensable for cellular responses against mitochondria-related oxidative stress.
The role of FXR1 in the regulation of mRNA turnover through its RNA-binding domain has been reported [27, 28]. In FXR1-overexpressing cells, FXR1 bound to and destabilized p21 mRNA, causing a decrease in its levels [28]. Furthermore, depletion of FXR1 in mouse adult neural stem cells caused deficient neural differentiation along with p21 reduction, but restoration of p21 mRNA levels partially rescued this abnormality [29]. MRE11 also shows mRNA-related regulatory roles. Inhibition of transcription progression following damage of template DNA generates R-loop. As this can repress completion of transcription leading to genomic instability, mechanisms to suppress R-loop also exist [30]. MRE11 was reported to cooperate with the Fanconi anemia pathway for suppression of R-loop [31]. R-loop accumulation was also reported in brain cells of fragile X syndrome and AOA2, and sSenataxin is also associated with R-loop suppression [7]. Although the relationship of FXR1 with R-loop is unknown, accumulation of R-loop in neural cells could contribute to neurodegeneration phenotypes such as cerebellar ataxia. Therefore, the functional interaction between MRE11 and FXR1 in R-loop suppression and their relationship with progressive cerebellar ataxia, in addition to oxidative stress responses, remain to be clarified.
We here identified the novel MRE11-binding protein FXR1, which could participate in the oxidative stress response with MRE11. As a possible cause of cerebellar ataxia might be related to oxidative stress, we need to clarify in detail the functional interaction between MRE11 and FXR1 in oxidative stress responses. However, we have to investigate their interaction in neural cells, because the sensitivity of neural cells to ROS is different from that of culture cells such as fibroblasts [11, 12]. Such research might supply significant clues to clarify the pathogenic mechanisms of other forms of neurodegeneration.
CONFLICT OF INTEREST
None declared.
FUNDING
This work was supported by JSPS KAKENHI (grant numbers JP25550026 and JP18H04978) and in part by the NIFS Collaborative Research Program (NIFS17KOCA002).

{kind=link}
{kind=link}
{kind=link}
{kind=link}